

Abstract
We generated a novel nonpeptidic protease inhibitor (PI), GRL-02031, by incorporating a stereochemically defined fused cyclopentanyltetrahydrofuran (Cp-THF) which exerted potent activity against a wide spectrum of human immunodeficiency virus type 1 (HIV-1) isolates, including multidrug-resistant HIV-1 variants. GRL-02031 was highly potent against laboratory HIV-1 strains and primary clinical isolates, including subtypes A, B, C, and E (50% effective concentration [EC50] range, 0.015 to 0.038 μM), with minimal cytotoxicity (50% cytotoxic concentration, >100 μM in CD4+ MT-2 cells), although it was less active against two HIV-2 strains (HIV-2EHO and HIV-2ROD) (EC50, ∼0.60 μM) than against HIV-1 strains. GRL-02031 at relatively low concentrations blocked the infection and replication of each of the HIV-1NL4-3 variants exposed to and selected by up to 5 μM of saquinavir, amprenavir, indinavir, nelfinavir, or ritonavir and 1 μM of lopinavir or atazanavir (EC50 range, 0.036 to 0.14 μM). GRL-02031 was also potent against multi-PI-resistant clinical HIV-1 variants isolated from patients who had no response to the conventional antiretroviral regimens that then existed, with EC50s ranging from 0.014 to 0.042 μM (changes in the EC50s were less than twofold the EC50 for wild-type HIV-1). Upon selection of HIV-1NL4-3 in the presence of GRL-02031, mutants carrying L10F, L33F, M46I, I47V, Q58E, V82I, I84V, and I85V in the protease-encoding region and G62R (within p17), L363M (p24-p2 cleavage site), R409K (within p7), and I437T (p7-p1 cleavage site) in the gag-encoding region emerged. GRL-02031 was potent against a variety of HIV-1NL4-3-based molecular infectious clones containing a single primary mutation reported previously or a combination of such mutations, although it was slightly less active against HIV-1 variants containing consecutive amino acid substitutions: M46I and I47V or I84V and I85V. Structural modeling analysis demonstrated a distinct bimodal binding of GRL-02031 to protease, which may provide advantages to GRL-02031 in blocking the replication of a wide spectrum of HIV-1 variants resistant to PIs and in delaying the development of resistance of HIV-1 to GRL-02031. The present data warrant the further development of GRL-02031 as a potential therapeutic agent for the treatment of infections with primary and multidrug-resistant HIV-1 variants.
The currently available combination therapy or highly active antiretroviral therapy (HAART) with two or more reverse transcriptase inhibitors and protease inhibitors (PIs) for human immunodeficiency virus (HIV) type 1 (HIV-1) infection and AIDS has been shown to suppress the replication of HIV-1 and extend the life expectancy of HIV-1-infected individuals (35, 38). However, the ability to provide effective long-term antiretroviral therapy for HIV-1 infection has become a complex issue, since those who initially achieved favorable viral suppression to undetectable levels have experienced treatment failure (11, 18, 28). In addition, it is evident that with these anti-HIV drugs, only partial immunologic reconstitution is attained in patients with advanced HIV-1 infection.
Nevertheless, recent analyses have revealed that the life expectancy of HIV-infected patients treated with HAART increased between 1996 and 2005, that the mortality rates for HIV-infected persons have become much closer to general mortality rates since the introduction of HAART, and that first-line HAART with boosted PI-based regimens results in less resistance within and across drug classes (2, 3, 18, 46).
In the development of new anti-HIV-1 therapeutics, we have faced a variety of challenges different from those faced during the design of the first-line drugs (7, 10, 39). The issue of the emergence of drug-resistant HIV-1 variants is one of the most formidable challenges in the era of HAART. Indeed, it is of note that the very features that contribute to the specificities and the efficacies of reverse transcriptase inhibitors and PIs provide the virus with a strategy to develop resistance (15, 19, 35), and it seems inevitable that this resistance issue will remain problematic for many years to come, although a few recently developed drugs, such as darunavir (DRV) and tipranavir, have been relatively successful as treatments for individuals carrying multidrug-resistant HIV-1 variants (5, 20).
In particular, a number of studies indicate that cross-resistance is a major obstacle to antiviral therapy with PIs (19, 24). Obviously, the emergence of viral resistance, difficulties with compliance with the complicated treatment protocols, and adverse side effects urge the development of new classes of PIs (i) that have potent activities against existing resistant HIV-1 variants and that do not allow or delay the emergence of resistance, (ii) that have improved pharmacokinetics parameters in humans, and (iii) that have less severe side effects (43).
The present paper represents the first demonstration of the results of antiviral analyses of a novel PI which contains cyclopentanyltetrahydrofuran (Cp-THF) and which is highly potent against a wide spectrum of HIV isolates, including a variety of multi-PI-resistant clinical strains, in vitro. In addition, we selected GRL-02031-resistant HIV-1 variants in vitro and characterized their virological properties and susceptibilities to other PIs. We also demonstrated that the emergence of HIV-1 variants resistant to GRL-02031 requires multiply accumulated amino acid substitutions in the protease-encoding region. Moreover, in an attempt to explain why GRL-02031 can exert potent activity against a wide spectrum of HIV-1 variants resistant to multiple PIs, we performed structural modeling and molecular docking and examined the interactions of GRL-02031 with HIV-1 protease.
MATERIALS AND METHODS
Cells and viruses.
MT-2 and MT-4 cells were grown in RPMI 1640-based culture medium supplemented with 10% fetal calf serum (JRH Biosciences, Lenexa, MD), 50 U/ml penicillin, and 50 μg/ml of streptomycin. The following HIV-1 strains were employed for the drug susceptibility assay (see below): HIV-1LAI, HIV-1Ba-L, HIV-1JRFL, HIV-1NL4-3, HIV-2EHO, and HIV-2ROD; two clinical HIV-1 strains isolated from drug-naive patients with AIDS, HIV-1ERS104pre and HIV-1MOKW (30, 45); and seven HIV-1 clinical isolates which were originally isolated from patients with AIDS who had received 9 to 11 anti-HIV-1 drugs over the past 32 to 83 months and which were genotypically and phenotypically characterized as multi-PI-resistant HIV-1 variants (47, 48). HIV-192UG037, HIV-197ZA003, and HIV-192TH019 were obtained from the NIH AIDS Reagent Program. All primary HIV-1 strains were passaged once or twice in 3-day-old phytohemagglutinin-activated peripheral blood mononuclear cells (PHA-PBMs), and the culture supernatants were stored at −80°C until use.
Antiviral agents.
GRL-02031 (Fig. 1), a novel nonpeptidic PI containing Cp-THF, was designed and synthesized. Detailed methods for the synthesis of GRL-02031 will be described elsewhere by A. K. Ghosh et al. 3′-Azido-2′,3′-dideoxythymidine (AZT; zidovudine) was purchased from Sigma (St. Louis, MO). Saquinavir (SQV) and ritonavir (RTV) were kindly provided by Roche Products Ltd. (Welwyn Garden City, United Kingdom) and Abbott Laboratories (Abbott Park, IL), respectively. Amprenavir (APV) was a kind gift from Glaxo-Wellcome, Research Triangle Park, NC. Nelfinavir (NFV) and indinavir (IDV) were kindly provided by Japan Energy Inc, Tokyo, Japan. Lopinavir (LPV) was synthesized by previously published methods (48). Atazanavir (ATV) was a kind gift from Bristol-Myers Squibb (New York, NY).
FIG. 1.
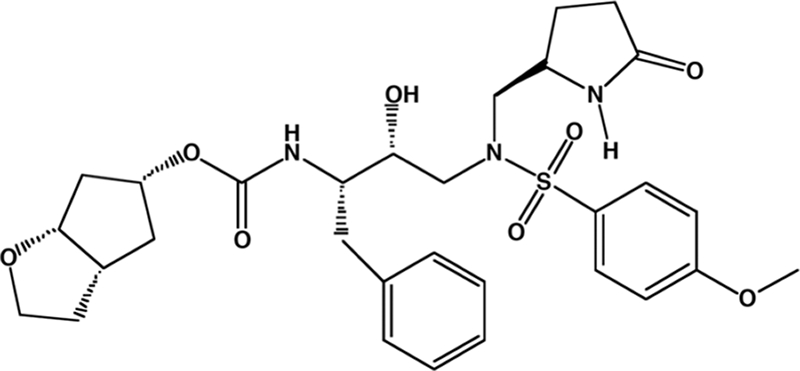
Structure of GRL-02031.
Drug susceptibility assay.
