

Abstract
High levels of expression of the human DEK gene have been correlated with numerous human malignancies. Intracellular DEK functions have been described in vitro and include DNA supercoiling, DNA replication, RNA splicing, and transcription. We have demonstrated DEK also suppresses cellular senescence, apoptosis and differentiation, thus promoting cell growth and survival in monolayer and organotypic epithelial raft models. Such functions are likely to contribute to cancer, but direct evidence to implicate DEK as an oncogene has remained elusive. Here we show that in line with an early role in tumorigenesis, murine papilloma formation in a classical chemical carcinogenesis model was reduced in DEK knockout mice. Additionally, HPV E6/E7, hRas and DEK cooperated in the transformation of keratinocytes in soft agar and xenograft establishment, thus also implicating DEK in tumor promotion at later stages. Finally, adenoviral DEK depletion via shRNA expression resulted in cell death in human tumor cells in vitro and in vivo, but did not significantly affect differentiated epithelial cells. Taken together, our data uncover oncogenic DEK activities as postulated from its frequent upregulation in human malignancies, and suggest that the targeted suppression of DEK may become a strategic approach to the treatment of cancer.
Introduction
The human DEK protein was originally identified as a fusion with the CAN/NUP214 nucleoporin in a subset of acute myeloid leukemia patients (1). DEK was subsequently reported as a gene that is frequently upregulated in aggressive human tumors such as glioblastoma, melanoma, and bladder carcinoma (2-4). DEK is an upregulated target of the HPV E7 oncogene in vitro (5), as well as in HPV E7 transgenic mouse epithelium and in human cervical cancer biopsies in vivo (6, 7). These studies had implicated retinoblastoma (RB) family members in the regulation of DEK, and transcriptional regulation of DEK through E2F1/E2F2 was later reported (4). Because RB tumor suppressor pathways are inactivated in most human malignancies, E2F-mediated expression is likely one relevant mechanism that drives DEK overexpression. In fact, DEK has been found to be highly upregulated in retinoblastoma and small cell lung cancers, both of which are strongly associated with RB loss ((8-11) & Supp. Fig. 1A).
A number of intracellular DEK activities have been studied extensively in vitro, and DEK was independently discovered as a protein that modulates the topology of SV40 minichromosomes (12). DEK has been proposed to function in replication (12), positive and negative regulation of transcription (13-19) as well as mRNA processing (20-22). DEK likely modulates DNA architecture with a high affinity for chromatin (23), and perhaps as a related property, DEK has recently been implicated in DNA damage responses (24). DEK inhibits senescence and apoptosis, at least in the latter case via the destabilization of p53 (5, 25, 26). The above functions, either individually or in combination, may well contribute to pro-carcinogenic DEK functions. Although DEK overexpression correlates strongly with carcinogenesis and although the above intracellular functions of DEK hint at a role in tumorigenesis, the question of whether DEK is indeed an oncogene has not been directly addressed. Based on Oncomine data and tumor cell line analyses, we report that DEK expression is transcriptionally upregulated in a wide variety of human tumors. This upregulation is not merely correlative as DEK overexpression in normal, immortalized keratinocytes (NIKs) cooperated with hRas, HPV E6 and E7 for anchorage independent growth and tumorigenesis in nude mice. In order to determine whether DEK was important for tumor formation in a murine epithelial cancer model, we subjected DEK knockout and control mice to a two-step DMBA/TPA protocol. Papilloma formation was significantly decreased in DEK knockout mice compared to wild type animals and heterozygote controls. Importantly, we show here that differentiated cells are almost completely resistant to DEK depletion compared to their undifferentiated counterparts. Our data are the first to demonstrate oncogenic DEK activities according to classical parameters, and strengthen the notion that targeting DEK may be a feasible approach for the treatment of cancer.
Materials and Methods
Cell Culture
U2OS human osteosarcoma cells were maintained in Dulbecco's modified Eagle medium (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS) and antibiotics. Normal immortalized keratinocytes (NIKs) were maintained on irradiated murine J2 3T3 feeder fibroblasts as described previously (27). Primary human foreskin keratinocytes (HFKs) were prepared from human foreskins (28) and maintained in Epilife Medium (Cascade Biologics, Portland, OR) with antibiotics. For the differentiation of HFKs, cells were overlaid with Epilife containing 10% FBS and 1uM CaCl.
Plasmids and viral constructs
The DEK open reading frame was amplified using the forward primer F- [5′ ATGTCCGCCTCGGCC 3′] and the reverse primer R- [5′ TCAAGAAATTAGCTCTTTTACAG 3′]. The cDNA was cloned into the pGEM-Teasy vector (Promega, Madison, WI). The DEK cassette was sequenced, digested with Not I and cloned into a Not I digested FMEV type vector pSF91-I-eGFP-PRE (R780) obtained from the Baum laboratory (29). FMEV retroviral vectors combine the long terminal repeat of Friend mink cell focus-forming virus with the 5′ untranslated leader region of the murine embryonic stem cell virus for optimal transgene expression. Vector particles were generated in the Viral Vector Core facility at CCHMC, and were pseudotyped with the feline endogenous virus (RD114) envelope protein. The pBABE-hRas vector encoding oncogenic H-RasV12 was a generous gift from Scott Lowe. The empty Ad as well as the AdDEKsh vector were described previously (25). Producer cell lines for empty LXSN retrovirus as well as retroviruses expressing HPV16 E6, E7 and E6/E7, respectively, were a generous gift from Dr. Denise Galloway, University of Washington, Seattle, Washington.
Adenoviral and retroviral infections
For adenoviral infections, the cells were washed with PBS and infected with the indicated infectious units (IUs)/cell of adenovirus stock in PBS containing 4% FBS for one hour as described previously (25). Virus was then aspirated and the cells were washed twice with PBS and overlaid with fresh media. For retroviral infections, NIKs were transduced with 4mls of R780 or R780-DEK retroviral supernatant containing 8ug/ml polybrene. The cells were washed with PBS after three hours, and overlaid with fresh media. Cell pools were sorted for GFP expression using a fluorescence-activated cell sorter (FACS VantageSEDiVa; Becton Dickinson, San Jose, CA). Cells were collected and maintained on plastic or feeder cells (for NIKs) as above. Cells infected with puromycin or neomycin resistant vectors were selected in 1μg/ml puromycin or 650μg/ml (for NIKs) or 200μg/ml (for HFKs) G418, respectively.
