

Abstract
OBJECTIVE:
TNFR1 plays a major role in rheumatoid arthritis (RA). Here we explore the relative importance of TNFR1 signaling in the hematopoietic tissue compartment for disease progression.
METHODS:
DBA/1 mice were lethally irradiated and rescued with bone marrow derived from either DBA/1 or TNFR1−/− animals. The mice were then input into the collagen induced arthritis (CIA) model and disease progression characterized.
RESULTS:
Surprisingly, TNFR1−/− transplant mice input into the CIA model develop increased disease as compared to controls. This could not be attributed to either an increased primary response to collagen or to the contribution of a non-DBA genetic background. Histological markers of advanced disease were evident in TNFR1−/− transplant mice shortly after initiation of the immune response to collagen and long before clinical evidence of disease. Serum TNFα was undetectable while serum IL-12p40 levels were increased in TNFR1−/− transplant mice at the end point of the study.
CONCLUSION:
These data raise the intriguing possibility of the existence of an anti-inflammatory TNFR1 mediated circuit in the hematopoietic compartment. This circuit bears a resemblance to emerging data delineating a switch in TNFα function observed in the resolution of bacterial infections. These data suggest that TNFR1 mediated signals in the radio-resistant tissues contributes to disease progression while TNFR1 mediated signals in the radio-sensitive tissues can contribute to protection from disease. We thus put forward the hypothesis that the degree of responce to TNFα blockade in RA is dependent, in part, on the relative genetic strengths of these two pathways.
Introduction
TNFα has long been appreciated as a plieotropic cytokine which plays important roles in homeostasis and inflammation. Indeed TNFα blockade now serves as a front line therapy for the treatment of rheumatoid arthritis (RA) (1). There are limitations to this therapy, however. Initial clinical trials of Etanercept, Infliximab, and Adalimumab, performed on patients who had failed earlier therapies resulted in ACR50 scores ranging from 22% - 40% of the population (1). More recently, the PREMIER study has demonstrated that early treatment results in improved response however a significant number of patients remain non-responders (2). Additionally, this therapy has been associated with a decreased ability to respond to bacterial infections as well as an increased incidence of anti-nuclear antibody production. In part these limitations may reflect the complexity of TNFα biology.
There exist two TNFα receptors; TNFR1 and TNFR2, which are widely expressed. Macrophages and NK cells express roughly equal quantities of both receptors (3, 4), while fibroblasts and epithelial cells express relatively more TNFR1 and activated lymphocytes express relatively more TNFR2 (5, 6). Consequently, TNFα mediates its effects through varying combinations of receptor signaling in different tissues. TNFα contributes to pathogenesis in RA synovial tissue through the induction of numerous inflammatory mediators which contribute to tissue destruction (7). Additionally, TNFα functions to modulate the immune response by recruiting and activating APCs and phagocytic cells, optimizing antibody responses, enhancing T cell priming, and promoting T cell proliferation (8).
Manipulation of TNFα expression or the expression of its receptors has a complex effect on autoimmune disease. Overexpression of human TNFα in mice results in the appearance of a spontaneous arthritis (9). Conversely, chronic treatment with low dose TNFα in either the NZW/B model of SLE or the NOD model of type I diabetes results in inhibition of autoimmunity (10, 11). If, however, TNFα expression is localized to the neonatal pancreas of NOD mice, diabetes is exacerbated (12). Similarly in multiple sclerosis (MS) and its animal models, TNFα plays a complex role. TNFα has been implicated in experimental autoimmune encephalomyelitis (EAE) disease progression in a number of studies (13-15). Treatment of MS patients by TNFα blockade, however, results in a worsening of the disease (16). This may correlate with the observation that within both MS and EAE, myelin reactivity regresses over time to be replaced by reactivity to other epitopes (17); a process recently found to require TNFα (18). Thus it appears that within the progression of several autoimmune diseases the effects of TNFα are strongly dependent on time and place.
The Collagen Induced Arthritis (CIA) model has proven to be a useful model in which to study TNFα signaling interactions. Mice expressing the H-2q MHC allele develop an inflammatory synovitis of the paws after injections of type II collagen (CII) in the presence of complete Freunds adjuvant (CFA), with synoviocyte proliferation, pannus formation, and cartilage destruction strikingly similar to that seen in RA (19). In this model, disease is both T and B cell dependent. Additionally, immune complex formation plays an important role as FcγR-deficient mice are protected from CIA (20). The role of TNFα was confirmed when administration of TNFα was found to exacerbate disease while administration of a TNFα blocking antibody protected against disease (21, 22). Given these results it was quite surprising that TNFR1 knockout mice developed a TNFα independent CIA with an incidence and pathology indistinguishable from control mice (23). These data would suggest that in the absence of TNFR1, mice undergo a developmental compensation leading to a relative increase in TNFα-independent inflammatory circuits.
In RA patients undergoing TNFα blockade therapy, short lived hematopoietic cells develop in a manner quite similar to what is observed in TNFR1−/− mice (24) and yet RA is ameliorated in responsive patients. Thus we would predict that while disruption of TNFR1 signaling in the entire mouse results in developmental compensation, disruption of TNFR1 signaling in the hematopoietic system alone would not. If TNFR1 signaling within hematopoietic cells plays an important role in disease progression we would further predict that selective disruption of TNFR1 in this compartment will result in decreased disease. Here we test this hypothesis by employing a bone marrow transplant system in which we sequester TNFR1 signaling to the radio-resistant tissue compartment. Using this system, we have found that surprisingly, TNFα signaling within the hematopoietic compartment does not promote disease progression, but rather, functions as a novel protective cytokine circuit.
Materials and Methods
Mice
DBA/1 LacJ mice and B6.129-Tnfrsf1atm1Mak (TNFR1−/−) mice were purchased from Jackson Labs (Bar Harbor, ME). The TNFR1 gene-targeted (−) allele was backcrossed 6 times into the DBA/1 LacJ background and then crossed to produce homozygous (TNFR1−/−) animals. Additionally, 6th backcross TNFR1+/− animals were used for certain control experiments.
