

Abstract
Genetic contributions to human cognition and behavior are clear but difficult to define. Williams syndrome (WS) provides a unique model for relating single genes to visual-spatial cognition and social behavior. We defined a ~1.5 Mb region of ~25 genes deleted in >98% of typical WS and then rare small deletions, showing that visual-spatial construction (VSC) in WS was associated with the genes GTF2IRD1 and GTF2I. To distinguish the roles of GTF2IRD1 and GTF2I in VSC and social behavior, we utilized multiple genomic methods (custom high resolution oligonucleotide microarray, multicolor FISH and somatic cell hybrids analyzed by PCR) to identify individuals deleted for either gene but not both. We analyzed genetic, cognitive and social behavior in a unique individual with WS features (heart defects, small size, facies), but with an atypical deletion of a set of genes that includes GTF2IRD1, but not GTF2I. The centromeric breakpoint localized to the region 72.32-72.38Mb and the telomeric breakpoint to 72.66 Mb, 10kb downstream of GTF2IRD1. Cognitive testing (WPPSI-R, K-BIT, and PLS-3) deomstrated striking deficits in VSC (Block Design, Object Assembly) but overall performance 1.5-3 SD above WS means. We have now integrated the genetic, clinical and cognitive data with previous reports of social behavior in this subject. These results combine with previous data from small deletions to suggest the gene GTF2IRD1 is associated with WS facies and VSC, and that GTF2I may contribute to WS social behaviors including increased gaze and attention to strangers.
Keywords: Williams syndrome, visual-spatial construction, social behavior, GTF2IRD1, GTF2I
INTRODUCTION
Williams syndrome (WS) is among the most compelling genetic disorders of human development and behavior and is due to a recurring deletion of about 1.5-1.8 megabases containing about 25-28 genes in chromosome band 7q11.23 [Ewart et al., 1993; Osborne, 1999], WS occurs once in approximately 7,500-20,000 live births [Greenberg, 1990; Stromme et al., 2002] and is associated with distinct physical, cognitive and behavioral features. Physical symptoms include small stature, a round face with small chin, flat malar region and thick lower lip, a hoarse voice, infantile transient hypercalcemia, hyperaccusis, high sensitivity to music and cardiovascular abnormalities, including supravalvular aortic stenosis (SVAS) and peripheral pulmonic stenosis [Morris, 2006]. A subset of these features, which may change with age, is apparent in most persons with WS.
In the neurocognitive domain, performance is impaired, with a typical IQ of 60 [Bellugi et al., 2000]. Intriguingly, the hallmark difficulties in WS are highly specific and virtually universal deficits in visual-spatial function that are most clearly illustrated by tests of visual-spatial construction, especially those involving block construction and copying measured by the Wechsler and its derivatives [Wechsler, 1974], by the equivalent pattern construction test of the Differential Ability Scales (DAS) [Elliott, 1990] and by the Visual-Motor Integration (VMI) [Beery and Buktenica, 1989]. In WS, these striking defects in visual spatial construction stand in contrast to more subtle defects in processing stimuli that are more specifically social, such as language and faces [Bellugi et al., 2000]. The convergence of different methodologies including integrated analyses of genetics and behavioral performance, are needed to help clarify puzzling findings of impairment in some visual-spatial tasks (pattern construction, copying, drawing) and relative proficiency in others (face and object recognition) found in people with WS.
Of further interest for the current work is that persons with WS appear hypersociable from early in life [Doyle et al., 2004; Järvinen-Pasley et al., 2008]. In addition to general overfriendliness, individuals with WS seem highly engaging, affectionate, interpersonally sensitive, empathic, and socially disinhibited [Udwin and Yule, 1991; Gosch and Pankau, 1994; Dykens and Rosner, 1999; Sullivan and Tager-Flusberg, 1999; Tager-Flusberg and Sullivan, 2000; Plesa-Skwerer et al., 2004; Dykens et al., 2005]. This behavior may be called gregarious, with affected individuals showing eagerness to make eye contact with, and increased approachability to strangers, even at ages when children are normally shy [Mervis and Klein-Tasman, 2000; Jones and Bellugi, 2001; Klein-Tasman and Mervis, 2003]. On the other hand, difficulties in peer interactions [Udwin and Yule, 1991; Gosch and Pankau, 1994, 1997] and in social judgment [Einfeld et al., 1997; Gosch and Pankau, 1997] are common, suggesting that the so-called ‘hypersociability’ does not translate into more advanced social cognitive abilities in WS [Sullivan and Tager-Flusberg, 1999; Tager-Flusberg and Sullivan, 2000]. Structural [Reiss et al., 2004] and functional [Meyer-Lindenberg et al., 2005] magnetic resonance imaging (MRI) and cross-cultural data [Zitzer-Comfort et al., 2007] have suggested that the WS deletion contributes to social behavior. Taken together, evidence to date suggests that the sociability associated with WS is complex, in part genetically based, and carefully designed studies are needed to further elucidate its nature.
We have proposed that genes affecting measureable cognitive features may be implicated by their occurrence in individuals with small deletions [Botta et al., 1999; Korenberg et al., 2000; Bayes et al., 2003; Gagliardi et al., 2003; Heller et al., 2003; Hirota et al., 2003]. For example, deletion of STX1A through GTF2I [Botta et al., 1999] has been associated with a subset of cognitive as well as typical cardiovascular and facial features and with further cases, the deletion of the genes GTF2IRD1 and GTF2I, located at the telomeric end of the deletion [Hirota et al., 2003; Tassabehji et al., 2005] has been implicated in WS defects of visual-spatial construction. Further parsing of the effects of GTF2IRD1 versus GTF2I on WS cognitive and social behavior has been hampered by the small number of cases with small deletions and the limited cognitive data.
Previously, the variability of social behavior in children with WS has been documented using the Salk Institute Sociability Questionnaire (SISQ) [Doyle et al., 2004; Järvinen-Pasley et al., 2008] and direct observation according to a pre-specified ethogram [Järvinen-Pasley et al., 2008]. These reports contrasted and quantified social behavior in toddlers with the typical WS deletion and compared these to a putatively atypical deletion case (5889), the molecular breakpoint analysis for which is reported here and was referred to incorrectly without data in the report of her social behavior in Järvinen-Pasley et al., 2008. Here we report on the clinical and developmental history, physical and cognitive features as well as high resolution molecular genetic analysis of the breakpoint in the atypical deletion Subject 5889 and integrate these data with the social behavior reported previously. We now report the specific WS features found in this subject from age 14 months through 7 years, including facies, cardiovascular (SVAS, PPS, renovascular stenoses, and hypertension), delayed growth, and deficits in visual-spatial construction but also atypical WS features including social behavior and normal developmental milestones in combination with somewhat higher cognitive function. Further, to determine the breakpoint, we have developed a custom high density oligonucleotide array of the human genomic sequence spanning the 14 Mb region that includes the WS deletion and applied this to show that the deletion in 5889 results in the complete hemizygosity of all genes in the typical WS region except one, GTF2I. This provides the capability for molecular definition of breakpoints at the level of single exons, and, in combination with previous data on atypical subjects, allows the further parsing of genes with phenotypes in WS: the gene for GTF2IRD1 with the visual-spatial constructive deficits, GTF2I with aspects of WS social behavior and both to abnormal cognitive development in WS.