The susceptibilities of HIV-1LAI, HIV-1Ba-L, HIV-2EHO, HIV-2ROD, and the primary HIV-1 isolates to various drugs were determined as described previously (26), with minor modifications. Briefly, MT-2 cells (2 × 104/ml) were exposed to 100 50% tissue culture infectious dose (TCID50s) of HIV-1LAI, HIV-1Ba-L, HIV-2EHO, or HIV-2ROD in the presence or the absence of various concentrations of drugs in 96-well microculture plates; and the plates were incubated at 37°C for 7 days. After 100 μl of the medium was removed from each well, 3-(4,5-dimetylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (10 μl, 7.5 mg/ml in phosphate-buffered saline) was added to each well in the plate, followed by incubation at 37°C for 2 h. After incubation, to dissolve the formazan crystals, 100 μl of acidified isopropanol containing 4% (vol/vol) Triton X-100 was added to each well and the optical density was measured in a kinetic microplate reader (Vmax; Molecular Devices, Sunnyvale, CA). All assays were performed in duplicate or triplicate.
To determine the sensitivities of the primary HIV-1 isolates to drugs, PHA-PBMs (106/ml) were exposed to 50 TCID50s of each primary HIV-1 isolate and cultured in the presence or the absence of various concentrations of drugs in 10-fold serial dilutions in 96-well microculture plates. To determine the drug susceptibilities of certain laboratory HIV-1 strains, MT-4 cells were employed as target cells, as described previously (26), with minor modifications. In brief, MT-4 cells (105/ml) were exposed to 100 TCID50s of drug-resistant HIV-1 strains in the presence or the absence of various concentrations of drugs and were incubated at 37°C. On day 7 of culture, the supernatants were harvested and the amounts of the p24 Gag protein were determined by using a fully automated chemiluminescent enzyme immunoassay system (Lumipulse F; Fujirebio Inc., Tokyo, Japan) (29). The drug concentrations that suppressed the production of p24 Gag protein by 50% (EC50) were determined by comparison of the amount of p24 Gag protein produced in drug-treated cell cultures with the level of p24 Gag protein produced in a drug-free control cell culture. All assays were performed in triplicate.
Generation of PI-resistant HIV-1 variants in vitro.
MT-4 cells (105/ml) were exposed to HIV-1NL4-3 (500 TCID50s) and cultured in the presence of various PIs at an initial concentration of 0.01 to 0.03 μM. Viral replication was monitored by determination of the amount of p24 Gag produced by MT-4 cells. The culture supernatants were harvested on day 7 and were used to infect fresh MT-4 cells for the next round of culture in the presence of increasing concentrations of each drug. When the virus began to propagate in the presence of the drug, the drug concentration was generally increased two- to threefold. Proviral DNA samples obtained from the lysates of infected cells were subjected to nucleotide sequencing. This drug selection procedure was carried out until the drug concentration reached 5 μM.
Determination of nucleotide sequences.
Molecular cloning and determination of the nucleotide sequences of HIV-1 isolates passaged in the presence of anti-HIV-1 agents were performed as described previously (26, 47). In brief, high-molecular-weight DNA was extracted from HIV-1-infected MT-4 cells by using the InstaGene matrix (Bio-Rad Laboratories, Hercules, CA) and was subjected to molecular cloning, followed by sequence determination. The primers used for the first round of PCR of the entire Gag- and protease-encoding regions of the HIV-1 genome were LTR-F1 (5′-GAT GCT ACA TAT AAG CAG CTG C-3′) and PR12 (5′-CTC GTG ACA AAT TTC TAC TAA TGC-3′). The first-round PCR mixture consisted of 5 μl of proviral DNA solution, 2.0 U of Premix Taq (Ex Taq version; Takara Bio Inc., Otsu, Japan), and 12.5 pmol of each of the first-round PCR primers in a total volume of 50 μl. The PCR conditions employed were as follows: an initial 2 min at 94°C, followed by 35 cycles of 30 s at 94°C, 30 s at 58°C, and 3 min at 72°C, with a final 8-min extension at 72°C. The first-round PCR products (1 μl) were used directly in the second round of PCR with primers LTR-F2 (5′-GAG ACT CTG GTA ACT AGA GAT C-3′) and Ksma2.1 (5′-CCA TCC CGG GCT TTA ATT TTA CTG GTA C-3′) under the same PCR conditions described above. The second-round PCR products were purified with spin columns (MicroSpin S-400 HR columns; Amersham Biosciences Corp., Piscataway, NJ), cloned directly, and subjected to sequencing with an ABI model 377 automated DNA sequencer (Applied Biosystems, Foster City, CA). The viral RNA in the selection culture should contain a number of noninfectious (or dead) virions due to randomly occurring amino acid substitutions, which could provide misleading results if the sequences of such noninfectious or dead virions were erroneously taken into account. The viral DNA extracted from the newly infected cells in the present cell-free transmission system represents the infectious virions in the previous culture.
Generation of recombinant HIV-1 clones.
The PCR products obtained as described above were digested with two enzymes, ApaI and SmaI; and the fragments obtained were introduced into pHIV-1NLSma, designed to have a SmaI site by changing two nucleotides (2590 and 2593) of pHIV-1NL4-3, as described previously (14, 25). To generate HIV-1 clones carrying the desired mutations, site-directed mutagenesis was performed with a QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA), and the mutation-containing genomic fragments were introduced into pHIV-1NLSma. Determination of the nucleotide sequences of the plasmids confirmed that each clone had the desired mutations but no unintended mutations. Each recombinant plasmid was transfected into 293T cells with Lipofectoamine 2000 transfection reagent (Invitrogen, Carlsbad, CA), and the infectious virions thus generated were harvested for 48 h after transfection and stored at −80°C until use.
Structural analysis of GRL-02031 interactions with wild-type HIV-1 protease.
The interactions of GRL-02031 with wild-type HIV-1 protease were examined by computational structural modeling and molecular docking on the basis of the published crystallographic data for protease complexed with PIs. Besides accounting for the conformational flexibility of the inhibitor, the polarization induced in the inhibitor by the protease was taken into consideration by employing polarizable quantum charges in the docking computations. The use of polarizable quantum charges has recently been shown to substantially improve the prediction of protein-ligand complex structures (4). The quantum mechanical polarized ligand docking protocol provided with the Glide (version 4.5), QSite (version 4.5), Jaguar (version 7.0), and Maestro (version 8.5) software (Schrödinger, LLC, New York, NY) was used as described below. The crystal structures 2FDE (protease-brecanavir complex) and 2IEN (protease-DRV complex) were used as templates in separate docking calculations to determine the binding mode of GRL-02031 with wild-type protease. The crystal coordinates were obtained from the Protein Data Bank (http://www.rcsb.org/). Hydrogens were optimized by placing constraints on the heavy atoms. The crystal water that mediates the interaction between PIs and the protease flap was retained, and all other crystal waters were deleted. Close interaction in the protease was annealed, and the docking grid was set up. Polarizable ligand charges were determined at the B3LYP/6-31G* level. The extraprecision mode of the Glide program (12, 13), which has a higher penalty for unphysical interactions, was used.
RESULTS
In vitro activity of GRL-02031 against laboratory and primary HIV strains and cytotoxicity of GRL-02031.
We designed and synthesized ∼80 different novel nonpeptidyl PIs containing a Cp-THF moiety and examined them for their anti-HIV activities and cytotoxicities in vitro. Among them, we found that GRL-02031 (Fig. 1) was the most potent against a laboratory HIV-1 strain, HIV-1LAI, and had a favorable cytotoxicity profile, as examined with target MT-2 cells. As shown in Table 1, GRL-02031 showed an anti-HIV-1 activity profile comparable to that of most of the Food and Drug Administration (FDA)-approved PIs examined in the present study, although its toxicity profile was apparently more favorable, with a 50% cytotoxic concentration (CC50) of >100 μM and a selectivity index (CC50/EC50) of >3,600.
TABLE 1.
Antiviral activity of GRL-02031 against HIV-1LAIa
| Compound | EC50 (μM) | CC50(μM) | Selectivity index |
|---|---|---|---|
| GRL-02031 | 0.028 ± 0.003 | >100 | >3,600 |
| SQV | 0.014 ± 0.005 | 9.9 ± 3.6 | 710 |
| APV | 0.033 ± 0.012 | >100 | >3,000 |
| IDV | 0.044 ± 0.007 | 69.8 ± 3.1 | 1,600 |
| RTV | 0.038 ± 0.004 | 21.3 ± 0.9 | 560 |
| NFV | 0.023 ± 0.006 | ND | ND |
| LPV | 0.032 ± 0.007 | ND | ND |
MT-2 cells (2 × 104/ml) were exposed to 100 TCID50s of HIV-1LAI and were cultured in the presence of various concentrations of PIs, and the EC50s were determined by using the MTT assay on day 7 of culture. All assays were conducted in duplicate. The data shown represent mean values (±1 standard deviation) derived from the results of three independent experiments. ND, not determined. Selectivity index, CC50/EC50.
