

To the Editor:
Hearing impairment is the most common genetic sensory defect in humans worldwide. It is estimated that profound hearing loss occurs in 4 out of every 10,000 children [Morton 1991]. Seventy percent of hereditary SNHL is nonsyndromic (DFN) and in at least 80% of these cases the inheritance is autosomal recessive (DFNB) [Smith et al., 2005]. Genetic studies have shown that mutations in 67 loci and 25 genes are associated with DFNB [Van Camp and Smith, 2008]. Further understanding of the molecular mechanisms associated with SNHL will assist in the development of therapeutics for hereditary hearing loss in the future.
Mutations in the RDX gene that encodes the radixin protein have recently been shown to cause DFNB24 hearing loss in consanguineous Pakistani and Indian families [Khan et al., 2007]. In this study, we report a novel splice site mutation in the RDX gene in a consanguineous Iranian family with autosomal recessive non-syndromic hearing loss (ARNSHL). This mutation is only the fourth to be identified at the rare DFNB24 ARNSHL locus and the second that is predicted to lead to nonsense mediated decay or a truncated version of the radixin protein.
A five-generation Iranian family with apparent congenital ARNSHL was studied (Fig.1). Family members in the fourth and fifth generations had severe-to-profound hearing loss suggesting autozygosity by descent (Fig.2). A complete physical examination and audiologic testing was completed on consenting family members and blood samples were obtained for DNA extraction. Liver function testing on affected individual V:2 showed total