MATERIALS AND METHODS
Subject
The family of Subject 5889 reported in this study was given informed consent by their geneticists, which was approved by the IRB of Cedars-Sinai Medical Center, Los Angeles. The clinical features were evaluated by a geneticist with experience in genetic disorders; a follow-up examination was performed when necessary.
Deletion Analysis
Fluorescence in situ hybridization (FISH)
The chromosomes were prepared from peripheral blood by following protocols described by Korenberg and Chen [1995; supplemental online materials]. The FISH study was performed in two-steps. First, three BAC probes that cover the entire common WS deleted and flanking duplicated regions were used to screen for the ends of the deletion (Figure 2A). The probes included BACs 1008H17, 592D8 and 1184P14, of which, 1008H17 includes the genes FKBP6 and FZD9, 592D8 includes ELN, and 1184P14 includes GTF2I. The probe 815K3 was used for an internal experimental control. Next, a series of eight additional BAC, PAC or cosmid probes were tested to define the breakpoint. These included BACs P195H06 (for STX1A), 1148G03 (for ELN), 054H15 and PACs 1186P10 (both overlapping GTF2IRD1 and GTF2I), 239C10 (marking the flanking duplicated regions), and cosmids 182B11 (for CYLN2) and 183E1 (for GTF2IRD1).
FIG. 2.

Deletion breakpoint analysis of Subject 5889. A: Physical map of BAC/PAC/cosmid probes used for molecular cytogenetics with fluorescence in situ hybridization (FISH) and summary of results defining deletion in 5889; B: Polymerase chain reaction (PCR) with somatic cell hybrids of the distal breakpoint region identified by FISH; C: High resolution array CGH analysis of a 14 megabase region on chromosome 7 (64-77Mb) using a custom isothermal oligonucleotide tiling array. The top panel shows the complete tiled region in a 10X window averaged view (200bp probe spacing). Bottom left and right panels show zoomed in views of the proximal and distal breakpoints for the deletion in which all data points (unaveraged data set with 385,013 probes and 20bp median spacing) were analyzed. Red line segments in both bottom panels denote copy number calls by the analysis algorithm (see Materials and Methods).
Somatic cell hybrids and PCR
An immortalized lymphoblast cell line was generated from Subject 5889, grown under standard conditions [Korenberg and Chen, 1995] and somatic cell hybrids were constructed to isolate the deleted and the non-deleted chromosome 7 (GMP Genetics) [Yan et al., 2000]. Chromosome 7 colonies were selected using PCR of polymorphic markers located outside the WS region and DNA was extracted from colonies 9, 11, 12, and 13. To identify the telomeric breakpoint, we utilized PCR with primers within the deleted region (established by FISH) and 14 primer sets flanking the predicted breakpoint region from GTF2IRD1 (73.661Mb, NCBI build 36.2) through the 5'end of GTF2I (73.671Mb) as shown in Figure 2B and SOM Table II.
Oligonucleotide isothermal high resolution DNA tiling microarray
A custom high resolution isothermal oligonucleotide DNA tiling array (Roche NimbleGen) was developed and used to examine both breakpoints (Figure 2C). All array CGH experiments were performed according to previously reported methods [Selzer et al., 2005] except that the copy number calling algorithm segMNT was used, which is available in NimbleScan software (version 2.4.27, www.nimblegen.com). DNA was isolated from both blood and cell lines generated from Subject 5889 (labeled with Cy3) and cohybridized to the array using the paternal DNA (labeled with Cy5) as a control as the deleted chromosome had been determined to be of paternal origin by using a panel of VNTR markers (raw data available upon request). The first step used DNA isolated from blood for screening on an initial array tiling all of chromosome 7. This analysis confirmed the 7q11.2 deletion, and showed that the centromeric breakpoint was located within the duplicated region, as found with BAC-FISH and as expected for typical WS deletions. To further define both breakpoints, a custom high density oligonucleotide array was generated that consisted of 385,000 probes spanning the region of 64-77Mb of chromosome 7 (NCBI Build 36.1, HG18), which includes the putative WS duplicated region, located at 70-71.5 Mb and the flanking duplicated regions. Probes on the array were spaced at a median distance of 20 bp and contained 45-85 bp oligonucleotides probes which were adjusted in their C+G content to yield similar optimum hybridization temperatures (isothermal array, Tm = 76C) [Urban et al., 2006; Korbel et al., 2007]. In contrast to previous arrays and to optimize the ability to define a WS breakpoint within the centromeric duplicated region, oligonucleotide probes were included that occurred up to nine-fold in the human genome. This would also provide the possibility to evaluate the contribution of pseudogenes and other copy number variation to WS phenotype. As the subject's deletion occurs on the chromosome of paternal origin, use of the father's DNA as the control provided the potential to precisely determine the subject's breakpoint with respect to single exons of the two related genes, GTF2IRD1 and GTF2I, located at the telomeric end of the typical WS deletion. The breakpoints determined by array CGH analysis were validated by FISH and somatic cell hybrids with PCR.
Cognitive-Behavioral Analysis
Subject 5889 was diagnosed at 14 months and evaluated at 2 ½ through 7 years at Cedars-Sinai Medical Center, and seen at 2 years 6 months and at 4 years 4 months at the Salk Institute for Biological Studies. Participants reported here in the comparison groups for cognitive performance were individuals from a cohort of children initially recruited for a collaborative project with the Center for Neurodevelopmental studies at the University of California-San Diego, and included children with full WS deletions, children with Down syndrome (DS), as well as typically developing children (TD).
For the Wechsler Preschool and Primary Scale of Intelligence - Revised (WPPSI-R) [Wechsler, 1989], the cohort included 7 WS aged 4-7 years, and 5889 was at age 4 years 4 months. For the Kaufman Brief Intelligence Test (K-BIT) [Kaufman and Kaufman, 1990], the cohort was 11 children withWS aged 4-10 years, and 5889 was at age 4 years 4 months. For the Preschool Language 3rd Edition (PLS-3) [Zimmerman, 1992], the cohort was 22 children with WS aged 1-5 years, and 5889 was at age 2 years 6 months and 4 years 4 months. No significant difference in ages was observed between the groups.
For the SISQ data used to map social features [Doyle et al., 2004; Järvinen-Pasley et al., 2008] versus gene deletion, the cohort included 34 WS aged 2-7 years, 18 DS aged 2-7 years, 18 TD aged 2-7 years and 5889 aged 2 years 6 months and 4 years 4 months. For the social observation study [Järvinen-Pasley et al., 2008], the cohort included 8 children with typical deletions for WS aged 3 years, mental age of approximately 16 months, and 8 chronological age- and mental age-matched children with DS, and 5889 aged 2 years 6 months.