GRL-02031 was further tested against two R5 laboratory HIV-1 strains (HIV-1Ba-L and HIV-1JRFL), three different subtypes of primary HIV-1 strains (HIV-192UG037 [subtype A], HIV-197ZA003 [subtype C], and HIV-192TH019 [subtype E]), and two HIV-2 strains (HIV-2ROD and HIV-2EHO). GRL-02031 was found to be potent against all these HIV-1 strains and had EC50s that ranged from 0.015 to 0.038 μM, as tested by the use of target PHA-PBMs, while GRL-02031 was moderately active against two HIV-2 strains (EC50, ∼0.60 μM), as tested by the use of MT-2 cells (data not shown).
GRL-02031 exerts potent activity against a wide spectrum of primary HIV-1 variants resistant to multiple PIs.
We next examined the activity of GRL-02031 against a variety of primary HIV-1 strains which were isolated from those with AIDS who had failed a number of anti-HIV therapeutic regimens after they had received 9 to 11 anti-HIV-1 drugs over the previous 32 to 83 months and who proved to be highly resistant to multiple PIs (47, 48). These primary strains contained 9 to 14 amino acid substitutions in the protease-encoding region of the HIV-1 genome which have been reported to be associated with HIV-1 resistance to various PIs (RTV, IDV, NFV, SQV, APV, and LPV) (8). The substitutions identified included Leu-10 → Ile (L10I; seven of seven isolates), M46I/L (six of seven isolates), I54V (five of seven isolates), L63P (seven of seven isolates), A71V/T (six of seven isolates), V82A or V82T (seven of seven isolates), and L90M (five of seven isolates) (see footnote a of Table 2).
TABLE 2.
Antiviral activity of GRL-02031 against clinical HIV-1 isolates in PHA-PBMsa
| Virus | Pheno- type | EC50 (μM)
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AZT | SQV | APV | IDV | NFV | RTV | LPV | DRV | GRL-02031 | ||
| HIV-1ERS104pre (wild type) | X4 | 0.005 ± 0.002 | 0.009 ± 0.005 | 0.025 ± 0.007 | 0.021 ± 0.008 | 0.016 ± 0.004 | 0.036 ± 0.008 | 0.032 ± 0.002 | 0.0035 ± 0.0003 | 0.027 ± 0.002 |
| HIV-1MOKW (wild type) | R5 | 0.016 ± 0.011 | 0.004 ± 0.002 | 0.015 ± 0.004 | 0.019 ± 0.005 | 0.023 ± 0.008 | 0.036 ± 0.008 | 0.029 ± 0.0005 | 0.003 ± 0.0005 | 0.026 ± 0.007 |
| HIV-1TM(MDR) | X4 | 0.89 ± 0.11 (178) | 0.27 ± 0.08 (30) | 0.35 ± 0.11 (14) | >1 (>48) | >1 (>63) | >1 (>30) | 0.23 ± 0.18 (7) | 0.004 ± 0.001 (1) | 0.026 ± 0.004 (1) |
| HIV-1MM(MDR) | R5 | 0.45 ± 0.08 (90) | 0.27 ± 0.06 (30) | 0.40 ± 0.10 (16) | >1 (>48) | >1 (>63) | >1 (>30) | 0.62 ± 0.28 (19) | 0.017 ± 0.008 (5) | 0.041 ± 0.004 (2) |
| HIV-1JSL (MDR) | R5 | 0.18 ± 0.11 (36) | 0.32 ± 0.13 (36) | 0.66 ± 0.17 (26) | >1 (>48) | >1 (>63) | >1 (>30) | 0.74 ± 0.32 (23) | 0.026 ± 0.005 (7) | 0.043 ± 0.003 (2) |
| HIV-1A(MDR) | X4 | 0.12 ± 0.11 (24) | 0.10 ± 0.07 (11) | 0.16 ± 0.08 (6) | >1 (>48) | 0.33 ± 0.07 (21) | >1 (>30) | 0.32 ± 0.003 (10) | 0.003 ± 0.0003 (1) | 0.014 ± 0.007 (1) |
| HIV-1B(MDR) | X4 | >1 (>200) | 0.30 ± 0.02 (33) | 0.31 ± 0.04 (12) | >1 (>48) | >1 (>63) | >1 (>30) | 0.25 ± 0.007 (8) | 0.026 ± 0.017 (7) | 0.029 ± 0.007 (1) |
| HIV-1C(MDR) | X4 | 0.28 ± 0.20 (56) | 0.033 ± 0.010 (4) | 0.22 ± 0.13 (9) | >1 (>48) | 0.36 ± 0.12 (23) | >1 (>30) | 0.46 ± 0.24 (14) | 0.007 ± 0.005 (2) | 0.027 ± 0.009 (1) |
| HIV-1G(MDR) | X4 | 0.60 ± 0.56 (120) | 0.027 ± 0.006 (3) | 0.23 ± 0.13 (9) | 0.34 ± 0.07 (16) | 0.23 ± 0.11 (14) | >1 (>30) | 0.13 ± 0.04 (4) | 0.007 ± 0.004 (2) | 0.028 ± 0.001 (1) |
The amino acid substitutions identified in the protease-encoding region of HIV-1ERS104pre, HIV-1TM, HIV-1MM, HIV-1JSL, HIV-1A, HIV-1B, HIV-1C, and HIV-1G compared to the consensus B sequence cited from the Los Alamos National Laboratory database include L63P, L10I/K14R/R41K/M46L/I54V/L63P/A71V/V82A/L90M/I93L, L10I/K43T/M46L/I54V/L63P/A71V/V82A/L90M/Q92K, L10I/L24I/L33F/E35D/M36I/N37S/M46L/I54V/R57K/I62V/L63P/A71V/G73S/V82A, L10I/I15V/E35D/N37E/K45R/I54V/L63P/A71V/V82T/L90M/I93L/C95F, L10I/K14R/L33I/M36I/M46I/F53L/K55R/I62V/L63P/A71V/G73S/V82A/L90M/I93L, L10I/I15V/K20R/L24I/M36I/M46L/I54V/I62V/L63P/K70Q/V82A/L89M, and L10I/V11I/T12E/I15V/L19I/R41K/M46L/L63P/A71T/V82A/L90M, respectively. HIV-1MOKW was confirmed to lack known drug resistance-associated amino acid substitutions. The EC50s were determined by employing PHA-PBMs as target cells and the inhibition of p24 Gag protein production as the endpoint. All values were determined in triplicate, and those shown were derived from the results of three independent experiments. Numbers in parentheses represent the fold changes in the EC50 for each isolate compared to the EC50s for HIV-1ERS104pre. MDR, multidrug resistant.
All drugs examined showed potent activity against two reference wild-type primary strains (X4 HIV-1ERS104pre [45] and R5 HIV-1MOKW [30]), with the EC50s ranging 0.004 to 0.036 μM (Table 2). However, all the primary strains examined were highly resistant to AZT, with the EC50s being from 24- to >200-fold greater than the EC50 against HIV-1ERS104pre. It was noted that SQV and LPV were still active against one or two of the seven strains and had EC50s that differed 3- to 4-fold from those for HIV-1ERS104pre; however, all the other FDA-approved PIs examined in this study except DRV failed to exert activity and had EC50s 6- to >63-fold greater than the EC50 for HIV-1ERS104pre. In contrast, GRL-02031, like DRV, potently blocked all seven primary strains and had EC50s that ranged from 0.014 to 0.043 μM. It should be noted that the change in the EC50 of GRL-02031 for all seven multi-PI-resistant isolates tested was less than twofold compared with the EC50 for a wild-type primary strain, HIV-1ERS104pre.
Selection of HIV-1NL4-3 with GRL-02031.
We then attempted to select a laboratory X4 HIV-1 strain (HIV-1NL4-3) by propagating it in MT-4 cells in the presence of increasing concentrations of APV, IDV, or GRL-02031, as described previously (47). The virus was initially exposed to 0.03 μM APV, 0.02 μM IDV, or 0.02 μM GRL-02031. At passages 21 and 27, HIV-1NL4-3 was capable of propagating in the presence of 167- and 250-fold greater concentrations of APV and IDV, respectively. At passage 26, HIV-1NL4-3 was capable of propagating in the presence of a 250-fold greater concentration of IDV; however, 37 passages were required until the virus became similarly resistant to GRL-02031 and capable of propagating in the presence of 5 μM (Fig. 2).
FIG. 2.

In vitro selection of HIV-1 variants resistant to GRL-02031. HIV-1NL4-3 was passaged in the presence of increasing concentrations of APV (open circles), IDV (solid circles), or GRL-02031 (open triangles) in MT-4 cells. The selection was carried out in a cell-free manner for a total of 21 to 37 passages with drug concentrations escalating from 0.02 to 5 μM. The nucleotide sequences of the proviral DNAs were determined by using cell lysates of HIV-1-infected MT-4 cells at the termination of each indicated passage.