PCR
The TNFR1 gene-targeted allele was identified by PCR using a protocol provided by Jackson Labs. DNA was extracted from tail tips using the DNeasy kit according to the manufacturer's instructions (Qiagen, Valencia, CA). Primers for amplifying the 470 bp wild type allele and 300 bp knockout allele were: olMR448, TGTGAAAAGGGCACCTTTACGGC (wild type); olMR449, GGCTGCAGTCCACGCACTGG (shared), and olMR450, ATTCGCCAATGACAAGACGCTGG (HSV-TK, knockout). PCR was performed with the initial conditions of melting at 94 °C, annealing at 64 °C, and extending at 72 °C, with a continual drop of 0.5 °C in the annealing temperature with each cycle until an annealing temperature of 58 °C was reached. An additional 12 cycles with annealing at 58 °C was then performed followed by a final extension of 2 min.
Bone Marrow Transplantation
Male mice were used for all experiments. Recipient groups aged 5 – 10 weeks (age matched to within 14 days) were irradiated with 1200 cGy using a 60Co source as a split dose separated by 4 hours. The mice were then rested overnight. The next morning, donor mice were sacrificed and femurs removed. Bone marrow was harvested from femurs by flushing 3 times in PBS supplemented with 1% FCS and filtering through a cell strainer. Cells were washed in PBS, counted by hemacytometer, and resuspended at a concentration of 107 cells/ ml. Tail vein injections of 106 donor cells (100μl) were then given to each of the recipient group and the animals rested for periods of time ranging from 5 – 12 weeks.
Collagen Induced Arthritis
Bovine type II collagen (CII, Elastin products, Owensville, MO) was prepared by dissolving to a concentration of 4 mg/ml in 0.01 M acetic acid at 4 °C overnight with tumbling rotation. The solution was then filter sterilized, aliquoted, and stored at −80 °C. Aliquots of collagen were thawed slowly on ice. Adjuvant was prepared by combining Mycobacterium tuberculosis (H37Ra; Difco, Detroit, MI), and Freunds incomplete adjuvant (Difco, Detroit, MI) to a concentration of 4 mg/ml. Emulsion was prepared by combining equal volumes of CII and adjuvant and stored on ice until use. Mice were anesthetized and injected intradermally with 100 μl emulsion at the base of the tail. A successful injection resulted in a white blister just under the skin. After 21 days, the process was repeated. Clinical arthritis was scored as described (25) using a scale of 0 – 4 for each paw by a trained observer (CWS, TR, or RIS) blinded to the treatment group. For any paw; 0 = no visible disease, 1 = edema or erythema of 1 joint, 2 = edema or erythema of 2 joints, 3 = edema or erythema of more than two joints, 4 = severe arthritis involving the entire paw and digits. The scores of the 4 paws are then added to create an arthritis score for the animal. Upon completion of the experiment, animals were sacrificed. Blood was obtained by cardiac puncture and paws were dissected and immediately placed in 10% buffered formalin (Biochemical Sciences, Swedesboro, NJ). Fixed paws were decalcified, sectioned, and stained with hematoxylin and eosin (H&E). Scoring for histological disease parameters was performed as described (26). Inflammation is scored as 0: normal, 1: Minimal infiltration of inflammatory cells in synovium and periarticular tissue of affected joints, 2: Mild infiltration, 3: Moderate infiltration with moderate edema, 4: Marked infiltration affecting most areas with marked edema, 5: Severe diffuse infiltration with severe edema. Pannus consists of that subset of infiltration that is localized to cartilage and subchondral bone and is scored as 0: Normal, 1: Minimal infiltration of pannus in cartilage and subchondral bone of marginal zone, 2: Mild infiltration of marginal zone with minor cortical and medullary bone destruction in affected joints, 3: Moderate infiltration with moderate hard tissue destruction in affected joints, 4: Marked infiltration with marked destruction of joint architecture, affecting most joints, 5: Severe infiltration associated with total or near total destruction of joint architecture, affects all joints. Cartilage damage is scored as 0: Normal, 1: Minimal to mild loss of toluidine blue staining with no obvious chondrocyte loss or collagen disruption in affected joints, 2: Mild loss of toluidine blue staining with focal mild (superficial) chondrocyte loss and/or collagen disruption in affected joints, 3: Moderate loss of toluidine blue staining with multifocal moderate (depth to middle zone) chondrocyte loss and/or collagen disruption in affected joints, 4: Marked loss of toluidine blue staining with multifocal marked (depth to deep zone) chondrocyte loss and/or collagen disruption in most joints, 5: Severe diffuse loss of toluidine blue staining with multifocal severe (depth to tide mark) chondrocyte loss and/or collagen disruption in all joints. Bone resorption is scored as 0: Normal, 1: Small areas of marginal zone/periosteal resorption, not readily apparent on low magnification, 2: More numerous areas of marginal zone/periosteal resorption, readily apparent on low magnification, minor overall cortical and medullary bone loss, 3: Obvious resorption of medullary trabecular and cortical bone without full thickness defects in entire cortex, loss of some medullary trabeculae, lesion apparent on low magnification, 4: Full thickness defects in cortical bone, often with distortion of profile of remaining cortical surface, marked loss of medullary bone, 5: Full thickness defects in cortical bone and destruction of joint architecture of all joints. The individual scoring the tissues was blinded to the experimental conditions. A sum of all histological measures is referred to as an all joints sum.
Flow Cytometry
Spleens were dissected and spleen cells prepared as described (27). Cells were stained for TNFR1 using a purified hamster anti-mouse antibody (BD Biosciences, San Jose, CA) as recommended by the manufacturer. Briefly, 106 spleen cells were placed in stain buffer (PBS plus 1% BSA) and incubated with Fc block (24G2, gift of M. Holers) for 15min, washed in stain buffer, incubated 30 min with anti-TNFR1 or isotype control (Purified Hamster IgG1 k, BD Biosciences, San Jose, CA), washed, incubated 30 min with a biotinylated anti-Hamster IgG (BD Biosciences, San Jose, CA), washed, and incubated 30 min with streptavidin PE in the presence of FITC conjugated anti-mouse CD3e (BD Biosciences, San Jose, CA). Following this final incubation, the cells were washed twice and fixed. Fixed cells were then analyzed on a FACScan. CD3+ cells were gated and TNFR1 mean fluorescence intensity (MFI) determined.