Western blot analyses
Western blot analyses were performed as described previously (25). Membranes were probed with either the DEK monoclonal antibody (BD Biosciences, San Diego, CA), a p53 (Ab-6) monoclonal antibody (Calbiochem, San Diego, CA), a cyclin A polyclonal antibody (Santa Cruz biotechnology, Santa Cruz, CA), PCNA antibody (BD Biosciences, San Diego, CA) p21 monoclonal antibody (Calbiochem, San Diego, CA), or an actin specific monoclonal antibody, a generous gift from James Lessard.
Soft agar assays
A total of 5×104 NIKs were submerged in 0.4% agarose in F media and plated on a 0.8% agarose/F media underlay. Cells were overlaid with 0.4% agarose/F media twice a week for four weeks. Colonies were quantified and pictures were taken at the end of the experiment.
Apoptosis Analysis by Flow Cytometry
Cells were infected with either empty Ad or AdDEKsh. Cells were harvested by trypsinization on the indicated days, washed in cold PBS, and fixed with BD cytofix/cytoperm (BD Pharmingen, San Diego, CA) for 20 minutes at room temperature. The cells were then washed with BD perm/wash buffer twice and 20ul/1×106 cells were incubated with biotinylated anti-active Caspase 3 antibody (BD Pharmingen, San Diego, CA) for one hour at room temperature in the dark. Cells were washed and incubated with 5ng per 1×106 cells of streptavidin-APC (BD Pharmingen, San Diego, CA) for 30 minutes at room temperature. The cells were washed and analyzed using BD Cell-Quest software on a Flow Cytometer (BD Biosciences, San Jose, CA).
Immunofluorescence Microscopy
Sections were deparaffinized in xylene, and rehydrated. Antigen retrieval was by heating the sections in 10mM NaCitrate, pH 6.0, in a rice cooker for twenty minutes and allowed to cool to room temperature. TUNEL staining was performed with the Cell Death Detection Kit reagents (Roche, Indianapolis, IN). Sections were washed with PBS, counterstained with DAPI Vector Vectashield mounting media (Vector laboratories, Burlingame, CA) and coverslipped. Immunofluorescence detection was via a Zeiss flourescence microscope (Zeiss, Thornwood, NY) and images were captured using 20x and 40x magnification with an axiovision camera (Lucas Microscope Service, Skokie, IL) driven by Axiovision software.
DMA/TPA murine skin carcinogenesis
A schematic of the targeting construct is shown in Fig. 1A. For generation of a Dek knockout mouse, an 8.6 kb SalI-BamHI fragment from a 129 SvJ phage l EMBL-3 genomic clone, containing the 5′ half of the DEK open reading frame, was subcloned into pBluescript. A 5 kb IRES-LacZ-Neo selectable marker (33) was inserted into the NsiI site in Dek exon 6 (Dek codon 184). The targeting vector was linearized with SalI and electroporated into E14 mouse ES cells. G418 resistant clones were isolated and homologous recombinants were identified by using Southern blot analysis of EcoRI-digested DNA, probed with a 5′ external 320 bp EcoRI-BamHI fragment. The wild type (9.5 kb) and mutant (8.4 kb) Dek fragments were easily distinguished (not shown) and 7 independent clones with normal karyotype were identified, 4 of which were microinjected into C57Black/6 blastocysts. All clones produced chimeric offspring, which gave germ line transmission of the targeted allele. Dek−/− mice were healthy and bred normally. Bone marrow of Dek−/− mice did not express DEK protein as determined by western blot analysis using a rabbit polyclonal antibody (12) (data not shown). All use and handling of mice were carried out in the American Association for Accreditation of Laboratory Animal Care-approved Cincinnati Children's Hospital Veterinary Care Facility according to an IACUC approved protocol to SIW. For genotyping, primer pair A (5′-CGA ACT CGT GAA GAG GAT CTT GA-3′, 5′-ATG TGT CAG GCT GCA TCT CCA ATG-3′) was utilized for the amplification of the wild type allele, primer pair B (5′-ATC CAT CAT GGC TGA TGC AAT GCG-3′, 5′-TGG AAG GTA AAG AGT GGC CCT TA-3′) was utilized for the amplification of the knockout allele. Mice were subjected to a two step carcinogenesis model adapted from (30). Briefly, 100μg of 9,10-dimethyl-1,2-benzanthrancene (DMBA, Sigma) in 100μl of acetone was applied to the shaved flank of mice once at the age of 2-3 months. One week after DMBA application, 30μg of the tumor promoter 12-O-tetradecanoyl-phorbol acetate (TPA) in 100μl of acetone was applied twice a week for twenty weeks. Papilloma counts were performed once a week and recorded. Tumors were harvested for analysis after 20 weeks of TPA treatment.
Fig. 1. Dek expression is important for the formation of papillomas in vivo.