We also determined the nucleic acid sequences of the protease-encoding region of the proviral DNA isolated from the cells exposed to GRL-02031 at passages 5, 15, 22, 30, and 37 (Fig. 3). At passage 5, no significant amino acid substitutions were identified; however, by passage 15, the virus had acquired the L10F substitution, which has been reported to be associated with PI resistance (6, 32). By passage 22, all eight clones of the virus examined had additionally acquired a flap mutation (I47V) and the primary substitution (V82I). By passage 30, in addition to L10F, I47V, and V82I, the virus had acquired L33F, M46I, and I85V. By passage 37, the virus had gained I84V and another Q58E substitution, an unusual mutation not associated with resistance to other HIV PIs. Genetic characterization of the protease-encoding gene of the HIV-1 variants selected with APV and IDV revealed that those variants had acquired previously reported mutations, such as L10F, V32I, M46I, and I84V in APV-selected HIV-1 (HIV-1APVr) and L10F, V32I, K43T, M46I, L63P, G73C, and V82A in HIV-1IDVr.
FIG. 3.

Sequence analysis of the protease-encoding region of HIV passaged in the presence of GRL-02031. The amino acid sequences of the proteases deduced from the nucleotide sequences of the protease-encoding region of HIV clones determined at five different passages are illustrated. The identity of each amino acid with that from pNL4-3 (top row) at each individual amino acid position is indicated by a dot.
Amino acid substitutions in Gag protein of HIV-1 exposed to GRL-02031.
We also determined the amino acid sequences of the Gag proteins of HIV-1NL4-3 exposed to GRL-02031. By passage 15, an unusual amino acid substitution, L363M, located at the p24-p2 cleavage site, had emerged. By passage 22, the virus had acquired another amino acid substitution, I437T, located close to the p7-p1 cleavage site. By passage 30 and beyond, G62R (within p17) and R409K (within p7) were seen in addition to L363M and I437T (data not shown).
Anti-HIV-1 profile of a GRL-02031-resistant HIV-1 variant.
We then examined the anti-HIV-1 profile of a GRL-02031-resistant HIV-1 variant. In this experiment, we employed eight HIV-1 variants which were selected in the presence of up to 1 or 5 μM of each of seven FDA-approved PIs and GRL-02031. Each of these eight variants contained a variety of previously known PI resistance-associated amino acid substitutions (see footnote a of Table 3). As shown in Table 3, when each HIV-1 variant was examined in target MT-4 cells, each HIV-1 variant except the variant selected with 1 μM LPV in vitro (HIV- 1LPV-1 μM) and the variant selected with 1 μM ATV in vitro (HIV-1ATV-1 μM), proved to be highly resistant to the PI with which the virus was selected, with EC50s exceeding 1 μM. However, GRL-02031 was potent against all the PI-resistant variants, although the compound was relatively less potent against the variant selected with 5 μM SQV in vitro (HIV-1SQV-5 μM) (a sixfold increase in the EC50 of SQV compared with that of GRL-02031).
TABLE 3.
Antiviral activities of GRL-02031 against laboratory PI-resistant HIV-1 variantsa
| Virus | EC50 (μM)
|
|||||||
|---|---|---|---|---|---|---|---|---|
| SQV | APV | IDV | NFV | RTV | LPV | ATV | GRL-02031 | |
| HIV-1NL4-3 | 0.008 ± 0.004 | 0.028 ± 0.009 | 0.014 ± 0.004 | 0.018 ± 0.009 | 0.021 ± 0.009 | 0.018 ± 0.001 | 0.0043 ± 0.0004 | 0.023 ± 0.008 |
| HIV-1SQV-5 μM | >1 (>125) | 0.21 ± 0.09 (8) | >1 (>71) | 0.32 ± 0.04 (18) | >1 (>48) | 0.70 ± 0.22 (39) | 0.32 ± 0.02 (74) | 0.14 ± 0.06 (6) |
| HIV-1APV-5 μM | 0.016 ± 0.010 (2) | >1 (>36) | 0.22 ± 0.15 (16) | 0.17 ± 0.10 (10) | >1 (>48) | 0.14 ± 0.05 (8) | 0.0032 ± 0.012 (1) | 0.037 ± 0.004 (2) |
| HIV-1IDV-5 μM | 0.022 ± 0.009 (3) | 0.26 ± 0.14 (9) | >1 (>71) | 0.65 ± 0.16 (36) | >1 (>48) | >1 (>56) | 0.063 ± 0.022 (15) | 0.047 ± 0.002 (2) |
| HIV-1NFV-5 μM | 0.028 ± 0.009 (4) | 0.078 ± 0.026 (3) | 0.27 ± 0.06 (19) | >1 (>56) | 0.08 ± 0.05 (4) | 0.29 ± 0.03 (16) | 0.024 ± 0.005 (6) | 0.036 ± 0.002 (2) |
| HIV-1RTV-5 μM | 0.010 ± 0.008 (1) | 0.43 ± 0.27 (15) | 0.30 ± 0.07 (21) | 0.24 ± 0.10 (13) | >1 (>48) | 0.11 ± 0.08 (6) | 0.021 ± 0.007 (5) | 0.077 ± 0.031 (3) |
| HIV-1LPV-1 μM | 0.033 ± 0.003 (4) | 0.32 ± 0.02 (11) | >1 (>71) | 0.51 ± 0.06 (28) | >1 (>48) | 0.30 ± 0.04 (17) | 0.041 ± 0.002 (10) | 0.038 ± 0.004 (2) |
| HIV-1ATV-1 μM | 0.034 ± 0.006 (4) | 0.18 ± 0.06 (6) | 0.33 ± 0.10 (24) | 0.21 ± 0.03 (12) | 0.14 ± 0.02 (7) | 0.025 ± 0.011 (1) | 0.31 ± 0.05 (72) | 0.033 ± 0.002 (1) |
| HIV-1GRL-02031-5 μM | 0.008 ± 0.001 (1) | 0.21 ± 0.02 (8) | 0.044 ± 0.015 (3) | 0.011 ± 0.004 (1) | 0.26 ± 0.10 (12) | 0.14 ± 0.09 (8) | 0.036 ± 0.005 (8) | >1 (>43) |
The amino acid substitutions identified in the protease-encoding region of HIV-1SQV-5 μM, HIV-1APV-5 μM, HIV-1IDV-5 μM, HIV-1NFV-5 μM, HIV-1RTV-5 μM, HIV-1LPV-1 μM, HIV-1ATV-1 μM, and HIV-1GRL-02031-5 μM compared to the consensus B sequence cited from the Los Alamos National Laboratory database include L10I/G48V/I54V/L90M, L10F/V32I/M46I/I54M//A71V/I84V, L10F/L24I/M46I/L63P/A71V/G73S/V82T, L10F/D30N/K45I/A71V/T74S, M46I/V82F/I84V, L10F/M46I/I54V/V82A, L23I/K43I/M46I/I50L/G51A/A71V, and L10F/L33F/M46I/I47V/Q58E/V82I/I84V/I85V, respectively. MT-4 cells (1 × 104) were exposed to each HIV-1 isolate (100 TCID50s), and the inhibition of p24 Gag protein production by the drug was used as the endpoint. The numbers in parentheses represent the fold changes in the EC50s for each isolate compared to the EC50 for HIV-1NL4-3. The data shown are mean values (±1 standard deviation) derived from the results of three independent experiments conducted in triplicate.
When the virus was selected with up to 5 μM GRL-02031 (HIV-1GRL-02031-5 μM) was examined in MT-4 cells, the EC50 of GRL-02031 turned out to be >1 μM although HIV- 1GRL-02031-5 μM remained susceptible to other PIs, in particular, SQV, IDV, and NFV. The HIV-1LPV-1 μM variant was substantially resistant to APV, IDV, NFV, RTV, LPV, and ATV; however, this variant was highly susceptible to GRL-02031 and had an EC50 of 0.038 μM (Table 3). HIV-1ATV-1 μM variant was also substantially resistant to IDV, NFV, and ATV; however, this variant was susceptible to LPV and GRL-02031. Of note, LPV, which has currently been widely used as a first-line therapeutic among HAART regimens, was not active against three HIV-1 variants (HIV-1SQV-5 μM, a variant selected with 5 μM IDV [HIV-1IDV-5 μM], and a variant selected with 5 μM NFV [HIV-1NFV-5 μM]), with the differences in the EC50s being more than 16-fold compared to the value for wild-type strain HIV-1NL4-3. This anti-HIV-1 profile of LPV greatly contrasted with that of GRL-02031. GRL-02031 was highly potent against all the variants examined except HIV-1SQV-5 μM (sixfold change in the EC50 compared to that for HIV-1NL4-3). It is also noteworthy that SQV, IDV, and NFV remained potent against HIV-1GRL-02031-5 μM, suggesting that the combination of GRL-02031 and SQV, IDV, or NFV could exert complementarily augmented activity against multi-PI-resistant HIV-1 variants.
Sensitivities of infectious molecular HIV-1 clones carrying various amino acid substitutions to GRL-02031.