Statistical Analysis
Descriptive statistics (mean, SEM) were used to characterize the level of disease for each subject at each measurement. Repeated measures analysis of variance (RM-ANOVA) models were used to test for differences in mean level of disease by treatment group and by time course. The non-parametric Mann Whitney U test was used to compare treatment group means at the endpoint of the experiment. Ninety five percent confidence intervals and a two-tailed alpha level of 0.05 was used for all comparisons. The SPSS statistical software package (SPSS, Chicago, IL, 2005) was used to perform all analyses.
ELISA assays
Serum was obtained by cardiac puncture in animals sacrificed at various times after CII injection. Dilutions of serum were then assessed in triplicate by an enzyme-linked immunosorbent assay (ELISA) specific for total anti-CII IgG as described (28). A standard curve was created using a pool of previously characterized sera from arthritic animals and data expressed as units relative to that standard curve. Serum dilutions were also assayed for IL-12 p40 immunoreactivity using the BD OptEIA Mouse IL-12 p40 ELISA Set (BD Biosciences, San Jose, CA) according to the manufacturer's instructions and for TNFα by ELISA performed by ELISA Tech, LLC (Aurora, CO).
Results
Establishing a bone marrow transplant CIA model
TNFR1−/− mice were backcrossed 6 times into the DBA background and then bred to create homozygous TNFR1−/− DBA mice. The TNFR1 genotype of each mouse was established by PCR as described in Materials and Methods. Arthritis was induced with 2 injections of CII plus CFA as described. Similar to the results of Tada et al. we found that disease incidence and intensity was indistinguishable in TNFR1+/− and TNFR1−/− animals (23) and demonstrates that in the absence of the TNFR1 gene product, mice undergo a genetic compensation event (data not shown).
A minimal lethal irradiation dose of 1200 cGy to destroy bone marrow was established for these animals. DBA/1 mice were then irradiated and rested for 16 hours. Bone marrow was prepared from femurs of either DBA or TNFR1−/− donors as described in Materials and Methods and transplanted to irradiated recipients by tail vein injection to create DBA → DBA mice and TNFR1−/− → DBA mice respectively. We determined that while hematopoietic compartments appeared repopulated by 6 weeks, a minimum of 12 weeks was required for complete reconstitution of disease (data not shown). Thus by irradiating recipients at 1200 cGy, rescuing them with donor bone marrow, and resting them for 12 weeks we are able to induce arthritis of identical clinical intensity to that seen in untransplanted animals.
Development of arthritis in TNFR1−/− → DBA transplant mice
By irradiating DBA mice and rescuing them with TNFR1−/− DBA bone marrow (TNFR1−/− → DBA) we created chimeric mice in which the majority of hematopoietically derived cells could no longer signal through TNFR1 while radio-resistant (primarily non-hematopoietic tissues) such as synovium and vascular endothelium were able to signal through TNFR1 normally. To demonstrate this, we stained spleen CD3+ spleen cells from DBA, TNFR1−/−, and TNFR1−/− → DBA for TNFR1 expression. TNFR1 staining intensity was low but measurable in DBA CD3+ spleen cells as compared to TNFR1−/− CD3+ spleen cells (Figure 1A). CD3+ spleen cells from numerous TNFR1−/− → DBA transplanted mice each produced a staining pattern identical with that of TNFR1−/− mice (Figure 1B). From this we can conclude that the majority of hematopoietic cells in this model derive from the TNFR1−/− donors.
1. Reconstituted spleen cells do not express TNFR1 after TNFR1−/− bone marrow transplant.

Upon termination of CIA experiments in TNFR1−/− → DBA mice, spleens were dissected, spleen cells isolated, and processed for TNFR1 and CD3 staining as described in Materials and Methods. Spleen cells from DBA mice were included as a positive control and spleen cells from TNFR1−/− mice were included as a negative control. A) Representative histograms for TNFR1 staining of CD3+ cells from DBA spleen cells (solid gray), TNFR1−/− spleen cells (dark line), and TNFR1−/− → DBA spleen cells (light line). B) Mean fluorescence intensities were calculated and graphed for each mouse.
Surprisingly, when CIA was induced, we found that these mice developed increased disease as compared to controls (Figure 2). Given the inherent variance in the CIA model, we performed this experiment numerous times to allow statistical analysis. Examples of large and small increases observed in individual experiments are shown in Figures 2A and 2B respectively. Increased disease was observed in TNFR1−/− → DBA mice as compared to either DBA or DBA → DBA controls in 9 out of 9 independent experiments. In Figure 2C endpoint disease intensity measures were analyzed non-parametrically. The entire population of DBA, DBA → DBA, and TNFR1−/− → DBA groups were pooled and the distribution of each pooled population expressed as a box plot. Median endpoint disease intensity in all pooled DBA and DBA → DBA mice were quite similar. In comparison, the median disease intensity for the TNFR1−/− → DBA group was significantly higher with a P value less than 0.001.