A. Targeting of the mouse Dek gene. The top line shows the 5′ half of the Dek gene comprising exons 1-6. The arrow indicates the direction of transcription. The Nsi1 site (N) in exon 6 is indicated as well as the EcoRI sites (E) used for Southern blotting. The middle line shows the targeting construct containing 8.6 kb of genomic DNA with the IRES-LacZ-Neo selectable marker inserted into the NsiI site in exon 6. The bottom line shows the targeted allele. P indicates the 320 bp EcoRI-BamHI fragment used as a probe for Southern blotting. B. DMBA/TPA carcinogenesis model. Dek wildtype versus heterozygote and knockout littermates (three per group) were subjected to 100ug DMBA on the flank, and starting at one week post tumor initiation, 30ug of the tumor promoter TPA was applied twice a week for 20 weeks. The graph represents the number of papillomas per mouse that were present at the indicated time points. The asterisk represents a p value of less than 0.05 between the knockout and heterozygous mice. H&E staining below depicts the morphology of a representative papilloma at 50x magnification. The boxed area in the left panel is shown at 200x magnification in the right panel. Different genotypes developed papillomas that were morphologically similar. C. Western blot analysis. Primary keratinocytes from the heterozygous and knockout Dek mice were harvested for the isolation of protein, and western blot analysis for DEK and actin was carried out. Primary murine keratinocytes were prepared from the skin of newborn DEK heterozygous and knockout mice, floated overnight at 4°C in 0.25% trypsin/EDTA. On the next day, the epidermis was separated from the dermis, minced, filtered and keratinocytes were plated in Defined Keratinocyte Serum-Free Media supplemented with FBS and antibiotics. After seven days, protein was harvested for western blot analysis. Bands shown were imaged from the same exposed film. Immunohistochemistry on normal, untreated skin of wild type, heterozygote and knockout Dek mice confirms DEK loss in the knockout mice at 1000x magnification. Tissue sections were deparaffinized, rehydrated, subjected to antigen retrieval and processed. Sections were incubated with IgG1 antibody as a control, or with DEK antibody. Sections were washed and incubated in ABC Reagent for 30 minutes, washed again and incubated with 3,3-diaminobenzidine (DAB). Sections were then washed and stained in Nuclear Fast Red prior to mounting and visualization.
Adenoviral injections into established tumors
For tumorigenesis studies using an IACUC approved protocol, the flanks of 4-8 wk old female athymic nude mice (Harlan laboratories, Indianapolis, IN) were injected with 5×106 cells. NIKs containing hRas vector, and either empty R780 backbone or the R780-DEK vector, together with either HPV16 E6, E7 or HPV 16 E6/E7 were used for the injections. Tumor growth was monitored for three months, at which time tumors were harvested, fixed in 4% paraformaldehyde overnight, washed in PBS and subsequently dehydrated in a series of alcohols. Tumors were embedded in paraffin and sectioned for analysis. For the in vivo DEK targeting studies, 1×106 U2OS cells were injected into each flank. Tumors were allowed to grow to 150-250mm3 and at that time, 108 IU of empty Ad or AdDEKsh were injected into the left or right flank tumors, respectively, for internal comparison. Tumors were either harvested at three days post viral delivery or were injected twice more at day three and six days post initial viral injection and harvested one day after the final injection. Tumors were fixed in paraformaldehyde and subsequently washed for OCT or paraffin embedding and sectioned prior to analysis.
Results
The human DEK proto-oncogene is upregulated in many human cancers
Using the oncomine research platform, we identified a large number of human malignancies in which DEK was significantly overexpressed (Supp. Fig. 1A). Interestingly, the degree of DEK upregulation often correlated with the severity of prognosis as indicated by histopathological determination of a higher stage and grade, or poor differentiation characteristics (data not shown). For several tumor types including glioblastoma and cervical cancer, DEK upregulation has already been emphasized in the literature (3, 6). Whereas DEK was highly overexpressed in most malignancies, its expression levels were decreased in prostate cancers, and in a subset of ovarian and adult bone marrow associated cancers. Decreased DEK expression in AML as observed here (Fig. 1A, see GEO website http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE7186) contrasts with a previous report by the Knuutila laboratory (31) describing increased DEK expression. Interestingly, a striking difference between the two reports is patient age: DEK downregulation was observed in pediatric AML, whereas DEK upregulation was observed in adult AML. Additional experiments are needed to determine whether these differences in DEK expression indeed distinguish pediatric from adult AML, perhaps even contributing to their known biological differences. Regardless, the expression data indicate that high DEK expression is not universally expected for all tumor types. DEK was overexpressed in cervical cancer and head and neck cancers, both of which have been correlated with human papilloma virus (HPV) infection. This association is in agreement with recent data wherein DEK was highly expressed in HPV positive cervical lesions compared to controls (6)(7). As shown in Supp. Fig. 1B, western blot analysis of a panel of epithelial primary and cancer cells demonstrated that DEK protein is upregulated in the majority of human cancer cell lines compared to primary keratinocytes and fibroblasts (compare lanes 4-13 to 1-3). DEK expression appeared to be especially high in cervical cancer cell lines (lanes 4-8) as well as in osteosarcoma cells (lanes 9-10). DEK expression was also strongly increased in 293 cells expressing the adenoviral E1A protein. E1A, like E7, is known for the potent inhibition of RB protein family members. Taken together, these data suggested that DEK is upregulated in many human malignancies and that it may promote multiple stages of tumor progression.
DEK is important for papilloma formation in vivo
Because these and other published data suggested an oncogenic role for DEK , we used a new Dek knockout mouse model to investigate a role for this molecule in the early stages of skin tumorigenesis in vivo. Generation of the Dek knockout mice is described in detail in the Materials and Methods, and a schematic of the targeting construct is shown in Fig. 1A. A classical two-step epithelial carcinogenesis model was employed (32), which consists of a one time application of DMBA for initiation, followed by TPA promotion twice a week for 20 weeks. We used littermate Dek knockout, heterozygous and wild type mice for these experiments. As depicted in Fig. 1B, there was a significant delay in the formation of papillomas in Dek knockout mice compared to wildtype and heterozygous mice. Papillomas ultimately formed in the Dek knockout mice, suggesting a role for DEK in tumor initiation in this model. DEK knockout in primary murine keratinocytes was confirmed by western blot analysis and by immunohistochemistry on normal, untreated mouse skin (Fig. 1C)
DEK overexpression promotes transformation in vitro and in vivo
The finding that DEK supported papilloma formation in vivo suggested but did not prove that it functions as an oncogene. In order to probe such transforming potential directly, we performed soft agar colony assays using normal human keratinocytes (NIKs) that stably expressed oncogenic hRas and HPV16 E6/E7 in the presence or absence of overexpressed DEK (Fig. 2B). NIKs are spontaneously immortalized cells, which are carried on feeder cells and have retained responsiveness to differentiation in organotypic raft culture (33). DEK overexpression dramatically increased colony formation, indicating transforming potential. Similar DEK cooperation was observed in NIKs which were transfected with the full length, E6/E7-expressing HPV16 genome (data not shown), and subsequently selected to yield a pure population of HPV positive cells. E7–induced p53 and p21 protein levels were suppressed by the coexpression of HPV E6, suggesting that DEK can promote transformation through p53-independent mechanisms (Fig. 2A).