Psychometric assessment
For the current study, we report on three measures of verbal and non-verbal intelligence (WPPSI-R, K-BIT and PLS-3; supplemental online materials). For all measures, individual scores from Subject 5889 were compared to those of the full deletion WS group and those of the typically developed children group by conversion to z-scores with a mean of zero and standard deviation of one (see Figure 4). Given the single-case contingency, we were forced to forgo inferential statistics and to rely on the base rates logic as a basis for comparison. Since z-scores reflect normal distributions, z-scores of 2.0 or higher were considered to be statistically significant (i.e., different from that population), as the probability of achieving such scores by chance is less than 0.05.
FIG. 4.
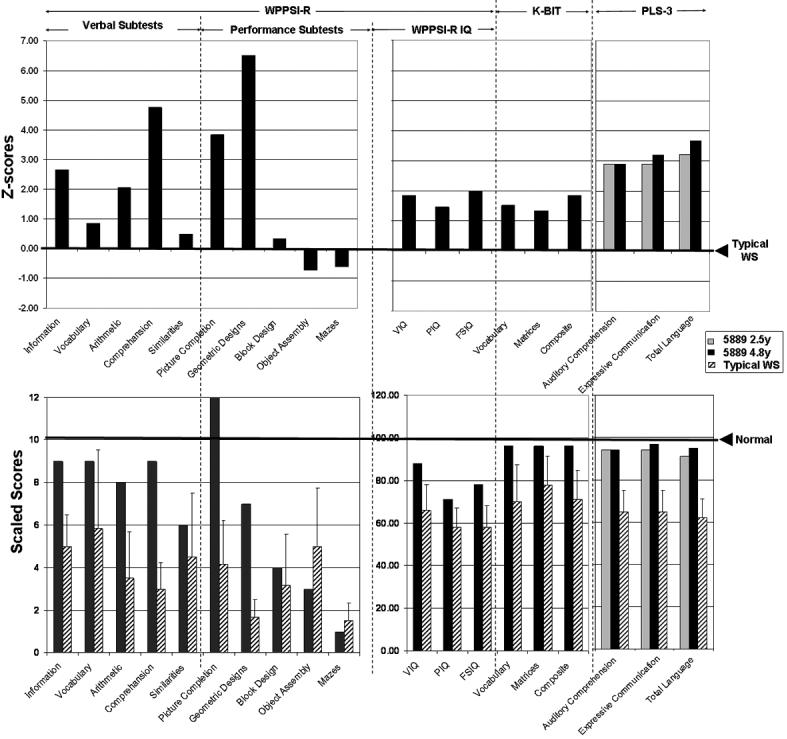
Subject 5889's scores on cognitive measures (WPPSI-R, K-BIT and PLS-3) as compared to mean WS performance (upper panel, Typical WS score denoted by right arrow) and compared to WS children with typical deletion (hatched bars) and typically developed children (lower panel, Normal score denoted by right arrow).
RESULTS
Physical and Developmental Features
A summary of the clinical features is shown in Table I and a complete clinical description of Subject 5889 is included in the supplementary on-line material (SOM). Briefly, 5889 was born at 38 wks gestation (weight at birth 10 centile, but at 4 4/12, 3 centile; height and head circumference <3 centile). She has the typical physical and developmental features of WS including growth delay through age 7, facial (Figure 1 and Table I), ophthalmologic (strabismus), cardiac (SVAS, PPS), vascular stenoses (renal artery stenosis) and hypertension (TableI).
FIG. 1.
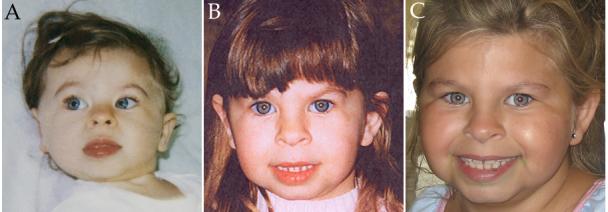
Facial photographs of Subject 5889 at 14 months (A), 2.6 years (B) and 7 years (C) old. Reproduced with parental permission.
Motor developmental milestones are within the normal range (sitting at 6-7 months; cruising at 10 months; walking at 13 months) and neural exam normal (strength, tone and gait) from 4 months. However, in contrast to typical WS, language was not delayed and she was “highly talkative” at 29 months; results on the MacArthur Toddler form of the CDI (Child Development Inventory) were at the mean for typically developing children in sentence complexity, at the 50% for 24 Months (vocabulary production) and at 50% for 26 months (use of irregular nouns and verbs). In summary, although her physical development and milestones were typical for WS, she did not demonstrate the typical delay in language, or the overly social behavior, characteristic of young children with WS.
Deletion Analysis
Subject 5889 was diagnosed with WS by clinical presentation and FISH (Oncor) at 14 months but was suspected to have an atypical deletion and was referred for molecular analysis in view of her normal developmental milestones.
The results of molecular cytogenetic analyses are shown in Figures 2 and 3. At the centromeric end, the probe BAC1008H17 was not present, suggesting that the breakpoint was located beyond FKBP6 and FZD9. At the telomeric end, however, the presence of a signal for probe BAC1184P14 but a decreased signal for BAC 1186P10 suggested that the break occurred in the region of GTF2IRD1-GTF2I and further analyses were necessary to define the precise breakpoint with respect to these two genes (Figure 2A). Therefore, somatic cell hybrids were generated and were analyzed with high density PCR probes spanning the region. PCR indicated that colonies 11 and 13 contained the deleted chromosomes 7 and colonies 9 and 12, the non-deleted. Further PCR probes narrowed the telomeric breakpoint to a 249 bp region located 10 kb downstream of the gene for GTF2IRD1, defined by probes RD 49 and RD50 (Figure 2B and SOM Table 2).
FIG. 3.

Molecular map of the Subject 5889. High resolution molecular determination of deletion breakpoints in Subject 5889 occur within 72.32-72.38Mb at centromeric end and and 10 kb downstream of GTF2IRD1 at telomeric end.
Chromosomal breakpoints were mapped by array CGH using a custom high resolution oligonucleotide isothermal DNA tiling array (Roche NimbleGen). While the custom array with a median probe spacing of 20bp and more complete coverage of the duplicated regions can potentially map breakpoints to the 100-200bp level, the centromeric breakpoint could not be further refined beyond the FISH analysis (Figure 2A) due to the complexity of this region (Figure 2C, lower left panel), which contains segmental duplications and other copy number variants. However, the telomeric breakpoint was localized to 73,665,076-73,666,056 bp, located 10 kb downstream of the 3'UTR of the gene GTF2IRD1, which is consistent with the PCR analysis of the somatic cell hybrids. Therefore, the deleted region in Subject 5889 possibly includes genes NSUN5 and TRIM50, most of FKBP6 and all of the other genes in the interval through GTF2IRD1, but not the neighboring gene, GTF2I (Figure 3).