bilirubin (0.4 mg/dl), direct bilirubin (0.1 mg/dl), alanine transaminase (41 U/ml), aspartate transaminase (35 U/ml) and alkaline phosphatase (229 U/L), all normal. Human Research Institutional Review boards at the Welfare Science and Rehabilitation University and Iran University of Medical Sciences and the University of Iowa approved all procedures.
Fig. 1.
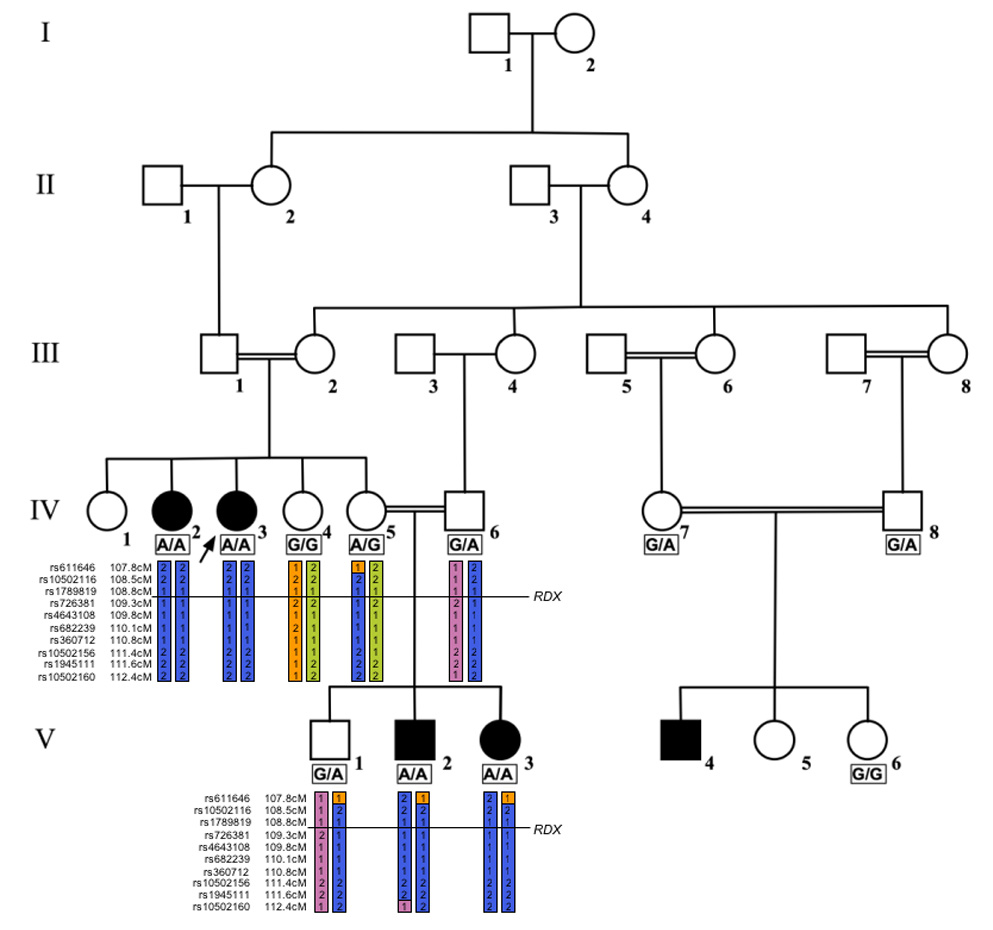
Pedigree of the Iranian family with ARNSHL. Open symbols = unaffected; filled symbols = ARNSHL. The genotypes and haplotypes for affected individuals and carriers are shown.
Fig. 2.
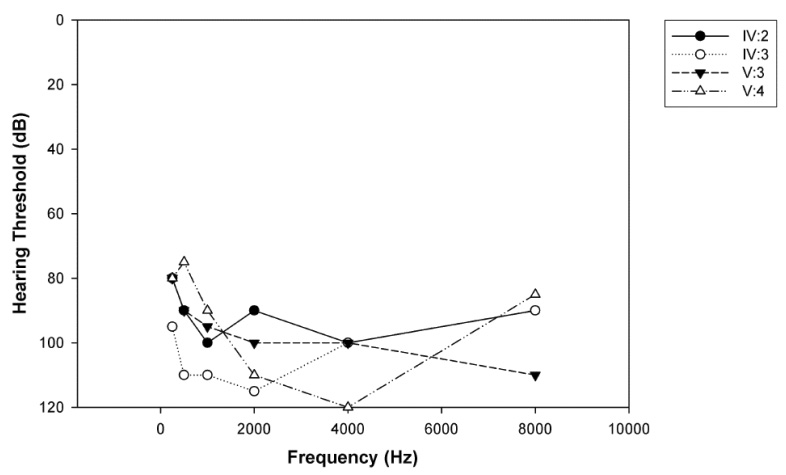
Audiograms from affected family members. All affected individuals displayed severe-to-profound bilateral ARNSHL.
Genomic DNA from individuals III:1, IV:3, IV:4, IV:5, IV:6, IV:7, IV:8, V:1, V:2, V:3, and V:6 (Fig.1) was genotyped for 50,000 SNPs using Affymetrix (Santa Clara, CA, USA) 50K XBA GeneChips at the Translational Genomics Research Institute (Pheonix, Arizona). All genotypes were determined using the BRLMM genotyping algorithm [Di et al., 2005; Rabbee and Speed 2006]. Genotyping data were examined with PEDSTATS [Wigginton and Abecasis 2005] for Mendelian inheritance errors and MERLIN [Abecasis et al., 2002] for errors based on inferred double recombination events between tightly linked markers. Because the size of the pedigree was above the threshold for efficient implementation of the Lander-Green multipoint linkage mapping algorithm, a smaller subset of the pedigree was used which only considered the descendents of individuals III:1, III:2, III:3, III:4 (Fig.1).