Fig. 2. Overexpression of DEK protein in normal immortalized keratinocytes stimulates transformation in vitro.
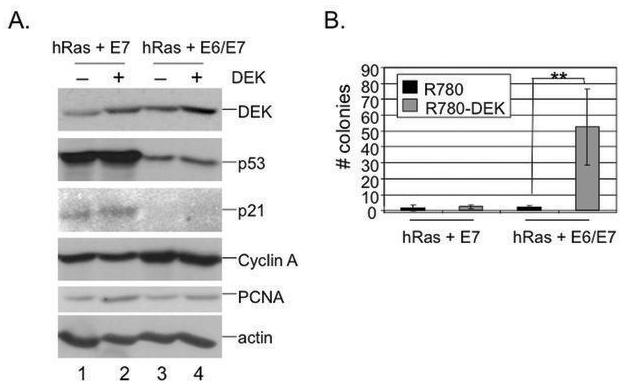
A. Western blot analyses. NIKs were transduced with either R780 or R780-DEK and sorted for GFP expression. Sorted NIKs were subsequently transduced with oncogenic hRas-V12 and either HPV E7 alone or HPV E6 and E7 in combination. Total protein lysates were subjected to DEK, cyclin A, p53, p21, PCNA and actin specific western blot analyses. B. Soft agar colony assay. Cells from (A) were submerged in 0.4% agarose, overlaid on top of a 0.8% agarose underlay and monitored for colony formation over three weeks, at which point colonies were counted. Two asterisks indicate a p value of less than 0.01.
We next investigated the ability of DEK to promote transformation in vivo. NIKs transduced with hRas, HPV E6 alone, HPV E7 alone or in combination with E6, and either the R780 empty vector or R780-DEK, were injected into the left and right flank of nude mice, respectively. Tumor formation was monitored for two months. No tumors formed in the presence of HPV E6 or E7 alone. However, DEK significantly increased the rate of tumor formation in the HPV E6/E7-expressing tumors from 25% to 75% (Fig. 3A). Tumors that formed from cells transduced with empty R780 vector or R780-DEK expressing virus were morphologically confirmed to be squamous cell carcinomas; however, DEK overexpressing tumors were significantly larger than tumors arising from vector treated control cells (Fig. 3B-C). Moreover, the mitotic index was increased in DEK expressing tumors compared to controls as assessed by phosphorylated histone H3 expression (Fig. 3D). Collectively, these data demonstrate that E6 or E7 is insufficient for inducing NIKs to form tumors, even in the presence of oncogenic ras. In contrast, combined E6 and E7 expression cooperates with ras in inducing tumorigenesis. Furthermore, the incidence and size of E6/E7 induced tumors is increased by DEK overexpression providing evidence that DEK cooperates with other oncogenes in tumor initiation and progression.
Fig. 3. DEK overexpression stimulates transformation in vivo in cooperation with the HPV oncoproteins and oncogenic ras.

A, B. Nude mouse injections. 5× 106 NIKs transduced with hRas, either E7 or E6/E7 and either empty R780 or R780-DEK vector were injected into the flanks of athymic nude mice. The R780 controls were injected into the left flank while DEK overexpressing cells were injected on the right flank of each mouse. Tumor formation (A) as well as tumor volume (B) were monitored over two months. Tumor volume was calculated using the following formula: length × width2 × π/6. One asterisk represents a p-value of < 0.05 while two asterisks represent a p-value of < 0.01. Experiments containing 3-4 mice per group were performed three independent times. C. Tumor morphology in the presence and absence of overexpressed DEK. Tumors from (A) were fixed, embedded in paraffin and sectioned. Sections were then stained with hematoxylin/eosin and pictures were taken at 50 and 200 fold magnification for the top and bottom panels, respectively. D. Immunohistochemistry for phosphorylated (Ser10) Histone H3 was performed on paraffin sections that were baked, deparaffinized, rehydrated and subjected to antigen retrieval. Sections were blocked using goat antiserum in phosphate buffered saline. Primary phosphorylated (Ser10) Histone H3 1:1000 (US Biological) antibodies were diluted in blocking solution, applied to tissue sections and incubated overnight at 4°C Antibody staining was detected with Vectastain Elite ABC and DAB Substrate Kits (Vector Laboratories, Inc.). Counts represent evaluation of 300 tumor cells representing 2-5 tumor sections/mouse and 3 mice per group. The asterisk represents a p value of less than 0.05, and pictures were taken at 1000 fold magnification.
Adenoviral delivery of DEK shRNA results in osteosarcoma cell death in vivo
We have shown previously that DEK-specific RNA interference results in cell death in vitro in tumor cell lines, and to a lesser extent in primary cells (25). Although DEK stimulated tumor growth in xenograft experiments (Fig. 3) and was important for murine papilloma formation in the DMBA/TPA model (Fig. 1), these experiments did not address whether sustained DEK expression was important for tumor maintenance in vivo. We therefore investigated whether DEK depletion might result in tumor cell death. Equal numbers of U2OS osteosarcoma cells were injected into both flanks of athymic nude mice. Tumors were allowed to grow to 200mm3 and then injected with GFP-expressing replication-deficient empty Ad or AdDEKsh virus using an established fractionated injection protocol at three different timepoints (34). The tumors were harvested one day after the final viral injection for analysis. After the first injection, GFP expression in the tumors was detectable by bioluminescence whole animal imaging (Fig. 4A), demonstrating that both tumors had received GFP-expressing adenovirus. GFP expression remained detectable even after tumor excision using a dissecting fluorescence microscope (data not shown). After image capture, the tumors were fixed and embedded for analysis. TUNEL staining revealed dramatic induction of apoptosis in AdDEKsh infected compared to Ad control infected tumors (Fig. 4B). Because these viruses are replication-deficient, thus only allowing for transient DEKsh expression, total tumor growth was not substantially different between the control and DEK-depleted state. Whether uniform DEK depletion will cause tumor regression will therefore await the development of more efficient delivery methods. Nonetheless, our data provide proof of concept that DEK depletion results in cell death in vivo and that the targeting of DEK in human tumors may therefore be a feasible approach to mediate disease regression.