2. Increased disease in TNFR1−/− → DBA mice.
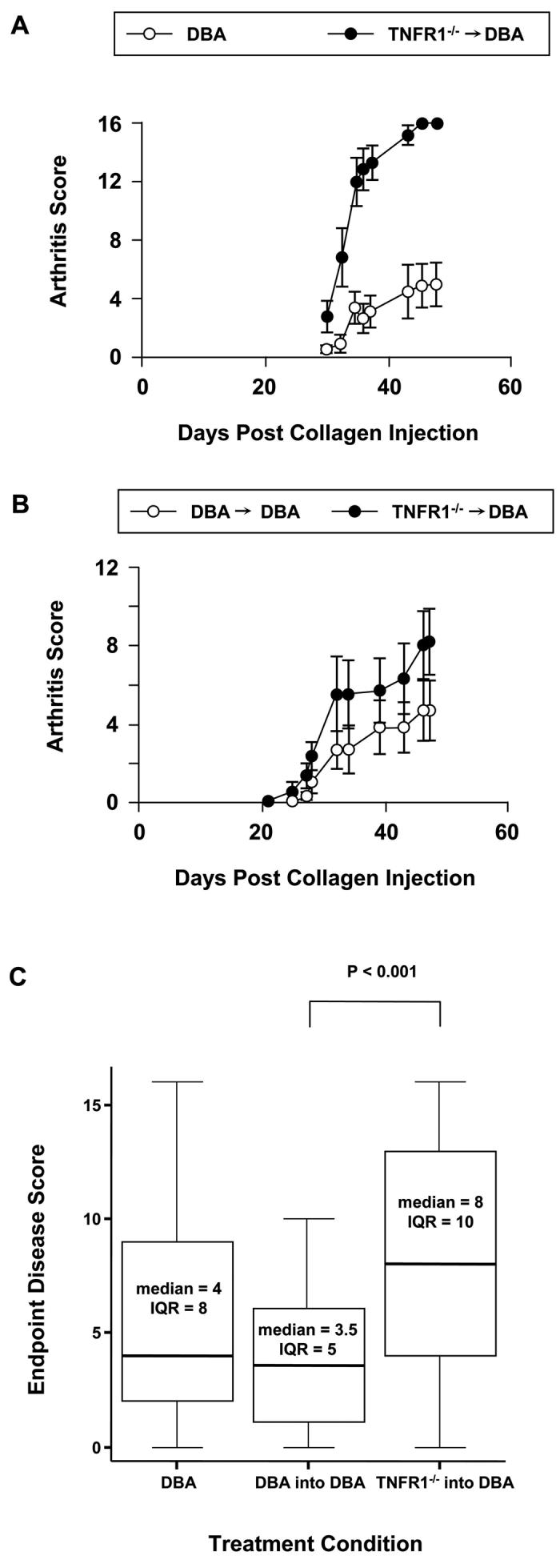
A, B) Representative experiments: A) DBA mice were irradiated at 5 weeks and rescued with TNFR1−/− bone marrow. The mice were rested for 12 weeks and then CIA induced in both transplant and age matched unmanipulated DBA controls. N = 6 for the transplant group. N = 16 for the control group. B) DBA mice were irradiated at 12 weeks and rescued with either TNFR1−/−or DBA bone marrow. The mice were rested for 12 weeks and then CIA induced in both transplant groups. N = 6 for both groups. C) Box and whisker diagrams for all pooled data of 9 experiments. DBA: N = 41. DBA → DBA: N = 32. TNFR1−/− → DBA: N = 57. Significance determined by Mann-Witney U test.
We wanted to compare clinical disease intensity with histological markers of disease, in part, to obtain an independent measure of disease and in part, to see if all parameters of disease were increased in our TNFR1−/− transplants. To this end, animals were sacrificed and paws were dissected and immediately fixed in neutral formalin for histology. Processing and staining were performed as described in Materials and Methods. Several representative joints are shown from both DBA and TNFR1−/− transplant animals in Figure 3 (panels A – I). Paws collected from 3 experiments were scored for inflammation, pannus, cartilage destruction, and bone destruction. Histological measures of disease correlated well with our clinical measurements and as shown all measures of disease were increased in our TNFR1−/− transplants (Figure 3, panels J – M).
3. Histological analysis of disease in transplanted animals.
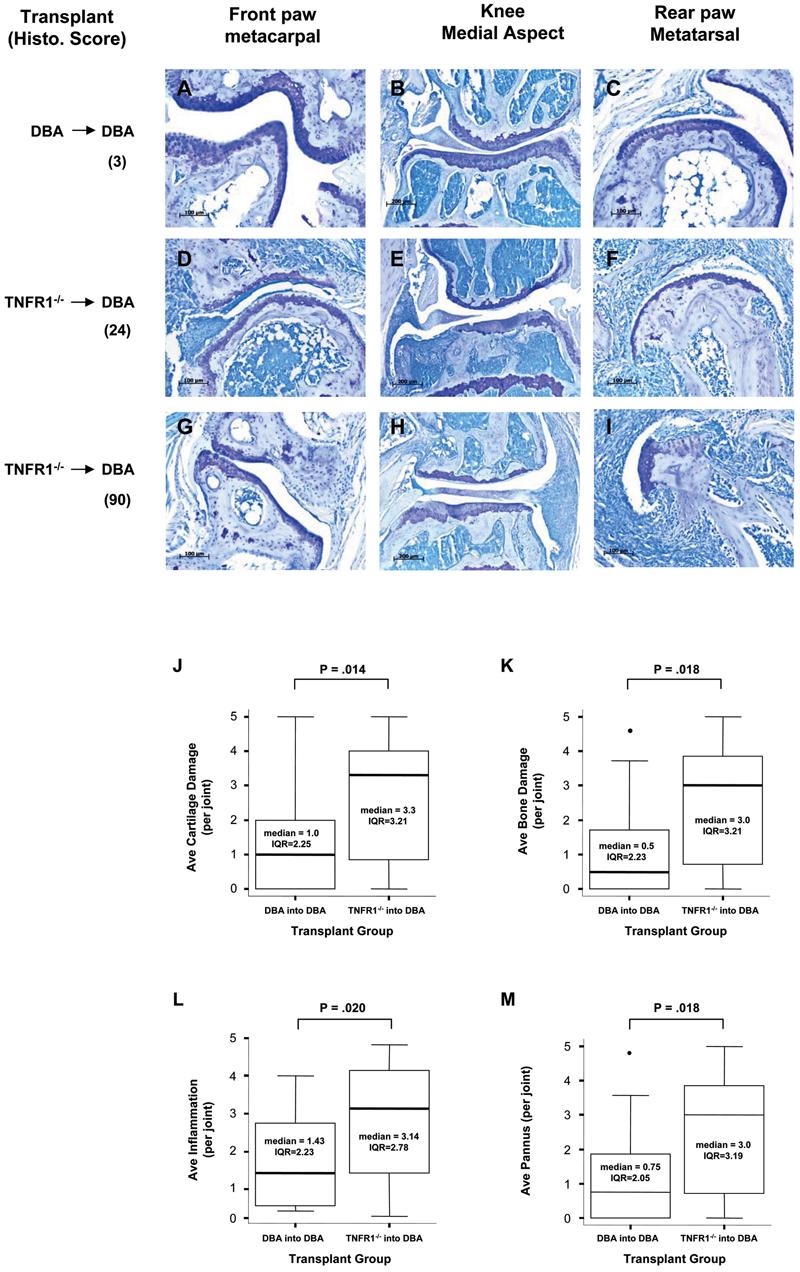
Panels A – I)Histology of representative joints from animals from one transplant experiment are shown. Each column represents a specific joint which is named at the top of the figure. Pannus, inflammation, bone damage, and cartilage damage were scored for the entire animal and are shown to the left as the sum of all joints analyzed. A-C) a DBA → DBA transplant mouse with very little disease (all joints sum = 3). D-F) a TNFR1−/−→ DBA transplant mouse with intermediate disease (all joints sum = 24). G-I) a TNFR1−/−→ DBA transplant mouse with intense disease (all joints sum = 90). Panels J – M) Histological analysis of 3 independent experiments. Paws and knees were processed and stained as described. Each joint was scored as described in Materials and Methods for cartilage damage: J, bone damage: K, inflammation: L, and pannus: M. Data from each transplant was pooled and expressed as a box and whisker plot. DBA → DBA, N = 17. TNFR1−/−→ DBA, N = 22. Significance was determined using a non-parametric Mann-Whitney test.