Finally, we attempted to determine the profile of the activity of GRL-02031 against a variety of HIV-1NL4-3-based molecular infectious clones containing a single primary mutation previously reported or a combination of such mutations (Table 4) (21, 31, 40-42). Interestingly, no significant changes in EC50s were observed when HIV-1 clones containing only one of the amino acid substitutions (L10F, L33F, M46I, I47V, Q58E, V82I, I84V, or I85V) which emerged in the selection process in the present work (Fig. 3) were tested with GRL-02031. We tested each of these one-mutation-containing infectious clones against a selected PI, and again, no significant changes in EC50s were seen (Table 4).
TABLE 4.
Sensitivities of infectious molecular HIV clones carrying various amino acid substitutions to GRL-02031a
| Recombinant HIV-1 clone | EC50 (μM) of GRL-02031 | Fold change in EC50 | Fold change in EC50 compared with that of other PIs | Fold change in EC50 of other PIs reported previously (reference) |
|---|---|---|---|---|
| pNL4-3 (wild-type) | 0.023 ± 0.008 | 1.0 | ||
| L10F | 0.037 ± 0.001 | 1.6 | 1.4 (APV) | 1.5 (IDV) (42) |
| L33F | 0.028 ± 0.005 | 1.2 | 1.0 (RTV) | 1.4 (APV) (31) |
| M46I | 0.028 ± 0.009 | 1.2 | 1.2 (RTV) | 1.0 (APV) (40) |
| I47V | 0.037 ± 0.006 | 1.6 | 1.2 (APV) | 2.2 (APV) (31) |
| Q58E | 0.033 ± 0.007 | 1.4 | 1.0 (APV) | Not previously reported |
| V82I | 0.035 ± 0.001 | 1.5 | 1.5 (RTV) | 1.9 (APV) (31) |
| I84V | 0.030 ± 0.0001 | 1.3 | 2.2 (IDV) | 10.6 (IDV) (41) |
| I85V | 0.024 ± 0.011 | 1.0 | 2.1 (RTV) | Not previously reported |
| M46I/I47V | 0.073 ± 0.009 | 3.2 | 1.3 (APV) | 1.0 (APV) (40) |
| V82I/I85V | 0.035 ± 0.002 | 1.5 | 1.6 (RTV) | Not previously reported |
| I84V/I85V | 0.097 ± 0.010 | 4.2 | 14.8 (RTV) | Not previously reported |
| L10F/I47V/V82I/I85V | 0.43 ± 0.06 | 18.7 | 1.9 (RTV) | Not previously reported |
| L10F/M46I/I47V/V82I/I85V | >1 | >43 | 10.0 (APV) | Not previously reported |
| D30N | 0.020 ± 0.009 | 0.9 | 5.6 (NFV) | 6.0 (NFV) (41) |
| G48V | 0.040 ± 0.0008 | 1.7 | 5.1 (SQV) | 7.0 (SQV) (21) |
| I50V | 0.015 ± 0.008 | 0.7 | 1.2 (APV) | 3.5 (APV) (31) |
| L90M | 0.032 ± 0.001 | 1.4 | 1.0 (SQV) | 3.0 (SQV) (21) |
MT-4 cells (1 × 104/ml) were exposed to 100 TCID50s of each infectious molecular HIV clone, and the inhibition of p24 Gag protein production by the drug was used as the endpoint on day 7 in culture. The fold change represents the ratio of the EC50 for each mutant clone to the EC50 for wild-type HIV-1NL4-3. All assays were performed in triplicate, and the values shown are mean values (±1 standard deviation) derived from the results of three independent experiments.
It was noted that increases in the EC50s of GRL-02031 were seen only when more than two amino acid substitutions were introduced into HIV-1NL4-3. A substantial reduction in susceptibility (differences in EC50s of more than threefold) was seen when the virus had two mutations (M46I/I47V or I84V/I85V). Further increases in the EC50s were seen when four substitutions (L10F/I47V/V82I/I85V) or five substitutions (L10F/M46I/I47V/V82I/I85V) were introduced. Moreover, we generated molecular clones containing a primary mutation with which HIV-1 is known to acquire substantial resistance to a PI(s) (such as D30N, G48V, I50V, and L90M) and determined the EC50s of GRL-02031 (Table 4). We found that GRL-02031 was potent against all such molecular clones with a primary mutation, with the differences in the EC50s being 0.7- to 1.7-fold in comparison with the EC50 for HIV-1NL4-3, although HIV-1D30N and HIV-1G48V showed moderate to substantial levels of resistance to NFV (5.6-fold change) and SQV (5.1-fold change) (Table 4). These data suggest that HIV-1 can acquire substantial resistance to GRL-02031 only when it gains multiple mutations in the protease, a potentially advantageous property of GRL-02031.
Structural analysis of GRL-02031 interactions with wild-type protease.
Finally, we conducted molecular and structural analyses of the interactions of GRL-02031 with protease (Fig. 4). By refined structural modeling based on the previously published crystal structures 2FDE (protease-brecanavir complex) and 2IEN (protease-DRV complex), we found that the oxygen atom of Cp-THF has a hydrogen bond interaction with Asp29 in the S-2 pocket of the protease. The hydrogen bond interactions of GRL-02031 with Asp25 and Gly27, which have been observed for various PIs, were also predicted to be present. In addition, GRL-02031 has hydrogen bond interactions mediated through a water molecule with flap residues Ile50 and Ile50′. Of note, GRL-02031 has an R configuration at the pyrrolidone stereocenter. Interestingly, the structural models demonstrated that for the R-stereochemical configuration, two distinct binding modes of GRL-02031 were found in the S-2′ pocket. The 2-pyrrolidone group and the methoxybenzene moiety can orient toward Asp29′ and Asp30′ for configuration 1 and configuration 2, respectively (Fig. 4A and B). In configuration 1, the 2-pyrrolidone oxygen has hydrogen bond interactions with Asp29′ in the S-2′ pocket. In configuration 2, the methoxybenzene orients toward the S-2′ pocket and forms tight hydrogen bonds with Asp30′. The interactions of PIs (48) with Asp29′ and/or Asp30′ have been reported to be mediated by water molecules. It is likely that the presence of water molecules may influence the relative abundance of configurations 1 and 2. The alternate bimodal binding feature observed in this molecular analysis should provide advantages to the PI in maintaining its antiviral potency when the HIV-1 protease either has a polymorphism or develops amino acid substitutions under drug pressure.
FIG. 4.

Molecular interactions of GRL-02031 with HIV-1 protease. (A) A model of the interaction of GRL-02031 with HIV protease. The bird's-eye view of the docked pose (inset) is presented along with a blown-up figure highlighting the important hydrogen bond interactions. The inhibitor is predicted to have hydrogen bond interactions with Asp25, Gly27, Asp29, Ile50, Asp29′, and Ile50′. Note that the pyrrolidone oxygen (red stick) interacts with the S-2′ subpocket and forms a hydrogen bond interaction with Asp29′. (B) Superimposed binding configurations of GRL-02031 with the HIV-1 protease. The carbons are shown in gray in configuration 1 and in green in configuration 2. Selected hydrogen bond interactions of configuration 2 are shown. In configuration 2, the methoxybenzene interacts with the S-2′ site and forms a hydrogen bond interaction with Asp30′. The interaction of the P-2 ligand Cp-THF is the same in both configurations. (C) The binding cavity of HIV protease with lipophilic potential is shown. GRL-02031 fits tightly in the binding cavity and has favorable polar and nonpolar interactions with the active-site residues of the HIV-1 protease. The van der Waals surfaces of Ile47 and Ile47′ (both in magenta) and of Ile84′ (in purple) demonstrate that they form tight nonpolar interactions with GRL-02031. The protease residues are shown in stick representation. The following atoms are indicated by designated colors: C, gray; O, red; N, blue; S, yellow; H, cyan. Both protease chains are shown in green. The figure was generated with the MOLCAD program (Sybyl, version 8.0; Tripos, L.P, St. Louis, MO).
We also examined the lipophilic potential of the computationally defined cavity for the binding of GRL-02031 within the HIV protease (Fig. 4C). It was revealed that GRL-02031 fits tightly in the binding cavity and has favorable polar and nonpolar interactions with the active-site residues of the HIV-1 protease. The van der Waals surfaces of Ile47 and Ile47′ and of Ile84′ demonstrate that they form tight nonpolar interactions with GRL-02031. Our antiviral data showing that the I47V substitution is associated with HIV-1 resistance to GRL-02031 (Tables 3 and 4) are in agreement with this structural finding, in that the substitution should reduce GRL-02031's interaction with protease and helps develop HIV-1 resistance to the inhibitor.