Neurocognitive Performance
The upper panel of Figure 4 summarizes Subject 5889's Z-scores on the WPPSI-R, K-BIT and PLS-3 subtests, as compared to WS children with typical deletions, while the lower panel of Figure 4 shows Subject 5889's scaled scores on these subtests relative to those of WS and the WPPSI-R, K-BIT and PLS3 typical norms. On the WPPSI-R, Subject 5889 obtained a Full Scale IQ (FSIQ) score of 78, with a Performance IQ (PIQ) score of 71 and Verbal IQ (VIQ) score of 88, which were 1-2 SD higher than typical WS children. Individuals with WS with classic deletions typically exhibit mean FSIQ of approximately 54-59 (SD = ~ 11) [Bellugi et al., 2000; Meyer-Lindenberg et al., 2006], with greater impairments seen in PIQ than in VIQ. Her verbal scores were significantly above that of other WS children on three out of five verbal subtests (Information, Arithmetic, Comprehension; Z-scores = 2-6 above the WS means). Supporting her superior language skills relative to typical WS, Subject 5889's performance on the K-BIT and PLS-3 was also significantly higher than that of her typical WS peers. In contrast, her scores on visual-spatial construction were striking, with high or normal performance on tests involving only recognition (Picture Completion SS 12; Geometric Design Recognition subcomponent raw score 6/7), but poor performance on all subtests that involve motor integration (Block Design 4, Object Assembly 3; Mazes 1, only 9/57 raw score on the drawing items of Geometric Design, and poor performance on forms 7-9 of the VMI).
Relationship between Neurocognitive Performance and Social Behavior
In order to investigate whether the behavioral dimensions of sociability may be independent of, and/or not easily explained, by the level of cognitive functioning in WS, we carried out correlations between the SISQ Approach Strangers subscale and IQ on data from individuals with WS using the cohort previously reported [Doyle et al., 2004]. Twenty children and adolescents with WS (11M, 9 F; M age = 13.47, SD = 2.31; range 7.9 - 16.7 years) were administered the SISQ and either the WISC-III (Wechsler Intelligence Scale for Children, 3rd. ed. 1991) or WISC-R [Wechsler, 1974]. Mean FSIQ = 52.0, SD = 7.7, range 42 - 66. SISQ Approach Strangers scores were not correlated with Full Scale IQ (r = .031, p = .90). Further, there were no differences between males and females (FSIQ; t (18) = 3.43, p = .74; Approach Strangers; t (18) = -.070, p = .95).
DISCUSSION
WS provides a unique model in which to link human behavior with human genetics. The role of specific genes in WS is emerging from the analysis of atypical deletions but at present, these are limited to a role for GTF2IRD1 and GTF2I in visual-spatial processing [Hirota et al., 2003] and suggested facial features [Hirota et al., 2003; Tassabehji et al., 2005] and further progress calls for high resolution genomic analyses combined with paradigms that differentiate cognitive and social features. Moreover, there are no previous reports that clearly distinguish the roles of GTF2IRD1 versus GTF2I. The current report provides the evidence to dissect genes for WS, particularly for GTF2IRD1 in a subset of facial features (ages 14 months-7 years) and visual-spatial construction, and for GTF2I in aspects of the social behavior of WS (Figure 6). Moreover, Subject 5889 is a child with many physical characteristics of WS including short stature, SVAS, PPS and other vascular stenoses. Taken together with her physical features, the deletion in 5889 shows that GTF2I may not play a necessary role in the short stature, facial features, SVAS, PPS and renal-vascular stenoses, but may play an important role in some aspects of cognition and social behavior seen in WS.
FIG. 6.


WS Cognition: Gene Map of Visual-Spatial Construction and Social Behavior. A: Summary of facial features, cognition test scores, and gene copy number analysis of GTF2IRD1 for WS patients with typical and atypical (AT) deletions. B: WS region gene map and deletion summary of typical and atypical WS patients and corresponding mapping of reported phenotypes.
At the beginning of study, it was a challenge to determine the breakpoint in the duplicated region as the assembled human genome was not available. In this report, we further refine the molecular breakpoints of Subject 5889 by combining high resolution oligonucleotide array CGH analysis with multicolor fluorescence hybridization and PCR with somatic cell hybrids. The centromeric breakpoint maps to the region 72.32-72.38Mb, which includes most of FKBP6 and possibly includes NSUN5 and TRIM50. Due to the presence of segmental duplications and copy number variants in this region, further molecular analysis (e.g., CGH with alternate reference DNAs) will be required to more precisely map this breakpoint. The telomeric breakpoint maps to a 249 bp region located between GTF2IRD1 and GTF2I, a region with no homology to the centromeric breakpoint. The gene for GTF2I is left intact, along with 45 kb of the upstream putative control region, which may contribute to more normal function of this non-deleted gene. The location of the breakpoints in non-homologous DNA sequences suggests that the break in 5889 may have occurred by non-homologous end joining (NHEJ) or other processes that do not require homology at the breakpoint, and support the proposal that other mechanisms predispose to genomic instability of the WS region [Korenberg et al., 2000; Korbel et al., 2007].
WS facial features are associated with atypical deletions but the genes have not been completely defined. The classic WS facial appearance has been associated with deletions that include CYLN2, GTF2IRD1 and GTF2I [Botta et al., 1999; Heller et al., 2003] but not with those that preserve this region [Tassabehji et al, 1999; Hirota et al.; 2003; Karmiloff-Smith et al., 2003; Tassabehji et al., 2005; Howald et al., 2006; Van Hagen et al., 2007]. Deletion of CYLN2 in combination with GTF2IRD1 and GTF2I has been associated with typical WS facial dysmorphism; for example, patients A and B [Botta et al., 1999] and cases 2 and 3 [Heller et al., 2003]. However, deletion of CYLN2 combined only with partial deletion of GTF2IRD1 (and preservation of GTF2I) has shown “mild” facial features. For example, family K3321 with four members [Morris et al., 2003] had long philtrum in all and in one of four, flat malar region, bitemporal narrowing or full cheeks reported; HR [Tassabehji et al., 2005] had “facial features…suggestive of, but not classical for, WBS”. A contribution of CYLN2 is suggested by the association of its partial deletion (with preservation of both GTF2IRD1 and GTF2I) with “mild” facial features (e.g., a case in Gagliardi et al., 2003 and IM3 in Howald et al., 2006). In contrast, WS deletions that do not affect CYLN2, GTF2IRD1 or GTF2I, are not reported to have WS facial features (e.g., cases 2 and 3 [Hirota et al., 2003], CS [Karmiloff-Smith et al., 2003; Tassabehji et al., 1999 and 2005], CO3 [Howald et al., 2006] and WS75 [Van Hagen et al., 2007]). Thus, the evidence from small deletions implicates a role for CYLN2 and GTF2IRD1 in mild facial features and the case reported here illustrates that full deletion of GTF2IRD1 is associated with typical WS facies (Figure 6). The significance of case HR [Tassabehji et al., 2005] (who quantitatively showed “mild” features) for determining the genetics of WS facial features is unclear as the deletion included only the first of 26 exons for GTF2IRD1. It is of interest that although decreased expression was detected, the gene region probed was not specified and may represent a transcript not decreased in full deletion as this decrease has not been confirmed by other groups [Merla et al., 2006; Edelmann et al., 2007]. The current Subject 5889 has the typical facial features of WS (shown in Table I and Figure 1) and these results allow the conclusion that deletion of the gene for GTF2IRD1 is associated with the WS facial features seen in 5889. Recent results showed that craniofacial tissues are one of the predominantly distributions of Gtf2ird1 in mouse [Palmer et al., 2007]. However, the null mutation of Gtf2ird1 produced by the targeted insertion of a nls-LacZ cassette into exon 2 of the gene do not exhibit craniofacial dysmorphology or dental abnormalities [Palmer et al., 2007]. This is different from the previous report by the other group, in which the null mutant mice contain a deletion of ~40 kb from downstream of Cyln2 to exon 1 of Gtf2ird1, and display craniofacial dysmorphology and dental abnormalities [Tassabehji et al., 2005]. A more complete and specific Gtf2ird1 null mutant mouse model may be required to explain the discrepancy.