Increased disease is not a consequence of an increased antibody response to collagen
One explanation for increased disease is that our TNFR1−/− transplant animals have an increased immune response to CII. To test this hypothesis, we prepared TNFR1−/− → DBA transplants and allowed them to rest for 12 weeks. These transplants, along with age matched DBA controls were injected with CII plus CFA as described above and then sacrificed at 1, 2, and 3 week intervals after the initial CII injection. Tissues and serum was harvested. Serum samples were then analyzed for anti-collagen antibodies by ELISA as described in Materials and Methods. As shown in Figure 4 (panels A and B), IgG2a as well as total IgG antibody levels increased in an identical fashion for the two groups.
4. Characterization of disease properties.

Panels A – B) DBA mice were irradiated at 8 weeks and rescued with TNFR1−/− DBA bone marrow (TNFR1−/− → DBA). The mice were rested for 12 weeks and given the first CII injection along with age matched untransplanted DBA controls. Mice were sacrificed and blood obtained by cardiac puncture at the time points indicated. Anti-collagen IgG2a (A) and Total anti-collagen IgG (B) were measured by ELISA as described in Materials and Methods and normalized to a standard curve of serum derived from arthritic mice. Dots represent individual mice. The bar represents the mean value. C) TNFR1−/− mice were irradiated at 8 weeks and rescued with TNFR1−/− bone marrow. The mice were rested for 12 weeks and then CIA induced in both transplant and age matched unmanipulated DBA controls. N = 6 mice for the transplant group. N = 8 mice for the control group. No significant differences between groups were detected at any time point. Data is representative of 2 independent experiments. D) DBA mice were irradiated at 8 weeks and rescued with either TNFR1−/− DBA bone marrow (TNFR1−/− → DBA) or with DBA bone marrow (DBA→ DBA). The mice were rested for 5 weeks and then CIA induced in both transplant groups. N = 10 for both transplant groups. Asterisks indicate a p value less than 0.05 when comparing transplant and control groups at that time point. Data is representative of 3 independent experiments.
Another possibility which might account for increased disease is the presence of non-DBA genes within the transplanted bone marrow in addition to the targeted disruption of the TNFR1 gene. Six backcrosses theoretically results in a residual 1.56% non-DBA genome. This was addressed by comparing disease intensity in TNFR1+/− → DBA transplants to DBA → DBA transplants. No increase in disease was observed (data not shown).
We then considered the possibility that the introduction of TNFR1−/− bone marrow into an empty compartment might some how affect disease progression. This was tested by preparing TNFR1−/− → TNFR1−/− transplants. After 12 weeks, the transplant group and age matched DBA controls were injected with CII plus CFA on days 0 and 21 to induce arthritis and disease progression was followed. Again, no difference in disease intensity was observed in the two groups (Figure 4C).
Finally, we also examined disease progression in TNFR1−/− → DBA transplants after a 5 week rest as compared to DBA→ DBA controls. Both transplant groups were treated as described above save that the groups were rested in this experiment for 5 weeks rather than 12 weeks. Interestingly, we still observed increased disease (Figure 4D). Thus the component of the immune system which is required for full disease in our transplant model and appears to require a full 12 weeks for reconstitution is not required for the appearance of increased disease mediated by a TNFR1−/− hematopoietic system.
Early disease in TNFR1−/− → DBA transplanted animals
As described above, TNFR1−/− → DBA transplant animals along with age matched DBA controls were sacrificed at varying times after the first CII injection but before the appearance of overt clinical disease. Paws were dissected, immediately fixed in neutral buffered formalin, and subsequently processed for histology. Interestingly, many TNFR1−/− → DBA paws showed some evidence of inflammation (representative examples are shown in Figure 5 panels A and B). All tissue sections were scored histologically for pannus, inflammation, bone damage, and cartilage damage as described above. The scores for all 4 paws were summed to create a total histological disease score for each mouse. Figure 5E shows the average scores for TNFR1−/− → DBA and age matched DBA controls at 1, 2, and 3 weeks post CII injection. The majority of the score was contributed by inflammation and when this alone was considered (Figure 5F) a significant difference between TNFR1−/− → DBA and DBA controls were seen at all time points. In addition, periosteal proliferation was observed in TNFR1−/− → DBA paws with early disease while this was never seen in the DBA controls (Figure 5 panels B and C). Periosteal proliferation, a feature unique to this model of RA, is a mark of advanced disease which, strikingly, is seen in these tissues long before the development of overt clinical disease.
5. Examination of early disease.
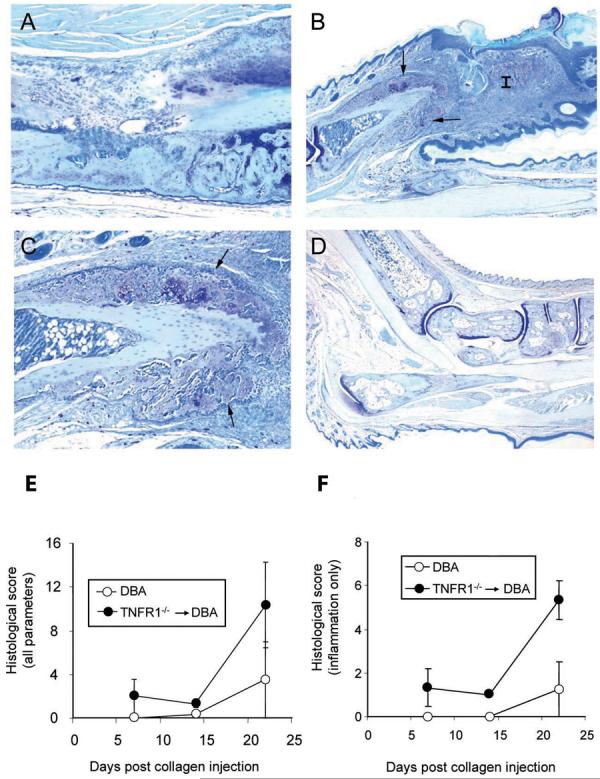
TNFR1−/− → DBA transplants were rested 12 weeks and, along with age matched DBA controls, were processed for histology 1, 2, and 3 weeks after the first CII injection. All images are from the 3 week time point. A) TNFR1−/− → DBA transplant ankle with mild inflammation. B) TNFR1−/− → DBA transplant rear digit with marked inflammation (I) and periosteal proliferation (arrows). C) Magnification of the region of periosteal proliferation within the previous image D) DBA control ankle with no detectable disease. E) Histology was scored for cartilage damage, bone damage, pannus, and inflammation and an all joints sum was established for each group. F) The inflammation score only is shown. Each point represents all paws from 3 animals (12 measures in total). Error bars represent SEM.