DISCUSSION
In the present work, we demonstrated that GRL-02031 suppresses the replication of a wide spectrum of HIV-1 isolates and is potent against a variety of HIV-1 variants highly resistant to multiple PIs, with the differences in the EC50s being less than twofold in comparison with the EC50 for wild-type strain HIV-1ERS104pre (Table 2). Additionally, when HIV-1NL4-3 was propagated in the presence of increasing concentrations of IDV, APV, or GRL-02031, the time of emergence of HIV-1 variants highly resistant to GRL-02031 was substantially delayed compared to that of IDV- or APV-resistant HIV-1 variants (Fig. 2). Indeed, 21, 27, and 37 passages were required for HIV-1 to acquire the ability to propagate in the presence of APV, IDV, and GRL-02031 at 5 μM, respectively. In this regard, when we generated a variety of PI-resistant HIV-1 variants by propagating laboratory strain HIV-1NL4-3 in the presence of increasing concentrations of a PI in MT-4 cells using the same procedure as that used in the present study, it required 27, 23, 22, 21, and 14 passages for the virus to propagate in the presence of 5 μM of SQV, APV, IDV, NFV, and RTV, respectively (26). However, it should be noted that the population size of HIV-1 in a culture is relatively small and that the viral acquisition of mutations can be affected by stochastic phenomena. For example, mutations take place at random and the rates of mutations in the HIV-1 genome may not be reproducible, although certain mutations that severely compromise viral replication would not remain in culture.
During the selection of HIV-1NL4-3 with GRL-02031, the L10F substitution, one of the secondary substitutions, first appeared. The L10F mutation occurs distal to the active site of the enzyme and is thought to act in concert with active-site mutations and compensate for a possible functional deficit caused by the latter (6, 32). Mutations at Leu-10 reportedly occur in 5 to 10% of HIV-1 isolates recovered from untreated HIV-1-infected individuals but increase in prevalence by 60 to 80% in heavily treated patients (19, 22). However, the virological and structural significance of the L10F substitution in HIV-1 resistance to GRL-02031 is presently unknown.
By passage 37, two active-site mutations (V82I and I84V) emerged. These V82 and I84 residues represent active-site residues whose side chains are involved in the formation of the protease substrate cleft and that make direct contact with certain PIs (48), and the V82I substitution has been shown to be effective in conferring resistance when it is combined with a second active-site mutation, such as V32I (23). Another active-site mutation (I85V) and two flap mutations (M46I and I47V) also emerged by passage 30. Both Met46 and Ile47 are located in the flap region of the enzyme; the I47V substitution is reported to be associated with viral resistance to APV and JE-2147 (40, 48). The lipophilic potential of the computationally defined cavity for the binding of GRL-02031 within the HIV protease seems to be related to a finding that the van der Waals surfaces of Ile47 and Ile47′ and of Ile84′ form tight nonpolar interactions with GRL-02031 (Fig. 4C). Our antiviral data showing that the I47V substitution is associated with HIV-1 resistance to GRL-02031 (Table 3) are in agreement with this structural finding. However, it is also of note that HIV-1 acquires substantial resistance to GRL-02031 when the virus gains multiple mutations in the protease (Table 4), as seen in the case of DRV (9). This resistance profile (i.e., the requirement of multiple mutations) of GRL-02031 may also confer certain advantage in the resistance profile of GRL-02031.
Two mutations at conserved residues, L33F and Q58E, also emerged by passage 37 and were present in 10 and 9 of 10 clones, respectively. L33F has primarily been reported in patients treated with RTV or APV (37). The L33F substitution alone did not change the susceptibility of HIV-1 to GRL-02031 (Table 4), although it has recently gained attention because of its association with resistance to the FDA-approved PI, tipranavir (33).
In the HIV-1 variants selected with GRL-02031, four amino acid substitutions in the Gag proteins (G62R, R409K, L363M, and I437T) were seen by passage 37. R409K within the p7 Gag seems to be associated with viral resistance to APV (14), although the significance of G62R within p17 is as yet unknown. The p7-p1 cleavage-site mutation I437T has been reported to be associated with ATV resistance (17). It is of note that by passage 15, an unusual amino acid substitution, L363M, emerged; this substitution has not previously been reported in relation to PI resistance. This L363M is located at the p24-p2 cleavage site, which represents the C terminus of the capsid (CA) p24 protein that is highly conserved and that is involved in virion assembly. The deletion of this cluster or the introduction of mutations such as L363A is known to cause significant impairment of particle formation and infectivity (34). It is noteworthy that L363M appears in HIV-1 variants resistant to a maturation inhibitor, PA-457 [3-O-(3′,3′-dimethylsuccinyl) betulinic acid], which binds to the CA-p2 cleavage site or its proximity, blocks the cleavage by protease during virion maturation, and exerts activity against HIV-1 (27, 44, 49).
It was noted that GRL-02031 and SQV remained active against most of the PI-selected HIV-1 variants and that SQV, IDV, and NFV remained potent against HIV-1GRL-02031-5 μM (Table 3), suggesting that the combination of GRL-02031, SQV, IDV, and NFV can exert complementarily augmented activity against multi-PI-resistant HIV-1 variants. Such a difference in the resistance profile of GRL-02031 when it is used with SQV and NFV may be due to the differences in binding and antiviral potency associated with the D30N and G48V mutations (Table 4).
In an attempt to explain why GRL-02031 can exert potent activity against a wide spectrum of HIV-1 variants resistant to multiple PIs, we performed structural modeling and molecular docking of the interactions of GRL-02031 with protease (Fig. 4). Interestingly, our structural modeling analysis demonstrated that there are two distinct binding modes of GRL-02031 in the S-2′ pocket of the protease. Either the 2-pyrrolidone group or the methoxybenzene moiety can orient toward Asp29′ and Asp30′ (configuration 1 and configuration 2, respectively) (Fig. 4B). It is presumed that such alternate binding modes provide distinct advantages to GRL-02031 in maintaining its antiviral activity against a wide spectrum of HIV-1 variants resistant to other currently available PIs. The alternate binding modes could explain the reason why the development of resistance to GRL-02031 is substantially delayed compared to the time to the development of resistance to APV or IDV (Fig. 2). In addition, the models of GRL-02031 indicated that it is capable of forming hydrogen bond interactions with the backbone atoms of Asp29, Asp29′, and/or Asp30′. Such backbone interactions have been shown to be important in maintaining potency not only against wild-type protease but also against drug-resistant mutant proteases (1, 15, 16, 36). This may also explain why GRL-02031 maintains its potency against a wide variety of drug-resistant mutant proteases.
It is of note that the difference seen with GRL-02031 (one- to twofold) seems substantially less than that seen with DRV (one- to sevenfold) (Table 2). Although this difference may not be translated into an actual difference in the clinical setting, it is worth noting that GRL-02031 may have certain advantages in its activity against highly drug-resistant HIV-1 variants. Considering that the acquisition of multiple amino acid substitutions is required for the emergence of HIV-1 resistance to GRL-02031, the profile of HIV-1 resistance to GRL-02031, which is apparently different from the profiles for the other PIs, might result in an advantage for GRL-02031, although further evaluations, including testing of the compound in the clinical setting, are required.
Taken together, GRL-02031 exerts potent activity against a wide spectrum of laboratory and clinical wild-type and multidrug-resistant HIV-1 strains without significant cytotoxicity in vitro and substantially delays the emergence of HIV-1 variants resistant to GRL-02031. These data warrant further consideration of GRL-02031 as a candidate as a novel PI for the treatment of AIDS.
Acknowledgments
We thank Shintaro Matsumi, Toshikazu Miyakawa, and Manabu Aoki for helpful discussion.
This work was supported in part by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (to D.D., H.N., and H.M.); a grant from the National Institutes of Health (grant GM 53386 to A.K.G.); a grant from a Research for the Future Program of the Japan Society for the Promotion of Science (grant JSPS-RFTF 97L00705 to H.M.); a Grant-in-Aid for Scientific Research (grant for priority areas to H.M.) from the Ministry of Education, Culture, Sports, Science, and Technology (Monbu-Kagakusho) of Japan; and a Grant for Promotion of AIDS Research from the Ministry of Health, Labor and Welfare (Kosei-Rodosho) of Japan (to H.M.).
This work utilized the computational resources of the Biowulf cluster at the NIH.
Footnotes
Published ahead of print on 27 October 2008.