An autosomal, genome-wide parametric linkage analysis was carried out as the pedigree showed affected males and females. All linkage analyses were performed with MERLIN. The Lander-Green multipoint linkage mapping algorithm assumes linkage equilibrium. The markers were too close together to be in linkage equilibrium, so a smaller marker set was created using an in-house program (linkdatagen.pl;http://bioinf.wehi.edu.au/ software/). The binsize was set to 0.5 cM (i.e. the highest heterozygosity marker was chosen in each 0.5 cM interval) and 6140 autosomal markers were selected. Linkage analyses were carried out using the following parameters for the disease allele a, and normal allele A - Pr(a)=0.0001, Pr(disease|aa)=0.999, Pr(disease|AA or Aa)=0.001 - reflecting a highly penetrant, autosomal recessive disease model. The genetic map used was generated by Affymetrix using interpolation based on the empirical deCode, Marshfield and SNP Linkage (SLM1) genetic maps [Broman et al., 1998; Kong et al., 2002]. The physical map used was derived from the UCSC database using the NCBI Build 36.1 (March 2006; http://genome.ucsc.edu/cgi-bin/hgGateway).
In the genome-wide analysis 38 Mendelian (.07% error rate) and 168 double recombinant errors were detected. A peak LOD score of 4.13 was achieved for a region on chromosome 11 (Fig.3) and there were no other regions where the LOD score exceeded 3. The critical region spanned approximately 3.6 cM between markers rs1789819 (109,185,632 bp) and rs10502160 (112,122,938 bp) (Fig.1). All four affected individuals included in the mapping were homozygous for a region between 108.814 cM and 112.368 cM on chromosome 11 (11q22.3-23.1).
Fig. 3.
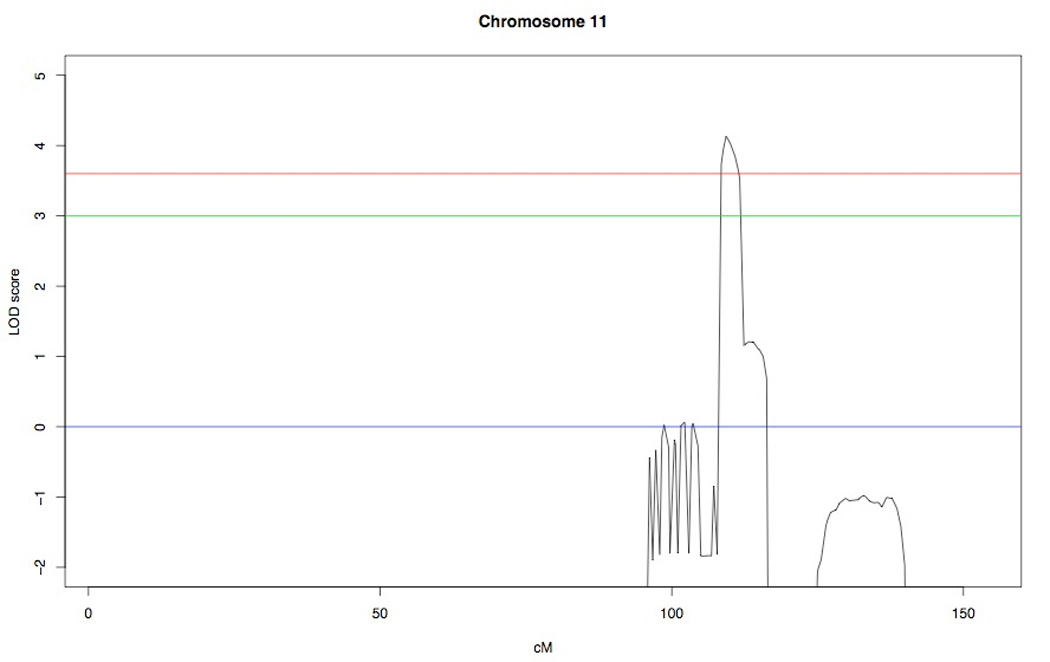
Graphic showing the only significant LOD score achieved in the genome-wide analysis on chromosome 11. The dashed line indicates threshold (LOD score of 3). The dotted line at LOD score of 3.6 represents the Lander-Kruglyak threshold. The peak LOD score was 4.13.