Cytokine Analysis
We considered two potential mechanisms which might contribute to increased disease. Firstly, the observation that TNFR1−/− → TNFR1−/− transplants did not develop increased disease as compared to DBA controls (Figure 4C) while TNFR1−/− → DBA transplants did would strongly suggest that TNFR1 must be present in the radio-resistant tissue compartment for this to occur, and by extension, that TNFα is driving disease. Indeed, others have shown that serum TNFα is increased in LPS treated TNFR1−/− and TNFR2−/− animals. To test for increased serum TNFα we analyzed serum samples from multiple experiments taken at various times relative to the injections of emulsion. Serum TNFα was measured by ELISA as described in Materials and Methods and was below the limits of detection in all samples: 4pg/ml - the lowest measurable TNFα standard. The second mechanism which we considered involves IL-12p40 expression. In studies of the role of TNFα in infection using TNFα knockout mice TNFa played an important role in both the induction of the immune response as well as its resolution (29). Importantly, it was shown that this resolution involves the TNFα mediated down regulation of IL-12. These data propelled us to analyze our serum samples for IL-12p40 by ELISA as described in Materials and Methods. As shown in Figure 6, for control groups, serum IL-12 levels rose between days 0 and 14 and remained constant through day 21. By day 45 – 50 (end), IL-12 levels had dropped back to base line. In TNFR1−/− → DBA animals IL-12p40 levels matched those seen in controls up to the time of the second CII injection. At that point, strikingly, IL-12 levels in TNFR1−/− → DBA continued to rise.
6. Serum IL-12 levels increase in TNFR1−/− → DBA animals.
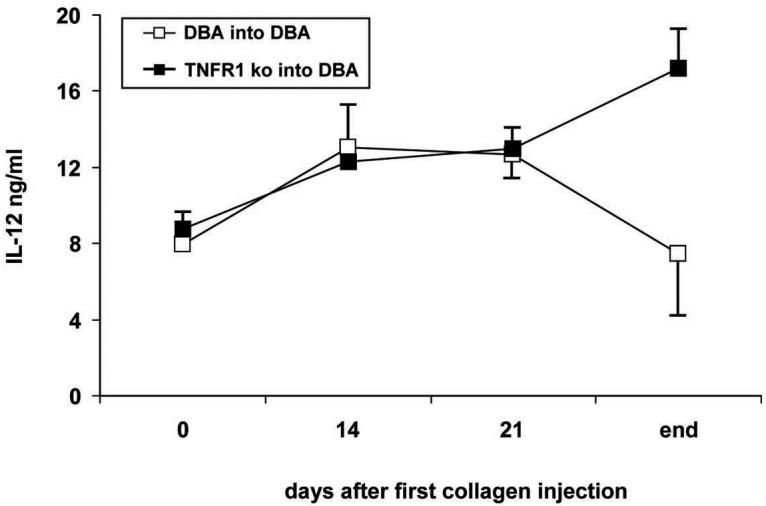
Serum was taken just before the first collagen injection, 14 days after the first injection, just before the second collagen injection, and at the end of the experiment IL-12 p40 subunit immunoreactivity was measured by ELISA as described. N for DBA → DBA transplant populations: day 0, 13; day 14, 3; day 21, 18; end, 13. N for TNFR1−/− → DBA transplants: day 0, 22; day 14, 3; day 21, 15; end, 18. For days 0, 21, and end, data was compiled from 3 independent experiments. The day 14 time point represents one experiment.
Discussion
Upon the initiation of this study, we expected that hematopoietic TNFa signaling would play a pathological role in arthritis. Our unmasking of a protective signaling pathway involving hematopoietic TNFR1 was completely unexpected. The transplant model that we have developed divides tissues into a bone marrow derived radio-sensitive compartment and a radio-resistant compartment. The radio-sensitive compartment which is repopulated by transplanted bone marrow within our animal model may be comparable to the population of cells arising from bone marrow in RA patients after TNF blockade therapy. In both TNFR1−/− mice (30) as in RA patients undergoing TNFa blockade therapy (24), germinal centers are decreased in numbers and sensitivity to intracellular bacterial infections is increased.
We expected to see decreased disease and yet we saw increased disease. What might be the mechanism? The requirement for TNFR1 within the radio-resistant compartment strongly argues for a role for TNFα signaling. Our measurements have found that, unlike LPS treated TNFR1−/− mice (31), we do not see a generalized increase in serum TNFα. These samples were unlikely to be degraded as IL-12p40 subunit was readily detectable. Further work is required to establish whether TNFα levels are increased within diseased joints from TNFR1−/− → DBA transplants and the extent to which TNFα blocking drugs can block disease in these animals.