REFERENCES
- 1.Amano, M., Y. Koh, D. Das, J. Li, S. Leschenko, Y. F. Wang, P. I. Boross, I. T. Weber, A. K. Ghosh, and H. Mitsuya. 2007. A novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI), GRL-98065, is potent against multiple-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 51:2143-2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antiretroviral Therapy Cohort Collaboration. 2008. Life expectancy of individuals on combination antiretroviral therapy in high-income countries: a collaborative analysis of 14 cohort studies. Lancet 372:293-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bhaskaran, K., O. Hamouda, M. Sannes, F. Boufassa, A. M. Johnson, P. C. Lambert, and K. Porter. 2008. Changes in the risk of death after HIV seroconversion compared with mortality in the general population. JAMA 300:51-59. [DOI] [PubMed] [Google Scholar]
- 4.Cho, A. E., V. Guallar, B. J. Berne, and R. Friesner. 2005. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 26:915-931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clotet, B., N. Bellos, J. M. Molina, D. Cooper, J. C. Goffard, A. Lazzarin, A. Wohrmann, C. Katlama, T. Wilkin, R. Haubrich, C. Cohen, C. Farthing, D. Jayaweera, M. Markowitz, P. Ruane, S. Spinosa-Guzman, and E. Lefebvre. 2007. Efficacy and safety of darunavir-ritonavir at week 48 in treatment-experienced patients with HIV-1 infection in POWER 1 and 2: a pooled subgroup analysis of data from two randomised trials. Lancet 369:1169-1178. [DOI] [PubMed] [Google Scholar]
- 6.Condra, J. H., W. A. Schleif, O. M. Blahy, L. J. Gabryelski, D. J. Graham, J. C. Quintero, A. Rhodes, H. L. Robbins, E. Roth, M. Shivaprakash, et al. 1995. In vivo emergence of HIV-1 variants resistant to multiple protease inhibitors. Nature 374:569-571. [DOI] [PubMed] [Google Scholar]
- 7.De Clercq, E. 2002. Strategies in the design of antiviral drugs. Nat. Rev. Drug Discov. 1:13-25. [DOI] [PubMed] [Google Scholar]
- 8.de Mendoza, C., and V. Soriano. 2004. Resistance to HIV protease inhibitors: mechanisms and clinical consequences. Curr. Drug Metab. 5:321-328. [DOI] [PubMed] [Google Scholar]
- 9.De Meyer, S., H. Azijn, D. Surleraux, D. Jochmans, A. Tahri, R. Pauwels, P. Wigerinck, and M. P. de Bethune. 2005. TMC114, a novel human immunodeficiency virus type 1 protease inhibitor active against protease inhibitor-resistant viruses, including a broad range of clinical isolates. Antimicrob. Agents Chemother. 49:2314-2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erickson, J. W., and S. K. Burt. 1996. Structural mechanisms of HIV drug resistance. Annu. Rev. Pharmacol. Toxicol. 36:545-571. [DOI] [PubMed] [Google Scholar]
- 11.Ferrer, E., D. Podzamczer, M. Arnedo, E. Fumero, P. McKenna, A. Rinehart, J. L. Perez, M. J. Barbera, T. Pumarola, J. M. Gatell, and F. Gudiol. 2003. Genotype and phenotype at baseline and at failure in human immunodeficiency virus-infected antiretroviral-naive patients in a randomized trial comparing zidovudine and lamivudine plus nelfinavir or nevirapine. J. Infect. Dis. 187:687-690. [DOI] [PubMed] [Google Scholar]
- 12.Friesner, R. A., J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis, and P. S. Shenkin. 2004. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47:1739-1749. [DOI] [PubMed] [Google Scholar]
- 13.Friesner, R. A., R. B. Murphy, M. P. Repasky, L. L. Frye, J. R. Greenwood, T. A. Halgren, P. C. Sanschagrin, and D. T. Mainz. 2006. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J. Med. Chem. 49:6177-6196. [DOI] [PubMed] [Google Scholar]
- 14.Gatanaga, H., Y. Suzuki, H. Tsang, K. Yoshimura, M. F. Kavlick, K. Nagashima, R. J. Gorelick, S. Mardy, C. Tang, M. F. Summers, and H. Mitsuya. 2002. Amino acid substitutions in Gag protein at non-cleavage sites are indispensable for the development of a high multitude of HIV-1 resistance against protease inhibitors. J. Biol. Chem. 277:5952-5961. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh, A. K., B. D. Chapsal, I. T. Weber, and H. Mitsuya. 2008. Design of HIV protease inhibitors targeting protein backbone: an effective strategy for combating drug resistance. Acc. Chem. Res. 41:78-86. [DOI] [PubMed] [Google Scholar]
- 16.Ghosh, A. K., P. R. Sridhar, S. Leshchenko, A. K. Hussain, J. Li, A. Y. Kovalevsky, D. E. Walters, J. E. Wedekind, V. Grum-Tokars, D. Das, Y. Koh, K. Maeda, H. Gatanaga, I. T. Weber, and H. Mitsuya. 2006. Structure-based design of novel HIV-1 protease inhibitors to combat drug resistance. J. Med. Chem. 49:5252-5261. [DOI] [PubMed] [Google Scholar]
- 17.Gong, Y. F., B. S. Robinson, R. E. Rose, C. Deminie, T. P. Spicer, D. Stock, R. J. Colonno, and P. F. Lin. 2000. In vitro resistance profile of the human immunodeficiency virus type 1 protease inhibitor BMS-232632. Antimicrob. Agents Chemother. 44:2319-2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gupta, R., A. Hill, A. W. Sawyer, and D. Pillay. 2008. Emergence of drug resistance in HIV type 1-infected patients after receipt of first-line highly active antiretroviral therapy: a systematic review of clinical trials. Clin. Infect. Dis. 47:712-722. [DOI] [PubMed] [Google Scholar]
- 19.Hertogs, K., S. Bloor, S. D. Kemp, C. Van den Eynde, T. M. Alcorn, R. Pauwels, M. Van Houtte, S. Staszewski, V. Miller, and B. A. Larder. 2000. Phenotypic and genotypic analysis of clinical HIV-1 isolates reveals extensive protease inhibitor cross-resistance: a survey of over 6000 samples. AIDS 14:1203-1210. [DOI] [PubMed] [Google Scholar]
- 20.Hicks, C. B., P. Cahn, D. A. Cooper, S. L. Walmsley, C. Katlama, B. Clotet, A. Lazzarin, M. A. Johnson, D. Neubacher, D. Mayers, and H. Valdez. 2006. Durable efficacy of tipranavir-ritonavir in combination with an optimised background regimen of antiretroviral drugs for treatment-experienced HIV-1-infected patients at 48 weeks in the Randomized Evaluation of Strategic Intervention in multi-drug reSistant patients with Tipranavir (RESIST) studies: an analysis of combined data from two randomised open-label trials. Lancet 368:466-475. [DOI] [PubMed] [Google Scholar]
- 21.Jacobsen, H., K. Yasargil, D. L. Winslow, J. C. Craig, A. Krohn, I. B. Duncan, and J. Mous. 1995. Characterization of human immunodeficiency virus type 1 mutants with decreased sensitivity to proteinase inhibitor Ro 31-8959. Virology 206:527-534. [DOI] [PubMed] [Google Scholar]
- 22.Kantor, R., W. J. Fessel, A. R. Zolopa, D. Israelski, N. Shulman, J. G. Montoya, M. Harbour, J. M. Schapiro, and R. W. Shafer. 2002. Evolution of primary protease inhibitor resistance mutations during protease inhibitor salvage therapy. Antimicrob. Agents Chemother. 46:1086-1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaplan, A. H., S. F. Michael, R. S. Wehbie, M. F. Knigge, D. A. Paul, L. Everitt, D. J. Kempf, D. W. Norbeck, J. W. Erickson, and R. Swanstrom. 1994. Selection of multiple human immunodeficiency virus type 1 variants that encode viral proteases with decreased sensitivity to an inhibitor of the viral protease. Proc. Natl. Acad. Sci. USA 91:5597-5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kemper, C. A., M. D. Witt, P. H. Keiser, M. P. Dube, D. N. Forthal, M. Leibowitz, D. S. Smith, A. Rigby, N. S. Hellmann, Y. S. Lie, J. Leedom, D. Richman, J. A. McCutchan, and R. Haubrich. 2001. Sequencing of protease inhibitor therapy: insights from an analysis of HIV phenotypic resistance in patients failing protease inhibitors. AIDS 15:609-615. [DOI] [PubMed] [Google Scholar]
- 25.Koh, Y., S. Matsumi, D. Das, M. Amano, D. A. Davis, J. Li, S. Leschenko, A. Baldridge, T. Shioda, R. Yarchoan, A. K. Ghosh, and H. Mitsuya. 2007. Potent inhibition of HIV-1 replication by novel non-peptidyl small molecule inhibitors of protease dimerization. J. Biol. Chem. 282:28709-28720. [DOI] [PubMed] [Google Scholar]