The critical region contained the known ARNSHL locus DFNB24. The RDX gene was amplified and sequenced using previously reported gene-specific primers [Khan et al., 2007]. A novel homozygous splice site mutation (c.698+1G>A) in intron 7 was found to segregate in affected family members (Fig.4). The +1G nucleotide of the 5’ donor splice site is invariant among all eukaryotes [Shapiro and Senapathy, 1987]. The c.698+1G>A mutation is predicted to result in read-through into intron 7 and a stop codon immediately following the amino acids encoded by exon 7. The last residue encoded by exon 7 would remain a lysine (K) due to the redundancy of the genetic code (AAG > AAA) and the next codon created would be a stop (TAA). This is predicted to result in a severely truncated protein lacking part of the FERM3 domain and the entire α- and c-terminal (CTD) domains (Fig.5), although the mutant mRNA may be subject to nonsense mediated decay and in this case no mutant protein would be produced. The c.698+1G>A mutation was not observed in 53 (106 chromosomes) Iranian and 133 (266 chromosomes) CEPH control individuals.
Fig. 4.
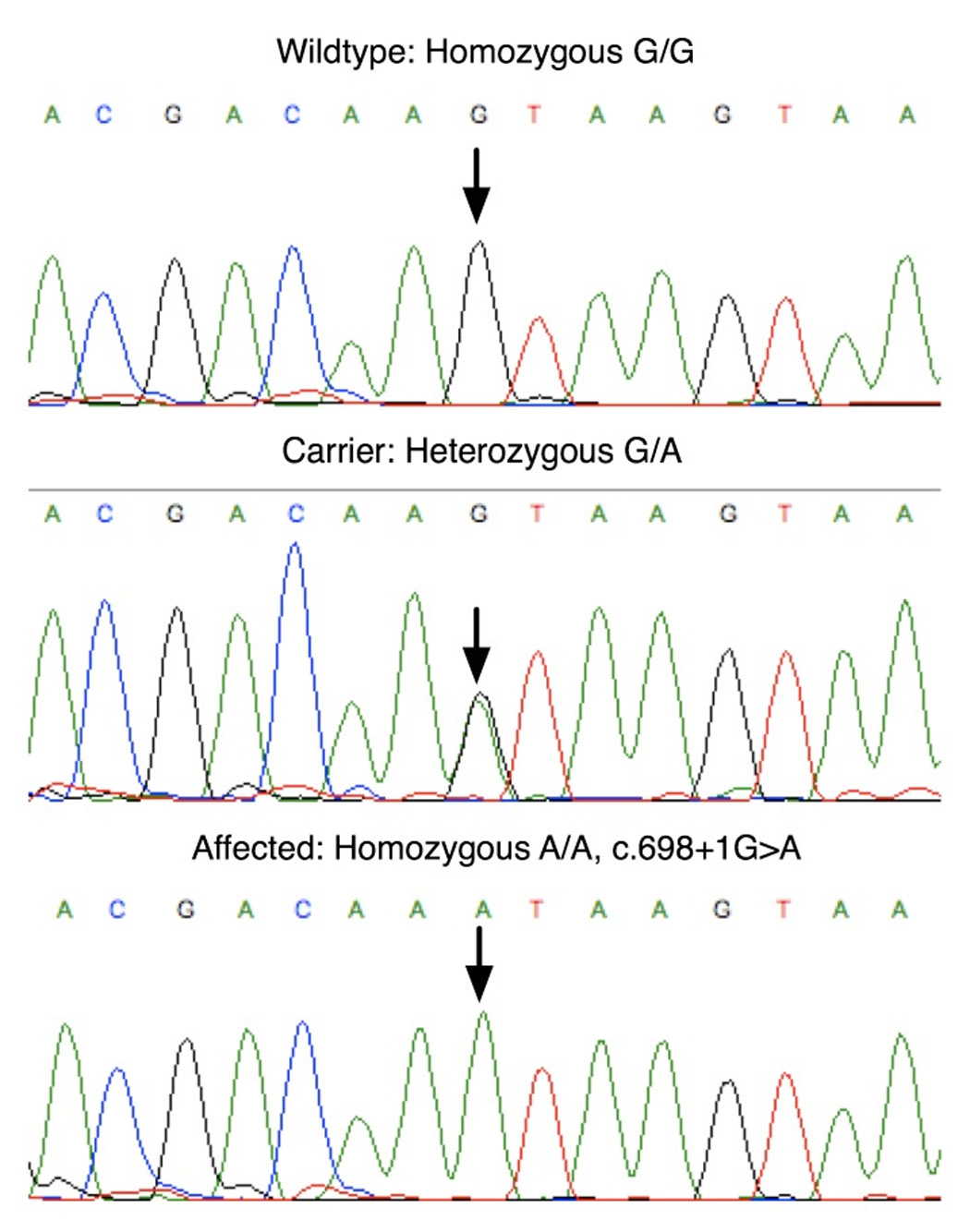
Sequence chromatograms showing the novel c.698+1G>A mutation in the RDX gene.