IL-12p40 expression points to a second interesting potential mechanism. The p40 subunit is shared by IL-12 (p40/p35), IL-23 (P40/p19), and a neutral inhibitor of IL-12 (p40/p40). IL-12 plays a crucial role in the response to intracellular bacteria. It is an important inducer of Th1 differentiation and a stimulator of NK activity (32). Additionally, IL-12 is a potent stimulator of TNFα and IFNγ production. IL-23 is an inducer of memory T cell proliferation (33) and Th17 differentiation (34). Indeed IL-23 mediated Th17 differentiation has been shown to play a critical role in CIA disease progression (35). Several studies have implicated TNFα in the inhibition of IL-12p40 production by monocytes and macrophages. While TNFα−/− mice infected with C. parvum die due to a runaway inflammatory response (30), addition of an IL-12 neutralizing antibody restored the ability of the animal to resolve the inflammation (36). Murine peritoneal macrophages cultured in the presence of TNFα were found to be much less capable of secreting either IL-12 or IL-23 in response to a variety of TLR stimuli (37). These data suggest that there exists a switch in cellular function that is engaged by a prolonged exposure to TNFα. At early times TNFα drives inflammation while at later times, TNFα appears to switch functions and promotes resolution. In our model we found serum IL-12 levels to rise and then fall back to baseline in control animals while in TNFR1−/− → DBA transplants IL-12 levels continued to rise. It is premature to interpret these data as demonstrating that either IL-12 or IL-23 plays a direct role in our observation of increased disease. Rather, we interpret the change in IL-12p40 secretion as indicating that a similar switch is being engaged within our system in control mice and is deficient in our transplant mice. It is likely that this switch alters much more than simply the regulation of IL-12 p40 expression.
In summary, we have shown that removal of TNFR1 signaling from the radio-sensitive hematopoietic compartment results in an increase in disease intensity in the collagen arthritis model and that this increase is not simply a result of increased stimulation of the anti-collagen response. Our observation bears a striking similarity to the observations of others concerning an anti-inflammatory role for TNFα and suggests that this role is unique to the hematopoietic compartment. Pathologic TNFα signaling would then occur through the synovium and other radio-resistant populations. Given the genetic variability of the human population it is likely that these two pathways are of variable strengths in different individuals and thus we put forth the proposal that these relative strengths will correlate with patient responses to TNFα blockade.
Acknowledgements
The authors would like to acknowledge the help of Dr. Ian McNeice in teaching us how to perform bone marrow transplants and Dr. Nirmal Banda and Kristine Kuhn for assistance with the CIA model. Dr. William Arend provided much critical experimental advice. In addition the authors would like to thank Dr. Michael Holers, Dr. Kristine Kuhn, and Dr. Katie Haskins for reading the manuscript and providing additional advice.
This work was supported by NIH AR48432 and the Arthritis Foundation.
References
- 1.Olsen NJ, Stein CM. New drugs for rheumatoid arthritis. N Engl J Med. 2004;350(21):2167–79. doi: 10.1056/NEJMra032906. [DOI] [PubMed] [Google Scholar]
- 2.Breedveld FC, Weisman MH, Kavanaugh AF, Cohen SB, Pavelka K, van Vollenhoven R, et al. The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum. 2006;54(1):26–37. doi: 10.1002/art.21519. [DOI] [PubMed] [Google Scholar]
- 3.Dembic Z, Loetscher H, Gubler U, Pan YC, Lahm HW, Gentz R, et al. Two human TNF receptors have similar extracellular, but distinct intracellular, domain sequences. Cytokine. 1990;2(4):231–7. doi: 10.1016/1043-4666(90)90022-l. [DOI] [PubMed] [Google Scholar]
- 4.Naume B, Shalaby R, Lesslauer W, Espevik T. Involvement of the 55- and 75-kDa tumor necrosis factor receptors in the generation of lymphokine-activated killer cell activity and proliferation of natural killer cells. J Immunol. 1991;146(9):3045–8. [PubMed] [Google Scholar]
- 5.Erikstein BK, Smeland EB, Blomhoff HK, Funderud S, Prydz K, Lesslauer W, et al. Independent regulation of 55-kDa and 75-kDa tumor necrosis factor receptors during activation of human peripheral blood B lymphocytes. Eur J Immunol. 1991;21(4):1033–7. doi: 10.1002/eji.1830210426. [DOI] [PubMed] [Google Scholar]
- 6.Ware CF, Crowe PD, Vanarsdale TL, Andrews JL, Grayson MH, Jerzy R, et al. Tumor necrosis factor (TNF) receptor expression in T lymphocytes. Differential regulation of the type I TNF receptor during activation of resting and effector T cells. J Immunol. 1991;147(12):4229–38. [PubMed] [Google Scholar]
- 7.Konttinen YT, Li TF, Hukkanen M, Ma J, Xu JW, Virtanen I. Fibroblast biology. Signals targeting the synovial fibroblast in arthritis. Arthritis Res. 2000;2(5):348–55. doi: 10.1186/ar111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kollias G, Kontoyiannis D. Role of TNF/TNFR in autoimmunity: specific TNF receptor blockade may be advantageous to anti-TNF treatments. Cytokine Growth Factor Rev. 2002;13(4-5):315–21. doi: 10.1016/s1359-6101(02)00019-9. [DOI] [PubMed] [Google Scholar]
- 9.Keffer J, Probert L, Cazlaris H, Georgopoulos S, Kaslaris E, Kioussis D, et al. Transgenic mice expressing human tumour necrosis factor: a predictive genetic model of arthritis. Embo J. 1991;10(13):4025–31. doi: 10.1002/j.1460-2075.1991.tb04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jacob CO, McDevitt HO. Tumour necrosis factor-alpha in murine autoimmune ‘lupus’ nephritis. Nature. 1988;331(6154):356–8. doi: 10.1038/331356a0. [DOI] [PubMed] [Google Scholar]
- 11.Satoh J, Seino H, Abo T, Tanaka S, Shintani S, Ohta S, et al. Recombinant human tumor necrosis factor alpha suppresses autoimmune diabetes in nonobese diabetic mice. J Clin Invest. 1989;84(4):1345–8. doi: 10.1172/JCI114304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green EA, Eynon EE, Flavell RA. Local expression of TNFalpha in neonatal NOD mice promotes diabetes by enhancing presentation of islet antigens. Immunity. 1998;9(5):733–43. doi: 10.1016/s1074-7613(00)80670-6. [DOI] [PubMed] [Google Scholar]