The cognitive performance of Subject 5889 differed significantly from that of age-matched WS children with typical deletion (Figure 4). Specifically, her performance on many language tasks (except Similarities) and some performance tasks (Picture Completion and the Spatial Recognition part of Geometric Designs) were well above the typical range for WS, suggesting that her normal copy number of GTF2I may have contributed to her increased function in both verbal and performance domains. However, her poor performance on some of the spatial tests (Block Design, Object Assembly, Mazes, Drawing part of Geometric Designs) is similar to that of typical WS, suggesting that GTF2IRD1 may be important for specific aspects of visual-spatial cognition, such as visual-motor integration. Previously, all atypical deletion subjects were deleted for only part of GTF2IRD1 or both GTF2IRD1 and GTF2I, preventing the dissection of their individual contribution to visual-spatial features. This is the first report parsing the parts of visual-spatial performance that were determined by the deletion of GTF2I versus GTF2IRD1. Taken together with data from the literature, we suggest that the deletion of GTF2IRD1 is not sufficient to disturb all visual-spatial performance but is more important for the typical motor deficits in visual-spatial construction, a hallmark of WS cognitive dysfunction (Figure 6).
In this report, atypical WS social behavior was associated with atypical deletion preserving GTF2I. The observational results (Figure 5B) previously reported [Järvinen-Pasley et al., 2008] closely mirrored the parental ratings (Figure 5A) [Doyle et al., 2004] in that 5889 did not exhibit the highly pronounced social drive that is characteristic of children with WS full deletion of her age. Her reduced indiscriminant approach to strangers relative to typical WS children, combined with her decreased tendency to maintain eye contact and her preference for non-social over social activities, differentiate her social behavior from that seen in age-matched typical WS. It is possible that Subject 5889's higher overall functioning may contribute to her social behavior, although the results reported here in the cohort of typical WS did not show a correlation between SISQ and IQ. Similar findings indicating a lack of association between sociability and intelligence in individuals with WS have been reported in the literature [Frigerio et al., 2006; Porter et al., 2007]. Combined with the genetic analyses reported here, the social data support the involvement of the gene for GTF2I in aspects of hypersociability, particularly the lack of indiscriminant approach seen in WS, and moreover, suggest that with additional atypical deletion cases, it may be possible to further parse social behavior in WS (Figure 6). However, other genes and interactions with GTF2I may clearly contribute and the roles for GTF2I and GTF2IRD1 will be better distinguished when patients with just one of these genes deleted are found. The individual roles and interactions of GTF2IRD1 and GTF2I in WS need to be clarified.
FIG. 5.
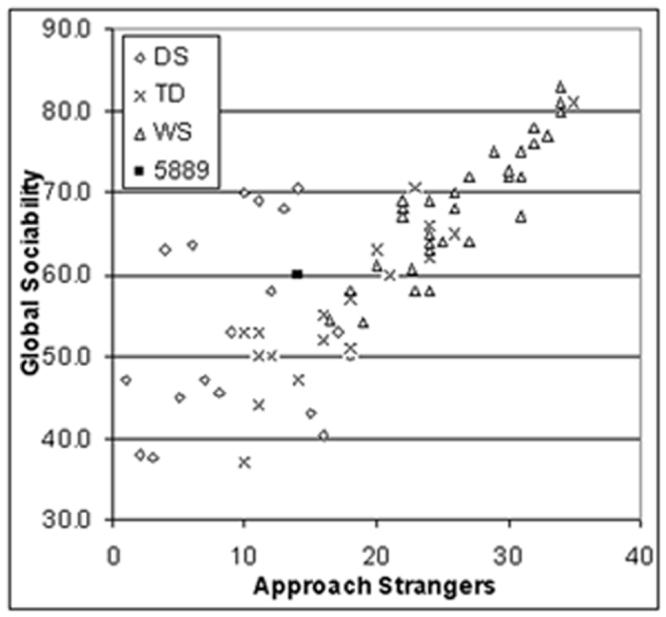
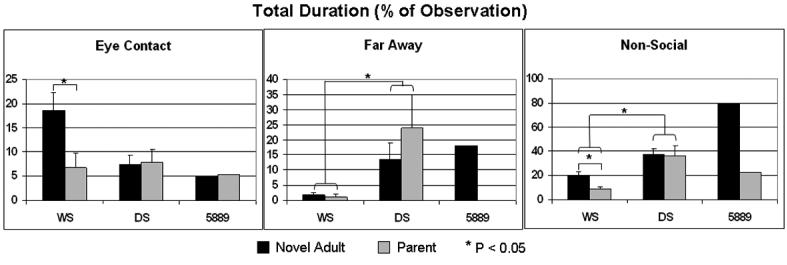
Quantification of social behavior in Subject 5889 versus WS, Down syndrome (DS) and controls (typically developed, TD). Data replotted from Doyle et al., 2004 and Järvinen-Pasley et al., 2008. A: The atypical social behavior of Subject 5889 was reflected in parental ratings of social behavior using the SISQ at age 2.55 [Doyle et al., 2004, versus WS only] and were repeated at age 4.6 years [Järvinen-Pasley et al., 2008 versus a single typical WS case only]. In contrast with age-matched typical WS group, as well as children with DS and TD, the parental ratings (converted to z-scores for comparison) revealed that 5889's Global Sociability and Approach Strangers scores both at 2.55 years (z = -2.59 and -2.39, respectively) and at 4.86 years (z = -2.6 and -3.2), were significantly different from her peers in the typical WS group. B: A number of key differences in direct social behavior were noted between the Subject 5889 and children with typical WS deletions [shown in Järvinen-Pasley et al., 2008], relative to control children with DS for the mean total durations of three ethogram behaviors. Overall, WS children with full deletions were significantly less likely than children with DS to spend time far away in proximity from (p=.007), and to engage in non-social activities with (p=.01), with adults, whether familiars (mothers) or novel (the experimenter), highlighting their tendency toward social over non-social activities. Furthermore, WS children with full deletions were significantly more likely to maintain eye contact with the novel adult than did children with DS (p=.038). Compared to the typical WS group, Subject 5889 (tested at age 2.5y) spent greater amount of time far away in proximity from the novel adult (z = 7.96), and engaged in non-social activities for greater amount of time with both the parent (z = 2.39) and the novel adult (z = 6.00). The difference in the amount of eye contact toward the unfamiliar adult failed to reach significance (z = 1.24), due to a large SD within the WS group. These results provide converging evidence for the SISQ data presented above, showing that Subject 5889 did not display the hypersociability characteristic of individuals with WS full deletions.