Fig. 5.
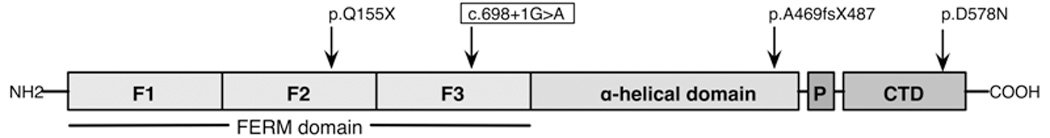
Domain structure of the RDX gene showing mutations known to cause DFNB24 hearing loss in humans. The novel splice site mutation described in this study (c.698+1G>A) is boxed.
Radixin and ezrin are present in the hair cell stereocilia of the mouse inner ear [Kitajiri et al., 2004; Pataky et al., 2004]. While it is known that radixin functions as a linker between sub-cellular actin and the plasma membrane [Niggli and Rossy 2008], the exact role of this protein in the stereocilia of hair cells is unclear. Current knowledge of the role of these proteins in the inner ear comes from study of a radixin knockout mouse model.
RDX −/− mice exhibit syndromic hearing impairment and hyperbilirubinemia [Kikuchi et al., 2002; Kitajiri et al., 2004]. The murine liver phenotype does not appear to model the human phenotype. One affected individual in the family reported here and another in a previously reported DFNB24 family have been shown to have normal liver function [Khan et al., 2007].
In conclusion, we have identified the first splice site mutation in the RDX gene and the first mutation known to cause DFNB24 hearing loss in the Iranian population. Our data also suggest that, in contrast to the murine model, liver dysfunction is not a component of the human phenotype. DFNB24 appears to be a rare form of ARSNHL as only four families have been reported. Further characterization of the molecular events triggered by this new mutation may increase our understanding of the function of radixin protein in the inner ear.
ACKNOWLEDGMENTS
The authors sincerely thank the family for their participation in this study. We are also grateful to T.B. Friedman and Z. Ahmed (Section on Human Genetics, Laboratory of Molecular Genetics, National Institute on Deafness and Other Communication Disorders, National Institutes of Health, Rockville, Maryland, USA) for generously providing oligonucleotides for this study. R.J.H. Smith is the Sterba Hearing Research Professor, University of Iowa College of Medicine, who supported the project with National Institutes of Health (NIH)-National Institute on Deafness and Other Communication Disorders (NIDCD) grant RO1 DCOO3544-09. M. Bahlo is supported by an Australian National Health and Medical Research Council Career Development Award.