Fig. 4. DEK depletion in xenograft tumors results in cell death in vivo.

A. In vivo imaging system (IVIS) analysis. Nude mice were injected with 5×10^5 U2OS tumor cells into each flank and injected with either empty Ad (left) or AdDEKsh (right) when tumors reached 200mm3. GFP expression in the tumors was determined and analyzed by IVIS software. Color bars represent minimum to maximum level of GFP expression. B. Immunofluorescence. Tumors from (A) were harvested, fixed and embedded in paraffin and sectioned for analysis. Sections were analyzed by immunofluorescence microscopy for the detection of cell death. The experiment was performed twice, using four mice for each experiment. Similar results were seen in both experiments.
Epithelial differentiation rescues DEK-depleted cells from apoptosis
Previous data demonstrated that DEK overexpression confers protection from apoptosis, and conversely, that acute DEK depletion by RNA interference results in apoptosis in p53-competent cancer cells and to a lesser extent in primary cells (25). Together with the above detection of apoptosis following DEK depletion in vivo (Fig. 4), this suggested that DEK targeting strategies may be therapeutically useful, but also that toxicity may be expected from such cancer treatments. To evaluate the degree of toxicity, and considering that differentiated cells comprise the majority of the epithelium, we next asked whether differentiated cells might be protected from cell death. As reported previously, primary keratinocytes were susceptible to a degree of DEK RNAi-associated apoptosis, albeit not to the level of U2OS osteosarcoma cells (Fig. 5A and B). We next infected primary keratinocytes with either empty Ad or AdDEKsh and subjected the cells to regular growth medium or to medium containing 1mM calcium and 10% FBS in order to induce differentiation (35). Differentiation was verified morphologically and reflected the typical tightening of cell-cell interactions (Fig. 5B). As reported previously, DEK depletion caused a 3-5% increase in apoptosis in the undifferentiated HFK population, compared to 15% apoptosis in osteosarcoma cells on day 3 post infection (Fig. 5A). However, there was only a 1% gain in apoptosis following DEK depletion in the differentiated HFKs, and the observed increase between Ad and AdDEKsh infected differentiated cells did not rise to the level of statistical significance (Fig. 5B). The expected decrease in the levels of DEK protein was confirmed by western blot analysis in both populations (Fig. 5B). Differentiation itself suppressed DEK protein expression slightly (compare lanes 1 and 3), even though the precise timing of DEK suppression was often donor dependent (data not shown). This repression occurs presumably through the upregulation of RB protein function during this process. It is possible that DEK overexpression in cancer renders cells more dependent upon DEK, thus supporting new cancer treatments via the targeting of DEK. Detection of the cyclin/cdk kinase inhibitor p21 in DEK positive and depleted keratinocytes revealed upregulation of p21 by DEK depletion in undifferentiated, but not differentiated cells. This was presumably a consequence of p53 activation as previously published (25), and indeed, p21 induction was preceded by increased p53 protein levels as predicted (data not shown). The extracts were also probed for expression of the p53 related p63 isoform ΔNp63, a stem and progenitor cell marker in mouse and human skin, and critical regulator of basal cell proliferation and differentiation (36). As expected for differentiating keratinocytes, ΔNp63 expression was distinctly downregulated, and interestingly, was reduced in response to DEK depletion in both differentiated and undifferentiated cells. These data indicate a role for DEK in the repression of squamous cell differentiation. Our recent findings describe the upregulation of ΔNp63 and decreased differentiation as a consequence of DEK overexpression (7). Whether ΔNp63 is a critical DEK mediator or simply marks differentiation inhibition in response to DEK overexpression remains to be determined. Our findings demonstrate that differentiated epithelial cells are less sensitive to DEKsh dependent apoptosis than are cancer cells, suggesting that targeting DEK may offer a therapeutic window for cancer treatment.
Fig. 5. Epithelial differentiation rescues cells from DEKsh-induced apoptosis.
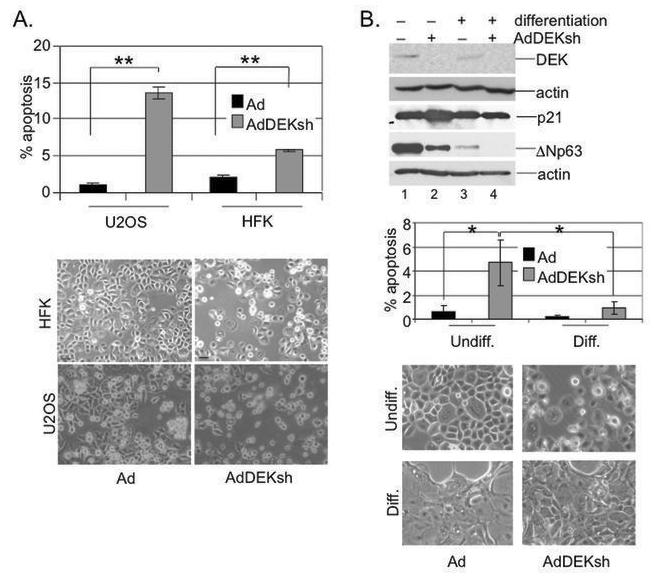
A. HFKs and U2OS cells were infected with either empty Ad or AdDEKsh and subjected to anti-active caspase 3 antibody and flow cytometry on day three. The cells were photographed after four days. B. HFK differentiation. HFKs were infected as in (A) and either subjected to normal medium, or differentiated upon the addition of 1mM calcium and 10% FBS. Cells were harvested for apoptosis assays as in A, subjected to DEK, p21, ΔNp63 and actin specific western blot analyses on day three, and analyzed for cellular morphology. One asterisk represents a p-value of < 0.05 while two asterisks represent a p-value of < 0.01. The Ad infected populations were not statistically different from each other nor were the Ad and DEKsh infected differentiated cell populations.