Our results suggest a possible role for GTF2I and GTF2IRD1 in WS social and cognitive behavior. GTF2IRD1 [Bayarsaihan et al., 2003] and GTF2I [Roy, 2001] may jointly contribute to WS behavior in that they are distantly related genes that encode transcriptional regulators and may each be related to different features or may act synergistically to determine distinct WS phenotypes. Further, previous studies have associated neuropsychiatric disorders with variations in neurohormones, including oxytocin and Arg-vasopressin, and their receptors [Witt et al., 1990; Witt and Insel, 1994; Modahl et al., 1998; Goldman et al., 2008]. It may be of interest to investigate GTF2I and GTF2IRD1 in their regulatory role in neurohormones and their receptors. Finally, recent reports of Gtf2ird1-targeted mice revealed reduced fear and aggression and altered serotonin metabolism [Young et al., 2007], suggesting that it may be of interest to examine these pathways in typical WS subjects and Subject 5889.
So, is it WS? With respect to facial features, small stature and aspects of visual-spatial construction, deletion sparing GTF2I resembles typical WS. In contrast, the typical tendency to indiscriminately approach strangers is not seen in this child and suggests an association of GTF2I with a part of the social behavior in WS. Finally, the involvement of GTF2I and GTF2IRD1 in regulation and development suggests that understanding their role in WS may provide insight into human brain development and adult function in the normal population.
Supplementary Material
Table 1.
Summary of frequencies of Clinical features of subject 5889 vs typical WS
| Typical WS | Subject 5889 | |
|---|---|---|
| Craniofacial | ||
| Bitemporal Narrowing | + | + |
| Stellate Iris | + | + |
| Epicanthal Folds | + | + |
| Periorbital Fullness | + | + |
| Medial Eyebrow | + | + |
| Malar hypoplasia (abnormal cheekbones) | + | + |
| flare Full Cheeks | + | + |
| Short Nose | + | + |
| Bulbous Nasal Tip | + | + |
| Long Philtrum | + | + |
| Prominent Lips | + | + |
| Small Jaw | + | + |
| Dental Abn/Malocclusion | 85 | + |
| CARDIOVASCULAR DEFFECTS | ||
| SVAS | 75 | + |
| PPS | 50 | + |
| VSD | 10 | - |
| Renal Artery Stenosis | + | + |
| Hypertension | 50 | no at infancy, yes at childhood |
| GENITOURINARY | ||
| Structural anomaly | ||
| Enuresis | ||
| Nephrocalcinosis | - | |
| Recurrent urinary tract infections | - | |
|
MUSCULOSKELETAL AND SUPPORTIVE
FEATURES | ||
| Joint hypermobility | 90 | - |
| Joint contractures | 50 | - |
| Radioulnar synostosis | - | |
| Long Neck or Sloped shoulders | + | |
| Joint limitation or laxity | - | |
| Rectal Prolapse | - | |
| Joint limitation | - | |
| Long trunk | + | |
| Hyperflexibility | + | |
| Abnormal Gait | - | |
| Preoccupation, spinning, rhythmic movements | - | |
| Hallux valgus | - | |
| Sacral Crease | - | |
| Frequent urination or delayed continence | - | |
| Strong attraction to music or perfect pitch | + | |
| Inguinal Hernia | 40/50 | - |
| OTHERS | ||
| Soft or velvetly skin | + | + |
| Hypercalcemia | 15 | - |
| Hypothyroidism | - | |
| Diatetes mellitus | - | |
| Strabismus | 50 | + |
| Esotropia | 50 | + |
| Hypersensitivity to sound | + | - |
| Hoarse Voice | + | + |
| Hyperacusis | 80 | - |
| Over friendly | + | - |
| Anxiety | + | - |
| Short attention span | + | - |
| Motor Delay | + | - |
| Language Delay | + | - |
ACKNOWLEDGMENTS
This work was supported by grants to J.R.K., A.L.R., D. M. and U.B. from the National Institute of Child Health and Human Development Grant P01 HD33113-12 and by the James S. McDonnell Foundation (U. B., J.R.K.). J.R.K. holds the Geri and Richard Brawerman Chair in Molecular Genetics. We acknowledge Rebecca R. Selzer and Todd A. Richmond contributed to the microarray design and analysis.
Footnotes
This work was performed in part at Cedars-Sinai Medical Center, Department of Pediatrics and Medical Genetics Institute.
REFERENCES
- Bayarsaihan D, Bitchevaia N, Enkhmandakh B, Tussie-Luna MI, Leckman JF, Roy A, Ruddle F. Expression of BEN, a member of TFII-I family of transcription factors, during mouse pre- and postimplantation development. Gene Expr Patterns. 2003;3:579–589. doi: 10.1016/s1567-133x(03)00118-2. [DOI] [PubMed] [Google Scholar]
- Bayes M, Magano LF, Rivera N, Flores R, Perez Jurado LA. Mutational mechanisms of Williams-Beuren syndrome deletions. Am J Hum Genet. 2003;73:131–151. doi: 10.1086/376565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beery K, Buktenica N. The Beery-Buktenica developmental test of visual-motor integration. Modern Curriculum Press; Cleveland, OH: 1989. [Google Scholar]
- Bellugi U, Lichtenberger L, Jones W, Lai Z, St George M. I. The neurocognitive profile of Williams Syndrome: a complex pattern of strengths and weaknesses. J Cogn Neurosci. 2000;1(12 Suppl):7–29. doi: 10.1162/089892900561959. [DOI] [PubMed] [Google Scholar]
- Botta A, Novelli G, Mari A, Novelli A, Sabani M, Korenberg J, Osborne LR, Digilio MC, Giannotti A, Dallapiccola B. Detection of an atypical 7q11.23 deletion in Williams syndrome patients which does not include the STX1A and FZD3 genes. J Med Genet. 1999;36:478–480. [PMC free article] [PubMed] [Google Scholar]
- Doyle TF, Bellugi U, Korenberg JR, Graham J. “Everybody in the world is my friend” hypersociability in young children with Williams syndrome. Am J Med Genet A. 2004;124:263–273. doi: 10.1002/ajmg.a.20416. [DOI] [PubMed] [Google Scholar]
- Dykens EM, Rosner BA. Refining behavioral phenotypes: personality-motivation in Williams and Prader-Willi syndromes. Am J Ment Retard. 1999;104:158–169. doi: 10.1352/0895-8017(1999)104<0158:RBPPIW>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Dykens EM, Rosner BA, Ly T, Sagun J. Music and anxiety in Williams syndrome: a harmonious or discordant relationship? Am J Ment Retard. 2005;110:346–358. doi: 10.1352/0895-8017(2005)110[346:MAAIWS]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Edelmann L, Prosnitz A, Pardo S, Bhatt J, Cohen N, Lauriat T, Ouchanov L, Gonzalez PJ, Manghi ER, Bondy P, Esquivel M, Monge S, Delgado MF, Splendore A, Francke U, Burton BK, McInnes LA. An atypical deletion of the Williams-Beuren syndrome interval implicates genes associated with defective visuospatial processing and autism. J Med Genet. 2007;44:136–143. doi: 10.1136/jmg.2006.044537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einfeld SL, Tonge BJ, Florio T. Behavioral and emotional disturbance in individuals with Williams syndrome. Am J Ment Retard. 1997;102:45–53. doi: 10.1352/0895-8017(1997)102<0045:BAEDII>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Elliott C. Differential ability scales. Harcourt, Brace; Jovanovich, San Deigo: 1990. [Google Scholar]