REFERENCES
- Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. Epub 2001 Dec 3. [DOI] [PubMed] [Google Scholar]
- Broman KW, Murray JC, Sheffield VC, White RL, Weber JL. Comprehensive human genetic maps: individual and sex-specific variation in recombination. Am J Hum Genet. 1998;63:861–869. doi: 10.1086/302011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di X, Matsuzaki H, Webster TA, Hubbell E, Liu G, Dong S, Bartell D, Huang J, Chiles R, Yang G, Shen MM, Kulp D, Kennedy GC, Mei R, Jones KW, Cawley S. Dynamic model based algorithms for screening and genotyping over 100 K SNPs on oligonucleotide microarrays. Bioinformatics. 2005;21:1958–1963. doi: 10.1093/bioinformatics/bti275. Epub 2005 Jan 18. [DOI] [PubMed] [Google Scholar]
- Khan SY, Ahmed ZM, Shabbir MI, Kitajiri S, Kalsoom S, Tasneem S, Shayiq S, Ramesh A, Srisailpathy S, Khan SN, Smith RJ, Riazuddin S, Friedman TB. Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Hum Mutat. 2007;28:417–423. doi: 10.1002/humu.20469. [DOI] [PubMed] [Google Scholar]
- Kikuchi S, Hata M, Fukumoto K, Yamane Y, Matsui T, Tamura A, Yonemura S, Yamagishi H, Keppler D, Tsukita S. Radixin deficiency causes conjugated hyperbilirubinemia with loss of Mrp2 from bile canalicular membranes. Nat Genet. 2002;31:320–325. doi: 10.1038/ng905. Epub 2002 Jun 17. [DOI] [PubMed] [Google Scholar]
- Kitajiri S, Fukumoto K, Hata M, Sasaki H, Katsuno T, Nakagawa T, Ito J, Tsukita S. Radixin deficiency causes deafness associated with progressive degeneration of cochlear stereocilia. J Cell Biol. 2004;166:559–570. doi: 10.1083/jcb.200402007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, Shlien A, Palsson ST, Frigge ML, Thorgeirsson TE, Gulcher JR, Stefansson K. A high-resolution recombination map of the human genome. Nat Genet. 2002;31:241–247. doi: 10.1038/ng917. Epub 2002 Jun 10. [DOI] [PubMed] [Google Scholar]
- Morton NE. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci. 1991;630:16–31. doi: 10.1111/j.1749-6632.1991.tb19572.x. [DOI] [PubMed] [Google Scholar]
- Niggli V, Rossy J. Ezrin/radixin/moesin: versatile controllers of signaling molecules and of the cortical cytoskeleton. Int J Biochem Cell Biol. 2008;40:344–349. doi: 10.1016/j.biocel.2007.02.012. Epub 2007 Feb 22. [DOI] [PubMed] [Google Scholar]
- Pataky F, Pironkova R, Hudspeth AJ. Radixin is a constituent of stereocilia in hair cells. Proc Natl Acad Sci U S A. 2004;101:2601–2606. doi: 10.1073/pnas.0308620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabbee N, Speed TP. A genotype calling algorithm for affymetrix SNP arrays. Bioinformatics. 2006;22:7–12. doi: 10.1093/bioinformatics/bti741. Epub 2005 Nov 2. [DOI] [PubMed] [Google Scholar]
- Shapiro MB, Senapathy P. RNA splice junctions of different classes of eukaryotes: sequence statistics and functional implications in gene expression. Nucleic Acids Res. 1987;15:7155–7174. doi: 10.1093/nar/15.17.7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Bale JF, Jr, White KR. Sensorineural hearing loss in children. Lancet. 2005;365:879–890. doi: 10.1016/S0140-6736(05)71047-3. [DOI] [PubMed] [Google Scholar]
- Van Camp G, Smith RJ. Hereditary Hearing Loss Homepage. 2008 http://webhost.ua.ac.be/hhh/
- Wigginton JE, Abecasis GR. PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics. 2005;21:3445–3447. doi: 10.1093/bioinformatics/bti529. Epub 2005 Jun 9. [DOI] [PubMed] [Google Scholar]