Discussion
The human DEK gene is widely referred to as a proto-oncogene in the literature because of its involvement in a chromosomal translocation in AML as well as its upregulated expression in multiple human malignancies. Together with reported intracellular DEK activities that inhibit senescence (20, 47), apoptosis (28) and cellular differentiation (7) oncogenic DEK activities have been suspected but never formally demonstrated. Because Dek knockout mice were relatively resistant to the formation of benign papillomas (Fig. 1), we suggest that DEK is an active contributor at early tumor stages. To directly assess such putative activities, we utilized normal immortalized keratinocytes (NIKs) to demonstrate that DEK i) contributes to human tumor formation, ii) cooperates with known oncogenes in transformation and iii) is required for the growth of primary proliferating, but not differentiated human keratinocytes. We chose NIKs for these studies since they are a widely utilized model for keratinocyte differentiation in two and three dimensional skin models. This spontaneously immortalized human keratinocyte cell line exhibits a near diploid, stable karyotype, wild type p53, normal responses to squamous differentiation in 3D organotypic rafts models, and is non-tumorigenic in athymic mice (37).
The cooperating high risk HPV E6 and E7 oncogenes exhibit well documented cellular immortalization and transformation properties (38). HPV E7 alone can support the immortalization of primary keratinocytes under certain circumstances, and co-expression of E6 greatly facilitates the process. E7 proteins bind to and degrade cellular RB family members, thus stimulating cellular proliferation (39, 40). Inappropriate proliferation in response to E7 expression is met by cellular defense mechanisms that at least in part involve upregulated p53 levels, thus predisposing cells to cell death and differentiation (41, 42). Coexpression of the high risk E6 proteins counteracts this reported trophic sentinel response through ubiquitin-mediated degradation of p53 (43, 44).
Given the presence of E6 in our transformation experiments (Fig. 2 and 3), an additional, and perhaps p53-unrelated role emerged for DEK. The HPV oncogenes alone were not sufficient for transformation (data not shown), and the V12 oncogenic form of the cellular Ras GTPase was therefore co-expressed. RasV12 represents a prominent cooperating oncogene in the multistep process of human carcinogenesis (45). Expression of E6/E7 and Ras was not sufficient for colony formation in soft agar over the course of 3 weeks, but was sufficient for causing tumors in 25% of immunodeficient mice over the course of 3 months. Importantly, the additional overexpression of DEK resulted in a dramatic stimulation of colony formation. This stimulation occurred despite maximal repression of p53 in the presence of E6 (Fig. 2A). DEK overexpression also increased tumor frequencies and volume (Fig. 3). Collectively, these data indicate DEK overexpression induces tumor growth through a mechanism that is independent of p53 repression. Moreover, the results further support a positive correlation between high levels of DEK expression and more aggressive later stage tumors in some human malignancies such as breast cancer (46).
High levels of DEK expression have been associated with numerous cancers including glioblastoma, hepatocellular carcinoma, AML, and breast cancer. Until recently, it was unclear whether this association was merely a correlative event, or whether DEK expression was functionally important in the genesis of such tumors (1, 3, 31, 46-48). We show here that DEK exhibits oncogenic activities at multiple stages of tumorigenesis. A need for sustained and high level DEK expression in many cancer cell lines is reflected by our previous data whereby DEK depletion resulted in cancer cell death in vitro (25). The finding that DEK depletion in preformed U2OS cell tumors results in apoptosis (Fig. 4) provides proof of concept for the treatment of cancer via DEK suppression. Future approaches may involve more efficacious ways to deliver DEK specific shRNA, alone and in combination with other treatments, and may explore the development of small molecules for the specific inhibition of oncogenic DEK activities.
DEK depletion resulted in the death of primary human keratinocytes in vitro (25), suggesting that a degree of toxicity would be expected after DEK targeting. Interestingly, differentiated keratinocytes were almost completely resistant to DEK depletion in comparison to their proliferating counterparts (Fig. 5). Thus, targeted DEK therapy may provide a therapeutic window between cancerous and normal cells. Taken together, our data provide the first evidence to implicate DEK as a bona fide oncogene, and suggest that DEK inhibition in human cancer may be a viable approach for the treatment of benign and malignant lesions.
Supplementary Material
Acknowledgements
We thank Monique Morrison for excellent technical assistance. We thank Dan Marmer and the Flow Cytometry and Cell Sorting Services at CCHMC, as well as Meredith Taylor, Mary Rolfes, and Pam Groen in the Department of Pathology for excellent technical assistance. We thank Han van der Loo and the CCHMC Viral Vector Core for retroviral production. We thank James Lessard for the monoclonal actin antibody. This research was supported by Public Health Service grants CA116316 to S. I. W., HL079193 to K. W-B, and CA114004 to T. P. C. T. W-D. was supported by a training grant T32 CA59268 from the National Cancer Institute and by an Illick fellowship from the Albert J. Ryan Foundation.
