

Abstract
In vitro replicas of bone marrow can potentially provide a continuous source of blood cells for transplantation and serve as a laboratory model to examine human immune system dysfunctions and drug toxicology. Here we report the development of an in vitro artificial bone marrow based on a 3D scaffold with inverted colloidal crystal (ICC) geometry mimicking the structural topology of actual bone marrow matrix. To facilitate adhesion of cells, scaffolds were coated with a layer of transparent nanocomposite. After seeding with hematopoietic stem cells (HSCs), ICC scaffolds were capable of supporting expansion of CD34+ HSCs with B-lymphocyte differentiation. Three-dimensional organization was shown to be critical for production of B cells and antigen specific-antibodies. Functionality of bone marrow constructs was confirmed by implantation of matrices containing human CD34+ cells onto the backs of severe combined immunodeficiency (SCID) mice with subsequent generation of human immune cells.
Keywords: Scaffolds, Tissue engineering, Bone marrow, Colloidal crystals, Stem cell
1. Introduction
In vitro production of human hematopoietic cells is imperative for the development of immune therapeutics such as human monoclonal antibodies for the treatment of many clinical disorders including a number of cancers. Artificial analogs of bone marrow can also greatly accelerate the testing of a variety of types of drugs. They can also significantly contribute to the understanding of hematopoiesis in humans because most information on HSC self-renewal and lineage commitment has been based primarily on murine studies. Although rodent studies have provided important fundamental insights into hematopoietic development, there is much that can be learned regarding human hematopoiesis through the use of in vitro human hematopoietic systems that are unavailable in non-human models.
Construction of such ex vivo analogs is quite challenging due to the complexity of the human bone marrow microenvironment [1]. Here, stromal cells play a critical role in both presenting membrane bound ligands and secreting soluble signaling molecules. HSC differentiation is strongly dependent on cellular interactions with stromal tissue and appropriate levels of signal proteins [1-10]. Although the native bone marrow environment can be recreated on 2D culture to some extent by the addition of proper cytokines and growth factors to the cell cultures and by the presence of co-cultured stromal or feeder cells [2-5], it is inefficient for the purpose of replicating hematopoiesis to produce functional leukocytes. Utilizing 3D cell cultures that induce more intensive cell-to-cell contacts between HSCs and stromal cells appears promising to produce the appropriate developmental niches for stem cells. For instance, generation of human T-lymphocytes from human CD34+ cells in vitro was made possible due to 3D organization of the cell scaffold [12]. The 3D scaffold microenvironment has been utilized to grow or guide the differentiation of different stem cell types. Among them include mesenchymal stem cells [13-17] hematopoietic stem cells [18] embryonic stem cells [19, 20].
One of the difficulties in the development of a bone marrow analog is the availability of a suitable 3D matrix, which must possess sufficiently large surface area for cell attachment, high porosity for cell migration and transport of nutrients, substantial transparency for inspection of constructs with optical techniques, and variability in scaffold structure to control cell-to-cell contacts. Even though various types of 3D matrices have been introduced for this purpose [21-31], their approaches remain largely empirical rather than systematic due to limited controllability and reproducibility of microscale structures. For that reason, there has been little attention to the design of pore geometry that can effectively mimic the function of bone marrow microenvironment.
Our approach for the replication of bone marrow started with de novo scaffold design and created a new type of versatile 3D matrix that clearly exhibited marked advantage of 3D organization for hematopoiesis and resulted in acquisition of structural requirements critical for a successful bone marrow analog. It was demonstrated that the new matrix seeded with feeder cells is capable of supporting substantial expansion of CD34+ HSCs and B-lymphocyte differentiation with production of functional B cells. The latter was substantiated by expression of key signal proteins, class switch of cells from immunoglobulin M (IgM) to IgG, secretion of influenza A specific antibody, and production of human immune cells in SCID mice after subcutaneous implantation of the designed scaffolds. These findings allow one to establish a fundamentally new model system for bone marrow suitable for both drug testing and ex vivo simulation of some aspects of human immune response.