- Ewart AK, Morris CA, Atkinson D, Jin W, Sternes K, Spallone P, Stock AD, Leppert M, Keating MT. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat Genet. 1993;5:11–16. doi: 10.1038/ng0993-11. [DOI] [PubMed] [Google Scholar]
- Frigerio E, Burt DM, Gagliardi C, Cioffi G, Martelli S, Perrett DI, Borgatti R. Is everybody always my friend? Perception of approachability in Williams syndrome. Neuropsychologia. 2006;44:254–259. doi: 10.1016/j.neuropsychologia.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Gagliardi C, Bonaglia MC, Selicorni A, Borgatti R, Giorda R. Unusual cognitive and behavioural profile in a Williams syndrome patient with atypical 7q11.23 deletion. J Med Genet. 2003;40:526–530. doi: 10.1136/jmg.40.7.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman M, Marlow-O'Connor M, Torres I, Carter CS. Diminished plasma oxytocin in schizophrenic patients with neuroendocrine dysfunction and emotional deficits. Schizophr Res. 2008;98:247–255. doi: 10.1016/j.schres.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosch A, Pankau R. Social-emotional and behavioral adjustment in children with Williams-Beuren syndrome. Am J Med Genet. 1994;53:335–339. doi: 10.1002/ajmg.1320530406. [DOI] [PubMed] [Google Scholar]
- Gosch A, Pankau R. Personality characteristics and behaviour problems in individuals of different ages with Williams syndrome. Dev Med Child Neurol. 1997;39:527–533. doi: 10.1111/j.1469-8749.1997.tb07481.x. [DOI] [PubMed] [Google Scholar]
- Greenberg F. Williams syndrome professional symposium. Am J Med Genet. 1990;6(Supplement):85–88. [Google Scholar]
- Heller R, Rauch A, Luttgen S, Schroder B, Winterpacht A. Partial deletion of the critical 1.5 Mb interval in Williams-Beuren syndrome. J Med Genet. 2003;40:e99. doi: 10.1136/jmg.40.8.e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota H, Matsuoka R, Chen XN, Salandanan LS, Lincoln A, Rose FE, Sunahara M, Osawa M, Bellugi U, Korenberg JR. Williams syndrome deficits in visual spatial processing linked to GTF2IRD1 and GTF2I on chromosome 7q11.23. Genet Med. 2003;5:311–321. doi: 10.1097/01.GIM.0000076975.10224.67. [DOI] [PubMed] [Google Scholar]
- Howald C, Merla G, Digilio MC, Amenta S, Lyle R, Deutsch S, Choudhury U, Bottani A, Antonarakis SE, Fryssira H, Dallapiccola B, Reymond A. Two high throughput technologies to detect segmental aneuploidies identify new Williams-Beuren syndrome patients with atypical deletions. J Med Genet. 2006;43:266–273. doi: 10.1136/jmg.2005.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Järvinen-Pasley A, Bellugi U, Reilly J, Mills DL, Galaburda A, Reiss AL, Korenberg JR. Defining the social phenotype in Williams syndrome: a model for linking gene, the brain, and behavior. Dev Psychopathol. 2008;20:1–35. doi: 10.1017/S0954579408000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones W, Bellugi U. Hypersociability: the social and affective phenotype of Williams Syndrome. Journey from Cognition to Brain to Gene The MIT Presswilliams syndrome. 2001:43–71. [Google Scholar]
- Jones W, Bellugi U, Lai Z, Chiles M, Reilly J, Lincoln A, Adolphs R. II. Hypersociability in Williams Syndrome. J Cogn Neurosci. 2000;1(12 Suppl):30–46. doi: 10.1162/089892900561968. [DOI] [PubMed] [Google Scholar]
- Karmiloff-Smith A, Grant J, Ewing S, Carette MJ, Metcalfe K, Donnai D, Read AP, Tassabehji M. Using case study comparisons to explore genotypephenotype correlations in Williams-Beuren syndrome. Am J Med Genet. 2003;40:136–140. doi: 10.1136/jmg.40.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman AS, Kaufman NL. Kaufman Brief Intelligence Test. American Guidance Service, Inc; Circle Pines, MN: 1990. [Google Scholar]
- Klein-Tasman BP, Mervis CB. Distinctive personality characteristics of 8-, 9-, and 10-year-olds with Williams syndrome. Dev Neuropsychol. 2003;23:269–290. doi: 10.1080/87565641.2003.9651895. [DOI] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Grubert F, Du J, Royce TE, Starr P, Zhong G, Emanuel BS, Weissman SM, Snyder M, Gerstein MB. Systematic prediction and validation of breakpoints associated with copy-number variants in the human genome. Proc Natl Acad Sci U S A. 2007;104:10110–10115. doi: 10.1073/pnas.0703834104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenberg JR, Chen XN. Human cDNA mapping using a high-resolution R-banding technique and fluorescence in situ hybridization. Cytogenet Cell Genet. 1995;69:196–200. doi: 10.1159/000133962. [DOI] [PubMed] [Google Scholar]
- Korenberg JR, Chen XN, Hirota H, Lai Z, Bellugi U, Burian D, Roe B, Matsuoka R. VI. Genome structure and cognitive map of Williams syndrome. J Cogn Neurosci. 2000;1(12 Suppl):89–107. doi: 10.1162/089892900562002. [DOI] [PubMed] [Google Scholar]
- Merla G, Howald C, Henrichsen CN, Lyle R, Wyss C, Zabot MT, Antonarakis SE, Reymond A. Submicroscopic deletion in patients with Williams-Beuren syndrome influences expression levels of the nonhemizygous flanking genes. Am J Hum Genet. 2006;79:332–341. doi: 10.1086/506371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mervis CB, Klein-Tasman BP. Williams syndrome: cognition, personality, and adaptive behavior. Ment Retard Dev Disabil Res Rev. 2000;6:148–158. doi: 10.1002/1098-2779(2000)6:2<148::AID-MRDD10>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Mervis CB, Berman KF. Neural mechanisms in Williams syndrome: a unique window to genetic influences on cognition and behaviour. Nat Rev Neurosci. 2006;7:380–393. doi: 10.1038/nrn1906. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Hariri AR, Munoz KE, Mervis CB, Mattay VS, Morris CA, Berman KF. Neural correlates of genetically abnormal social cognition in Williams syndrome. Nat Neurosci. 2005;8:991–993. doi: 10.1038/nn1494. [DOI] [PubMed] [Google Scholar]