References cited
- 1.von Lindern M, Fornerod M, van Baal S, et al. The translocation (6;9), associated with a specific subtype of acute myeloid leukemia, results in the fusion of two genes, dek and can, and the expression of a chimeric, leukemia-specific dek-can mRNA. Mol Cell Biol. 1992;12(4):1687–97. doi: 10.1128/mcb.12.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu Q, Hoffmann MJ, Hartmann FH, Schulz WA. Amplification and overexpression of the ID4 gene at 6p22.3 in bladder cancer. Mol Cancer. 2005;4(1):16. doi: 10.1186/1476-4598-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kroes RA, Jastrow A, McLone MG, et al. The identification of novel therapeutic targets for the treatment of malignant brain tumors. Cancer Lett. 2000;156(2):191–8. doi: 10.1016/s0304-3835(00)00462-6. [DOI] [PubMed] [Google Scholar]
- 4.Carro MS, Spiga FM, Quarto M, et al. DEK Expression is controlled by E2F and deregulated in diverse tumor types. Cell Cycle. 2006;5(11):1202–7. doi: 10.4161/cc.5.11.2801. [DOI] [PubMed] [Google Scholar]
- 5.Wise-Draper TM, Allen HV, Thobe MN, et al. The human DEK proto-oncogene is a senescence inhibitor and an upregulated target of high-risk human papillomavirus E7. J Virol. 2005;79(22):14309–17. doi: 10.1128/JVI.79.22.14309-14317.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu Q, Li Z, Lin H, Han L, Liu S, Lin Z. DEK overexpression in uterine cervical cancers. Pathology international. 2008;58(6):378–82. doi: 10.1111/j.1440-1827.2008.02239.x. [DOI] [PubMed] [Google Scholar]
- 7.Wise-Draper TM, Morreale RJ, Morris TA, Mintz RA, Hoskins EEB,SJ, Husseinzadeh N, Witte DP, Wikenheiser-Brokamp KAL,PF, Wells SI. DEK proto-oncogene expression interferes with the normal epithelial differentiation program. American Journal of Pathology. 2008 doi: 10.2353/ajpath.2009.080330. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orlic M, Spencer CE, Wang L, Gallie BL. Expression analysis of 6p22 genomic gain in retinoblastoma. Genes Chromosomes Cancer. 2006;45(1):72–82. doi: 10.1002/gcc.20263. [DOI] [PubMed] [Google Scholar]
- 9.Corson TW, Gallie BL. One hit, two hits, three hits, more? Genomic changes in the development of retinoblastoma. Genes Chromosomes Cancer. 2007;46(7):617–34. doi: 10.1002/gcc.20457. [DOI] [PubMed] [Google Scholar]
- 10.Dimaras H, Khetan V, Halliday W, et al. Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum Mol Genet. 2008;17(10):1363–72. doi: 10.1093/hmg/ddn024. [DOI] [PubMed] [Google Scholar]
- 11.Wikenheiser-Brokamp KA. Retinoblastoma regulatory pathway in lung cancer. Current molecular medicine. 2006;6(7):783–93. doi: 10.2174/1566524010606070783. [DOI] [PubMed] [Google Scholar]
- 12.Alexiadis V, Waldmann T, Andersen J, Mann M, Knippers R, Gruss C. The protein encoded by the proto-oncogene DEK changes the topology of chromatin and reduces the efficiency of DNA replication in a chromatin-specific manner. Genes Dev. 2000;14(11):1308–12. [PMC free article] [PubMed] [Google Scholar]
- 13.Sammons M, Wan SS, Vogel NL, Mientjes EJ, Grosveld G, Ashburner BP. Negative regulation of the RelA/p65 transactivation function by the product of the DEK proto-oncogene. J Biol Chem. 2006;281(37):26802–12. doi: 10.1074/jbc.M600915200. [DOI] [PubMed] [Google Scholar]
- 14.Ko SI, Lee IS, Kim JY, et al. Regulation of histone acetyltransferase activity of p300 and PCAF by proto-oncogene protein DEK. FEBS Lett. 2006;580(13):3217–22. doi: 10.1016/j.febslet.2006.04.081. [DOI] [PubMed] [Google Scholar]
- 15.Hu HG, Illges H, Gruss C, Knippers R. Distribution of the chromatin protein DEK distinguishes active and inactive CD21/CR2 gene in pre- and mature B lymphocytes. Int Immunol. 2005;17(6):789–96. doi: 10.1093/intimm/dxh261. [DOI] [PubMed] [Google Scholar]
- 16.Gamble MJ, Fisher RP. SET and PARP1 remove DEK from chromatin to permit access by the transcription machinery. Nat Struct Mol Biol. 2007;14(6):548–55. doi: 10.1038/nsmb1248. [DOI] [PubMed] [Google Scholar]
- 17.Campillos M, Garcia MA, Valdivieso F, Vazquez J. Transcriptional activation by AP-2alpha is modulated by the oncogene DEK. Nucleic Acids Res. 2003;31(5):1571–5. doi: 10.1093/nar/gkg247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Faulkner NE, Hilfinger JM, Markovitz DM. Protein phosphatase 2A activates the HIV-2 promoter through enhancer elements that include the pets site. J Biol Chem. 2001;276(28):25804–12. doi: 10.1074/jbc.M006454200. [DOI] [PubMed] [Google Scholar]
- 19.Hollenbach AD, McPherson CJ, Mientjes EJ, Iyengar R, Grosveld G. Daxx and histone deacetylase II associate with chromatin through an interaction with core histones and the chromatin-associated protein Dek. J Cell Sci. 2002;115(Pt 16):3319–30. doi: 10.1242/jcs.115.16.3319. [DOI] [PubMed] [Google Scholar]
- 20.Le Hir H, Gatfield D, Izaurralde E, Moore MJ. The exon-exon junction complex provides a binding platform for factors involved in mRNA export and nonsense-mediated mRNA decay. Embo J. 2001;20(17):4987–97. doi: 10.1093/emboj/20.17.4987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McGarvey T, Rosonina E, McCracken S, et al. The acute myeloid leukemia-associated protein, DEK, forms a splicing-dependent interaction with exon-product complexes. J Cell Biol. 2000;150(2):309–20. doi: 10.1083/jcb.150.2.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Soares LM, Zanier K, Mackereth C, Sattler M, Valcarcel J. Intron removal requires proofreading of U2AF/3′ splice site recognition by DEK. Science. 2006;312(5782):1961–5. doi: 10.1126/science.1128659. [DOI] [PubMed] [Google Scholar]
- 23.Kappes F, Burger K, Baack M, Fackelmayer FO, Gruss C. Subcellular localization of the human proto-oncogene protein DEK. J Biol Chem. 2001;276(28):26317–23. doi: 10.1074/jbc.M100162200. [DOI] [PubMed] [Google Scholar]