- 13.Korner H, Riminton DS, Strickland DH, Lemckert FA, Pollard JD, Sedgwick JD. Critical points of tumor necrosis factor action in central nervous system autoimmune inflammation defined by gene targeting. J Exp Med. 1997;186(9):1585–90. doi: 10.1084/jem.186.9.1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Probert L, Akassoglou K, Kassiotis G, Pasparakis M, Alexopoulou L, Kollias G. TNF-alpha transgenic and knockout models of CNS inflammation and degeneration. J Neuroimmunol. 1997;72(2):137–41. doi: 10.1016/s0165-5728(96)00184-1. [DOI] [PubMed] [Google Scholar]
- 15.Kassiotis G, Pasparakis M, Kollias G, Probert L. TNF accelerates the onset but does not alter the incidence and severity of myelin basic protein-induced experimental autoimmune encephalomyelitis. Eur J Immunol. 1999;29(3):774–80. doi: 10.1002/(SICI)1521-4141(199903)29:03<774::AID-IMMU774>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 16.van Oosten BW, Barkhof F, Truyen L, Boringa JB, Bertelsmann FW, von Blomberg BM, et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology. 1996;47(6):1531–4. doi: 10.1212/wnl.47.6.1531. [DOI] [PubMed] [Google Scholar]
- 17.Tuohy VK, Yu M, Yin L, Kawczak JA, Kinkel RP. Spontaneous regression of primary autoreactivity during chronic progression of experimental autoimmune encephalomyelitis and multiple sclerosis. J Exp Med. 1999;189(7):1033–42. doi: 10.1084/jem.189.7.1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kassiotis G, Kollias G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med. 2001;193(4):427–34. doi: 10.1084/jem.193.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wooley PH. Collagen-induced arthritis in the mouse. Methods Enzymol. 1988;162:361–73. doi: 10.1016/0076-6879(88)62091-x. [DOI] [PubMed] [Google Scholar]
- 20.Kleinau S, Martinsson P, Heyman B. Induction and suppression of collagen-induced arthritis is dependent on distinct fcgamma receptors. J Exp Med. 2000;191(9):1611–6. doi: 10.1084/jem.191.9.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thorbecke GJ, Shah R, Leu CH, Kuruvilla AP, Hardison AM, Palladino MA. Involvement of endogenous tumor necrosis factor alpha and transforming growth factor beta during induction of collagen type II arthritis in mice. Proc Natl Acad Sci U S A. 1992;89(16):7375–9. doi: 10.1073/pnas.89.16.7375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Piguet PF, Grau GE, Vesin C, Loetscher H, Gentz R, Lesslauer W. Evolution of collagen arthritis in mice is arrested by treatment with anti-tumour necrosis factor (TNF) antibody or a recombinant soluble TNF receptor. Immunology. 1992;77(4):510–4. [PMC free article] [PubMed] [Google Scholar]
- 23.Tada Y, Ho A, Koarada S, Morito F, Ushiyama O, Suzuki N, et al. Collagen-induced arthritis in TNF receptor-1-deficient mice: TNF receptor-2 can modulate arthritis in the absence of TNF receptor-1. Clin Immunol. 2001;99(3):325–33. doi: 10.1006/clim.2001.5027. [DOI] [PubMed] [Google Scholar]
- 24.Anolik JH, Ravikumar R, Barnard J, Owen T, Almudevar A, Milner EC, et al. Cutting edge: anti-tumor necrosis factor therapy in rheumatoid arthritis inhibits memory B lymphocytes via effects on lymphoid germinal centers and follicular dendritic cell networks. J Immunol. 2008;180(2):688–92. doi: 10.4049/jimmunol.180.2.688. [DOI] [PubMed] [Google Scholar]
- 25.Zalevsky J, Secher T, Ezhevsky SA, Janot L, Steed PM, O'Brien C, et al. Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J Immunol. 2007;179(3):1872–83. doi: 10.4049/jimmunol.179.3.1872. [DOI] [PubMed] [Google Scholar]
- 26.Bendele AM, Chlipala ES, Scherrer J, Frazier J, Sennello G, Rich WJ, et al. Combination benefit of treatment with the cytokine inhibitors interleukin-1 receptor antagonist and PEGylated soluble tumor necrosis factor receptor type I in animal models of rheumatoid arthritis. Arthritis Rheum. 2000;43(12):2648–59. doi: 10.1002/1529-0131(200012)43:12<2648::AID-ANR4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 27.Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, et al. Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest. 2006;116(4):961–73. doi: 10.1172/JCI25422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Banda NK, Vondracek A, Kraus D, Dinarello CA, Kim SH, Bendele A, et al. Mechanisms of inhibition of collagen-induced arthritis by murine IL-18 binding protein. J Immunol. 2003;170(4):2100–5. doi: 10.4049/jimmunol.170.4.2100. [DOI] [PubMed] [Google Scholar]
- 29.Kanaly ST, Nashleanas M, Hondowicz B, Scott P. TNF receptor p55 is required for elimination of inflammatory cells following control of intracellular pathogens. J Immunol. 1999;163(7):3883–9. [PubMed] [Google Scholar]
- 30.Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, et al. Characterization of tumor necrosis factor-deficient mice. Proc Natl Acad Sci U S A. 1997;94(15):8093–8. doi: 10.1073/pnas.94.15.8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, et al. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J Immunol. 1998;160(2):943–52. [PubMed] [Google Scholar]
- 32.Trinchieri G. Interleukin-12: a proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu Rev Immunol. 1995;13:251–76. doi: 10.1146/annurev.iy.13.040195.001343. [DOI] [PubMed] [Google Scholar]
- 33.Belladonna ML, Renauld JC, Bianchi R, Vacca C, Fallarino F, Orabona C, et al. IL-23 and IL-12 have overlapping, but distinct, effects on murine dendritic cells. J Immunol. 2002;168(11):5448–54. doi: 10.4049/jimmunol.168.11.5448. [DOI] [PubMed] [Google Scholar]
- 34.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278(3):1910–4. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 35.Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, et al. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med. 2003;198(12):1951–7. doi: 10.1084/jem.20030896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hodge-Dufour J, Marino MW, Horton MR, Jungbluth A, Burdick MD, Strieter RM, et al. Inhibition of interferon gamma induced interleukin 12 production: a potential mechanism for the anti-inflammatory activities of tumor necrosis factor. Proc Natl Acad Sci U S A. 1998;95(23):13806–11. doi: 10.1073/pnas.95.23.13806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zakharova M, Ziegler HK. Paradoxical anti-inflammatory actions of TNF-alpha: inhibition of IL-12 and IL-23 via TNF receptor 1 in macrophages and dendritic cells. J Immunol. 2005;175(8):5024–33. doi: 10.4049/jimmunol.175.8.5024. [DOI] [PubMed] [Google Scholar]