- Modahl C, Green L, Fein D, Morris M, Waterhouse L, Feinstein C, Levin H. Plasma oxytocin levels in autistic children. Biol Psychiatry. 1998;43:270–277. doi: 10.1016/s0006-3223(97)00439-3. [DOI] [PubMed] [Google Scholar]
- Morris CA. Williams-Beuren syndrome: Research, evaluation, and treatment. Johns Hopkins University Press; 2006. [Google Scholar]
- Morris CA, Mervis CB, Hobart HH, Gregg RG, Bertrand J, Ensing GJ, Sommer A, Moore CA, Hopkin RJ, Spallone PA, Keating MT, Osborne L, Kimberley KW, Stock AD. GTF2I hemizygosity implicated in mental retardation in Williams syndrome: genotype-phenotype analysis of five families with deletions in the Williams syndrome region. Am J Med Genet A. 2003;123:45–59. doi: 10.1002/ajmg.a.20496. [DOI] [PubMed] [Google Scholar]
- Osborne LR. Williams-Beuren syndrome: unraveling the mysteries of a microdeletion disorder. Mol Genet Metab. 1999;67:1–10. doi: 10.1006/mgme.1999.2844. [DOI] [PubMed] [Google Scholar]
- Palmer SJ, Tay ES, Santucci N, Cuc Bach TT, Hook J, Lemckert FA, Jamieson RV, Gunnning PW, Hardeman EC. Expression of Gtf2ird1, the Williams syndrome-associated gene, during mouse development. Gene Expr Patterns. 2007;7:396–404. doi: 10.1016/j.modgep.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Plesa-Skwerer D, Sullivan K, Joffre K, Tager-Flusberg H. Self concept in people with Williams syndrome and Prader-Willi syndrome. Res Dev Disabil. 2004;25:119–138. doi: 10.1016/j.ridd.2003.05.003. [DOI] [PubMed] [Google Scholar]
- Porter MA, Coltheart M, Langdon R. The neuropsychological basis of hypersociability in Williams and Down syndrome. Neuropsychologia. 2007;45:2839–2849. doi: 10.1016/j.neuropsychologia.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Eckert MA, Rose FE, Karchemskiy A, Kesler S, Chang M, Reynolds MF, Kwon H, Galaburda A. An experiment of nature: brain anatomy parallels cognition and behavior in Williams syndrome. J Neurosci. 2004;24:5009–5015. doi: 10.1523/JNEUROSCI.5272-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy AL. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I. Gene. 2001;274:1–13. doi: 10.1016/s0378-1119(01)00625-4. [DOI] [PubMed] [Google Scholar]
- Selzer RR, Richmond TA, Pofahl NJ, Green RD, Eis PS, Nair P, Brothman AR, Stallings RL. Analysis of chromosome breakpoints in neuroblastoma at sub-kilobase resolution using fine-tiling oligonucleotide array CGH. Genes Chromosomes Cancer. 2005;44:305–19. doi: 10.1002/gcc.20243. [DOI] [PubMed] [Google Scholar]
- Stromme P, Bjornstad PG, Ramstad K. Prevalence estimation of Williams syndrome. J Child Neurol. 2002;17:269–271. doi: 10.1177/088307380201700406. [DOI] [PubMed] [Google Scholar]
- Sullivan K, Tager-Flusberg H. Second-order belief attribution in Williams syndrome: intact or impaired? Am J Ment Retard. 1999;104:523–532. doi: 10.1352/0895-8017(1999)104<0523:SBAIWS>2.0.CO;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tager-Flusberg H, Sullivan K. A componential view of theory of mind: evidence from Williams syndrome. Cognition. 2000;76:59–90. doi: 10.1016/s0010-0277(00)00069-x. [DOI] [PubMed] [Google Scholar]
- Tassabehji M, Metcalfe K, Karmiloff-Smith A, Carette MJ, Grant J, Dennis N, Reardon W, Splitt M, Read AP, Donnai D. Williams syndrome: use of chromosomal microdeletions as a tool to dissect cognitive and physical phenotypes. Am J Hum Genet. 1999;64:118–125. doi: 10.1086/302214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tassabehji M, Hammond P, Karmiloff-Smith A, Thompson P, Thorgeirsson SS, Durkin ME, Popescu NC, Hutton T, Metcalfe K, Rucka A, Stewart H, Read AP, Maconochie M, Donnai D. GTF2IRD1 in craniofacial development of humans and mice. Science. 2005;310:1184–1187. doi: 10.1126/science.1116142. [DOI] [PubMed] [Google Scholar]
- Udwin O, Yule W. A cognitive and behavioural phenotype in Williams syndrome. J Clin Exp Neuropsychol. 1991;13:232–244. doi: 10.1080/01688639108401040. [DOI] [PubMed] [Google Scholar]
- Urban AE, Korbel JO, Selzer R, Richmond T, Hacker A, Popescu GV, Cubells JF, Green R, Emanuel BS, Gerstein MB, Weissman SM, Snyder M. High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays. Proc Natl Acad Sci U S A. 2006;103:4534–4539. doi: 10.1073/pnas.0511340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wechsler D. Manual for the Wechsler Intelligence Scale for Children-Revised. Psychological Coeporation; New York: 1974. [Google Scholar]
- Wechsler D. Wechsler Preschool and Primary Scale of Intelligence-Revised. Psychological Corporation; London: 1989. [Google Scholar]
- Witt DM, Insel TR. Increased Fos expression in oxytocin neurons following masculine sexual behavior. J Neuroendocrinol. 1994;6:13–18. doi: 10.1111/j.1365-2826.1994.tb00549.x. [DOI] [PubMed] [Google Scholar]
- Witt DM, Carter CS, Walton DM. Central and peripheral effects of oxytocin administration in prairie voles. Microtus ochrogaster. Pharmacol Biochem Behav. 1990;37:63–69. doi: 10.1016/0091-3057(90)90042-g. [DOI] [PubMed] [Google Scholar]
- Yan H, Papadopoulos N, Marra G, Perrera C, Jiricny J, Boland CR, Lynch HT, Chadwick RB, de la Chapelle A, Berg K, Eshleman JR, Yuan W, Markowitz S, Laken SJ, Lengauer C, Kinzler KW, Vogelstein B. Conversion of diploidy to haploidy. Nature. 2000;403:723–724. doi: 10.1038/35001659. [DOI] [PubMed] [Google Scholar]
- Young EJ, Lipina T, Tam E, Mandel A, Clapcote SJ, Bechard AR, Chambers J, Mount HT, Fletcher PJ, Roder JC, Osborne LR. Reduced fear and aggression and altered serotonin metabolism in Gtf2ird1-targeted mice. Genes Brain Behav. 2007 doi: 10.1111/j.1601-183X.2007.00343.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman IL. Preschool Language Scale. 3rd edition The Psychological Corporation; San Antonio, TX: 1992. [Google Scholar]
- Zitzer-Comfort C, Doyle T, Masataka N, Korenberg J, Bellugi U. Nature and nurture: Williams syndrome across cultures. Dev Sci. 2007;10:755–762. doi: 10.1111/j.1467-7687.2007.00626.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.