- 24.Kappes F, Fahrer J, Khodadoust MS, et al. DEK is a Poly(ADP-ribose) acceptor in apoptosis and mediates resistance to genotoxic stress. Mol Cell Biol. 2008;28(10):3245–57. doi: 10.1128/MCB.01921-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol. 2006;26(20):7506–19. doi: 10.1128/MCB.00430-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johung K, Goodwin EC, DiMaio D. Human papillomavirus E7 repression in cervical carcinoma cells initiates a transcriptional cascade driven by the retinoblastoma family, resulting in senescence. J Virol. 2007;81(5):2102–16. doi: 10.1128/JVI.02348-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakahara T, Peh WL, Doorbar J, Lee D, Lambert PF. Human papillomavirus type 16 E1circumflexE4 contributes to multiple facets of the papillomavirus life cycle. J Virol. 2005;79(20):13150–65. doi: 10.1128/JVI.79.20.13150-13165.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wise-Draper TM, Allen HV, Jones EE, Habash KB, Matsuo H, Wells SI. Apoptosis inhibition by the human DEK oncoprotein involves interference with p53 functions. Mol Cell Biol. 2006;26(20) doi: 10.1128/MCB.00430-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klump H, Schiedlmeier B, Vogt B, Ryan M, Ostertag W, Baum C. Retroviral vector-mediated expression of HoxB4 in hematopoietic cells using a novel coexpression strategy. Gene Ther. 2001;8(10):811–7. doi: 10.1038/sj.gt.3301447. [DOI] [PubMed] [Google Scholar]
- 30.Langowski JL, Zhang X, Wu L, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442(7101):461–5. doi: 10.1038/nature04808. [DOI] [PubMed] [Google Scholar]
- 31.Casas S, Nagy B, Elonen E, et al. Aberrant expression of HOXA9, DEK, CBL and CSF1R in acute myeloid leukemia. Leuk Lymphoma. 2003;44(11):1935–41. doi: 10.1080/1042819031000119299. [DOI] [PubMed] [Google Scholar]
- 32.Reddig PJ, Dreckschmidt NE, Ahrens H, et al. Transgenic mice overexpressing protein kinase Cdelta in the epidermis are resistant to skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1999;59(22):5710–8. [PubMed] [Google Scholar]
- 33.Lambert PF, Ozbun MA, Collins A, Holmgren S, Lee D, Nakahara T. Using an immortalized cell line to study the HPV life cycle in organotypic “raft” cultures. Methods Mol Med. 2005;119:141–55. doi: 10.1385/1-59259-982-6:141. [DOI] [PubMed] [Google Scholar]
- 34.Currier MA, Adams LC, Mahller YY, Cripe TP. Widespread intratumoral virus distribution with fractionated injection enables local control of large human rhabdomyosarcoma xenografts by oncolytic herpes simplex viruses. Cancer gene therapy. 2005;12(4):407–16. doi: 10.1038/sj.cgt.7700799. [DOI] [PubMed] [Google Scholar]
- 35.Dotto GP. Signal transduction pathways controlling the switch between keratinocyte growth and differentiation. Crit Rev Oral Biol Med. 1999;10(4):442–57. doi: 10.1177/10454411990100040201. [DOI] [PubMed] [Google Scholar]
- 36.Truong AB, Khavari PA. Control of keratinocyte proliferation and differentiation by p63. Cell Cycle. 2007;6(3):295–9. doi: 10.4161/cc.6.3.3753. [DOI] [PubMed] [Google Scholar]
- 37.Allen-Hoffmann BL, Schlosser SJ, Ivarie CA, Sattler CA, Meisner LF, O'Connor SL. Normal growth and differentiation in a spontaneously immortalized near-diploid human keratinocyte cell line, NIKS. The Journal of investigative dermatology. 2000;114(3):444–55. doi: 10.1046/j.1523-1747.2000.00869.x. [DOI] [PubMed] [Google Scholar]
- 38.Munger K, Baldwin A, Edwards KM, et al. Mechanisms of human papillomavirus-induced oncogenesis. J Virol. 2004;78(21):11451–60. doi: 10.1128/JVI.78.21.11451-11460.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gonzalez SL, Stremlau M, He X, Basile JR, Munger K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J Virol. 2001;75(16):7583–91. doi: 10.1128/JVI.75.16.7583-7591.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Helt AM, Galloway DA. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J Virol. 2001;75(15):6737–47. doi: 10.1128/JVI.75.15.6737-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eichten A, Rud DS, Grace M, Piboonniyom SO, Zacny V, Munger K. Molecular pathways executing the “trophic sentinel” response in HPV-16 E7-expressing normal human diploid fibroblasts upon growth factor deprivation. Virology. 2004;319(1):81–93. doi: 10.1016/j.virol.2003.11.008. [DOI] [PubMed] [Google Scholar]
- 42.Jones DL, Thompson DA, Munger K. Destabilization of the RB tumor suppressor protein and stabilization of p53 contribute to HPV type 16 E7-induced apoptosis. Virology. 1997;239(1):97–107. doi: 10.1006/viro.1997.8851. [DOI] [PubMed] [Google Scholar]
- 43.Huibregtse JM, Scheffner M, Howley PM. Cloning and expression of the cDNA for E6-AP: A protein that mediates the interaction of the human papillomavirus E6 oncoprotein with p53. Mol Cell Biol. 1993;13:775–84. doi: 10.1128/mcb.13.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scheffner M, Huibregtse JM, Vierstra RD, Howley PM. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquination of p53. Cell. 1993;75:495–505. doi: 10.1016/0092-8674(93)90384-3. [DOI] [PubMed] [Google Scholar]
- 45.Hahn WC, Weinberg RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2(5):331–41. doi: 10.1038/nrc795. [DOI] [PubMed] [Google Scholar]
- 46.Abba MC, Sun H, Hawkins KA, et al. Breast cancer molecular signatures as determined by SAGE: correlation with lymph node status. Mol Cancer Res. 2007;5(9):881–90. doi: 10.1158/1541-7786.MCR-07-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kondoh N, Wakatsuki T, Ryo A, et al. Identification and characterization of genes associated with human hepatocellular carcinogenesis. Cancer Res. 1999;59(19):4990–6. [PubMed] [Google Scholar]
- 48.Larramendy ML, Niini T, Elonen E, et al. Overexpression of translocation-associated fusion genes of FGFRI, MYC, NPMI, and DEK, but absence of the translocations in acute myeloid leukemia. A microarray analysis. Haematologica. 2002;87(6):569–77. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.