2. Materials and methods
2.1. ICC scaffolds fabrication
Colloidal crystals (CCs) were prepared following the previously reported method [32]. Briefly, a plastic mold (D=6.5mm) was connected to a long Pasteur glass pipette and glued on a glass beaker that was partially immersed in the ultrasonic water bath. Two to three drops of polystyrene beads suspension (D=100μm, Duke Scientific) was released through the Pasture pipette in 20min time intervals under gentle agitation generated by the ultrasonic water bath until the thickness approximately reaching 0.5-1mm. After completely dried at 60°C for overnight, the CCs were annealed at 120°C for 4 hours. Final dimensions of free-standing CCs were 6.5mm diameter and 0.5-1mm thickness, respectively.
Sodium silicate solution (14% NaOH, 27% SiO2) (Sigma) was diluted 1:1 ratio with deionized water, and then infiltrated into the colloidal crystals by centrifugation at 5800 rpm for 30min. After that, the composite colloidal crystals were dried overnight in the air to let the sol become a gel. Final silicate ICC scaffolds were obtained after removing the colloidal crystal template either burning out at 600°C for 30min or dissolving by tetrahydrofuran (THF) [33]. Hydrogel precursor solution composed of 30% (w/w) acrylamide monomer, 5% (w/w) bis-acrylamide cross-linker, and 0.1% (v/v) N,N,N,N-tetramethylethylenediamine accelerator was prepared with nitrogen gas purged deionized water. The precursor solution was infiltrated into the colloidal crystal by centrifugation at 5800 rpm for 10min, and 1% (w/w) potassium peroxide initiator solution was added. After complete polymerization within 2-3 hours at room temperature, the colloidal crystal containing hydrogel part was cut out and immersed in THF for 24 hours to remove the template. Finally the hydrogel ICC scaffolds were washed and equilibrated in deionized water. All chemicals were purchased from Sigma
2.2. Layer-by-layer surface modification
The surface of polyacrylamide hydrogel ICC scaffolds was coated with sequential layers of negatively charged 0.5% (w/w) clay platelets (average dimension of 1nm thickness and 70-150nm in diameter, Southern Clay Products) dispersion and positively charged 0.5% (w/w) poly (diallyldimethylammonium chloride) (PDDA, MW=200,000) (Sigma) solution utilizing a layer-by-layer (LBL) surface coating technique. The LBL coating was started with a PDDA layer. At first, hydrogel ICC scaffolds were immersed in PDDA solution for 15min and rinsed in deionized water for 30min. Then the scaffolds were immersed in clay solution for 15min and rinsed with deionized water for 30min. This LBL coating cycle was repeated five times. The outermost layer was clay nanoparticles. To demonstrate 3D LBL coating on hydrogel ICC scaffolds, FITC conjugated albumin (Sigma) which has negative charge was used instead of clay nanoparticles. A hydrogel ICC scaffold coated with ten bilayers of FITC-albumin and clay nanoparticles was imaged using the confocal microscope with 10x objective. Confocal series images taking 160μm depth were three dimensionally reconstructed using imaging software.
2.3. Bone marrow stromal cell culture
Human bone marrow stromal cell line HS-5 (CRL-11882) and human fetal osteoblast (hFOB) 1.19 cell line (CRL-11372) were purchased from ATCC (Manassas, VA). Culture media for HS-5 was composed of Dulbecco’s modified Eagle’s medium (DMEM) with 4mM L-glutamine, 4.5g/L glucose, 1.5g/L sodium bicarbonate, 10% (v/v) fetal bovine serum (FBS), and 1% (v/v) penicillin-streptomycin (all reagents purchased from ATCC). The hFOB cells were cultured with 45% Ham’s F12 medium, 45% DMEM, 10% FBS and 1% antibiotic. The cultures were maintained in a T-25 culture flask at 37°C with 5% CO2 in high humidity condition. Once cell growth reached approximately 80% confluence, they were detached from the culture flask using 0.25% (v/v) trypsin-EDTA solution and 105 cells were seeded on top of UV sterilized scaffolds which were placed in a 96 well-plate. Culture volume was maintained 250μL and media was changed every other day.