- 26.Koh, Y., H. Nakata, K. Maeda, H. Ogata, G. Bilcer, T. Devasamudram, J. F. Kincaid, P. Boross, Y. F. Wang, Y. Tie, P. Volarath, L. Gaddis, R. W. Harrison, I. T. Weber, A. K. Ghosh, and H. Mitsuya. 2003. Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI) UIC-94017 (TMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro. Antimicrob. Agents Chemother. 47:3123-3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li, F., R. Goila-Gaur, K. Salzwedel, N. R. Kilgore, M. Reddick, C. Matallana, A. Castillo, D. Zoumplis, D. E. Martin, J. M. Orenstein, G. P. Allaway, E. O. Freed, and C. T. Wild. 2003. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc. Natl. Acad. Sci. USA 100:13555-13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Little, S. J., S. Holte, J. P. Routy, E. S. Daar, M. Markowitz, A. C. Collier, R. A. Koup, J. W. Mellors, E. Connick, B. Conway, M. Kilby, L. Wang, J. M. Whitcomb, N. S. Hellmann, and D. D. Richman. 2002. Antiretroviral-drug resistance among patients recently infected with HIV. N. Engl. J. Med. 347:385-394. [DOI] [PubMed] [Google Scholar]
- 29.Maeda, K., H. Nakata, Y. Koh, T. Miyakawa, H. Ogata, Y. Takaoka, S. Shibayama, K. Sagawa, D. Fukushima, J. Moravek, Y. Koyanagi, and H. Mitsuya. 2004. Spirodiketopiperazine-based CCR5 inhibitor which preserves CC-chemokine/CCR5 interactions and exerts potent activity against R5 human immunodeficiency virus type 1 in vitro. J. Virol. 78:8654-8662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maeda, K., K. Yoshimura, S. Shibayama, H. Habashita, H. Tada, K. Sagawa, T. Miyakawa, M. Aoki, D. Fukushima, and H. Mitsuya. 2001. Novel low molecular weight spirodiketopiperazine derivatives potently inhibit R5 HIV-1 infection through their antagonistic effects on CCR5. J. Biol. Chem. 276:35194-35200. [DOI] [PubMed] [Google Scholar]
- 31.Maguire, M., D. Shortino, A. Klein, W. Harris, V. Manohitharajah, M. Tisdale, R. Elston, J. Yeo, S. Randall, F. Xu, H. Parker, J. May, and W. Snowden. 2002. Emergence of resistance to protease inhibitor amprenavir in human immunodeficiency virus type 1-infected patients: selection of four alternative viral protease genotypes and influence of viral susceptibility to coadministered reverse transcriptase nucleoside inhibitors. Antimicrob. Agents Chemother. 46:731-738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mammano, F., V. Trouplin, V. Zennou, and F. Clavel. 2000. Retracing the evolutionary pathways of human immunodeficiency virus type 1 resistance to protease inhibitors: virus fitness in the absence and in the presence of drug. J. Virol. 74:8524-8531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCallister, S., V. Kohlbrenner, K. Squires, A. Lazzarin, P. Kumar, E. DeJesus, J. Nadler, J. Gallant, S. Walmsley, P. Yeni, J. Leith, C. Dohnanyi, D. Hall, J. Sabo, T. MacGregor, W. Verbiest, P. McKenna, and D. Mayers. 2003. Characterization of the impact of genotype, phenotype, and inhibitory quotient on antiviral activity of tipranavir in highly treatment-experienced patients. Antivir. Ther. 8:S15. [Google Scholar]
- 34.Melamed, D., M. Mark-Danieli, M. Kenan-Eichler, O. Kraus, A. Castiel, N. Laham, T. Pupko, F. Glaser, N. Ben-Tal, and E. Bacharach. 2004. The conserved carboxy terminus of the capsid domain of human immunodeficiency virus type 1 Gag protein is important for virion assembly and release. J. Virol. 78:9675-9688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mitsuya, H., and J. Erickson. 1999. Discovery and development of antiretroviral therapeutics for HIV infection, p. 751-780. In T. C. Merigan, J. G. Bartlet, and D. Bolognesi (ed.), Textbook of AIDS medicine. The Williams & Wilkins Co., Baltimore, MD.
- 36.Mitsuya, H., K. Maeda, D. Das, and A. K. Ghosh. 2008. Development of protease inhibitors and the fight with drug-resistant HIV-1 variants. Adv. Pharmacol. 56:169-197. [DOI] [PubMed] [Google Scholar]
- 37.Molla, A., M. Korneyeva, Q. Gao, S. Vasavanonda, P. J. Schipper, H. M. Mo, M. Markowitz, T. Chernyavskiy, P. Niu, N. Lyons, A. Hsu, G. R. Granneman, D. D. Ho, C. A. Boucher, J. M. Leonard, D. W. Norbeck, and D. J. Kempf. 1996. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat. Med. 2:760-766. [DOI] [PubMed] [Google Scholar]
- 38.Murphy, E. L., A. C. Collier, L. A. Kalish, S. F. Assmann, M. F. Para, T. P. Flanigan, P. N. Kumar, L. Mintz, F. R. Wallach, and G. J. Nemo. 2001. Highly active antiretroviral therapy decreases mortality and morbidity in patients with advanced HIV disease. Ann. Intern. Med. 135:17-26. [DOI] [PubMed] [Google Scholar]
- 39.Murphy, R. L. 2000. New antiretroviral drugs in development. AIDS 14(Suppl. 3):S227-S234. [PubMed] [Google Scholar]
- 40.Partaledis, J. A., K. Yamaguchi, M. Tisdale, E. E. Blair, C. Falcione, B. Maschera, R. E. Myers, S. Pazhanisamy, O. Futer, A. B. Cullinan, C. M. Stuver, R. A. Byrn, and D. J. Livingston. 1995. In vitro selection and characterization of human immunodeficiency virus type 1 (HIV-1) isolates with reduced sensitivity to hydroxyethylamino sulfonamide inhibitors of HIV-1 aspartyl protease. J. Virol. 69:5228-5235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Patick, A. K., M. Duran, Y. Cao, D. Shugarts, M. R. Keller, E. Mazabel, M. Knowles, S. Chapman, D. R. Kuritzkes, and M. Markowitz. 1998. Genotypic and phenotypic characterization of human immunodeficiency virus type 1 variants isolated from patients treated with the protease inhibitor nelfinavir. Antimicrob. Agents Chemother. 42:2637-2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Prado, J. G., T. Wrin, J. Beauchaine, L. Ruiz, C. J. Petropoulos, S. D. Frost, B. Clotet, R. T. D'Aquila, and J. Martinez-Picado. 2002. Amprenavir-resistant HIV-1 exhibits lopinavir cross-resistance and reduced replication capacity. AIDS 16:1009-1017. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez-Barrios, F., and F. Gago. 2004. HIV protease inhibition: limited recent progress and advances in understanding current pitfalls. Curr. Top. Med. Chem. 4:991-1007. [DOI] [PubMed] [Google Scholar]
- 44.Salzwedel, K., R. Goila-Gaur, C. Adamson, F. Li, A. Castillo, N. Kilgore, M. Reddick, C. Matallana, D. Zoumplis, D. Martin, G. Allaway, E. Freed, and C. Wild. 2004. Selection for and characterization of HIV-1 isolates resistant to the maturation inhibitor PA-457. Antivir. Ther. 9:S8. [Google Scholar]
- 45.Shirasaka, T., M. F. Kavlick, T. Ueno, W. Y. Gao, E. Kojima, M. L. Alcaide, S. Chokekijchai, B. M. Roy, E. Arnold, R. Yarchoan, et al. 1995. Emergence of human immunodeficiency virus type 1 variants with resistance to multiple dideoxynucleosides in patients receiving therapy with dideoxynucleosides. Proc. Natl. Acad. Sci. USA 92:2398-23402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Walensky, R. P., A. D. Paltiel, E. Losina, L. M. Mercincavage, B. R. Schackman, P. E. Sax, M. C. Weinstein, and K. A. Freedberg. 2006. The survival benefits of AIDS treatment in the United States. J. Infect. Dis. 194:11-19. [DOI] [PubMed] [Google Scholar]
- 47.Yoshimura, K., R. Kato, M. F. Kavlick, A. Nguyen, V. Maroun, K. Maeda, K. A. Hussain, A. K. Ghosh, S. V. Gulnik, J. W. Erickson, and H. Mitsuya. 2002. A potent human immunodeficiency virus type 1 protease inhibitor, UIC-94003 (TMC-126), and selection of a novel (A28S) mutation in the protease active site. J. Virol. 76:1349-1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshimura, K., R. Kato, K. Yusa, M. F. Kavlick, V. Maroun, A. Nguyen, T. Mimoto, T. Ueno, M. Shintani, J. Falloon, H. Masur, H. Hayashi, J. Erickson, and H. Mitsuya. 1999. JE-2147: a dipeptide protease inhibitor (PI) that potently inhibits multi-PI-resistant HIV-1. Proc. Natl. Acad. Sci. USA 96:8675-8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou, J., X. Yuan, D. Dismuke, B. M. Forshey, C. Lundquist, K. H. Lee, C. Aiken, and C. H. Chen. 2004. Small-molecule inhibition of human immunodeficiency virus type 1 replication by specific targeting of the final step of virion maturation. J. Virol. 78:922-929. [DOI] [PMC free article] [PubMed] [Google Scholar]