In order to characterize stromal cell growth in 3D scaffolds, HS-5 cells were stained with 5μM long term cell tracing fluorescent dye, carboxyfluorescein diacetate succinimidyl ester (CFSE) (Molecular Probes). 105 pre-stained HS-5 cells were seeded on silicate scaffolds and cultured for 5 days. The cells growing within silicate scaffolds were imaged under a Leica SP2 confocal microscope with 20x objective. A 488nm laser was used for excitation and emission signals were collected within 515-550nm window. Transmission image was overlaid with fluorescent image to designate the location of cells within pores.
2.4. Human CD34+ HSCs isolation
Peripheral blood “buffy coats” were obtained from Gulf Coast Blood Bank or from donors (18-50 years of age) after informed consent under protocols that were reviewed by the Institutional Review Board of The University of Texas Medical Branch. Cord blood and bone marrow were purchased from Lonza Inc. (Allendale, NJ). The mononuclear leukocyte (MNL) fraction was isolated from these three CD34+ HSC sources using Ficoll density gradient separation medium as described by the manufacturer (Amersham-Biosciences, Piscataway, NJ). CD34+ HSCs were enriched by counter current centrifugal elutriation of MNLs in a Beckmann J6M elutriator (Beckman Instruments, USA) using a Sanderson chamber. A Masterplex peristaltic pump (Cole Parmer Instruments) was used to provide the counter current flow. RPMI 1640 supplemented with 2 mM glutamine, 100 units penicillin G and 100 μg/mL streptomycin and 10% heat inactivated defined fetal calf serum (Hyclone, Logan, UT) was used as elutriation medium. 3-6 × 106 cells were loaded at 3000 rpm and HSCs were isolated using a step-wise reduction of rotor speed until the appropriate cell diameter population, 6-7 μm, of CD34+ enriched cells was collected.
Immunophenotypic analysis of elutriation with 5-7 μm diameter HSCs at the time of isolation and seeding of the scaffold showed that these cells were lin-1 mature leukocyte marker negative, CD34+ cells were further purified by negative selection of any remaining lin-1 positive or mature cell types using Dynall magnetic beads or by flow cytometric cell sorting using a FACSaria cell sorter.
2.5. Human CD34+ HSCs expansion
CD34+ cells were labeled with the CFSE dye as previously described [34]. Scaffolds were then seeded with 1-4 × 104 CFSE labeled CD34+ HSCs in 50 μL of DMEM containing 5 % pluronic F-127 on the bottom of a 24 well tissue culture plate. A total of 3-5 scaffolds were placed in a standard 2-10 mL plastic bioreactor chambers mounted on a rotary bioreactor (Synthecon, Houston, TX) with a rotational speed of 5 rpm in the same media. In a subset of experiments matrices were seeded with stromal support cells from human bone marrow aspirates and non-autologous HSCs were added after 2 days of stromal culture. HSC/stromal cell cultures were also incubated in plastic Petri dishes in order to allow us to evaluate 2D versus 3D cell culture.
2.6. Inducing B Cell differentiation
To induce HSC differentiation, a co-culture of HS-5 and hFOB1.19 cell line was initially used. Although differentiation was successful these cell lines quickly overgrew the matrix without irradiation and thus were somewhat inconvenient. Later, we created primary cell lines from bone marrow aspirates which included cells positive for CD105 (100%), CD166 (100%), CD44 (95%), CD14 (1%), CD34 (1%) and CD45 (<1%) suggesting they were part of the stromal cell population; at least one cell type in the support cell mix was of osteoblast lineage as it was positive for osteonectin. These primary support cells formed densely populated layers similar to natural bone marrow and replicated the actual bone marrow environment better than the feeder layer made from a single cell type. Growth factors used to promote hematopoiesis included: interleukin (IL)-2 (5 ng/mL, Calbiochem), IL-7 (20 ng/mL, R&D Systems), Flt3 ligand (20 ng/mL, Chemicon), stem cell factor-1 (SDF-1 20 ng/mL, Calbiochem), BMP-4 (4ng/mL, R&D Systems), and IL-3 (10 ng/mL, Chemicon). Additives used to promote development of a B lymphocyte lineage included: soluble CD40L (5 ng/mL, Invitrogen), IL-4 (10 ng/mL, Calbiochem), IL-5 (10 ng/mL, Chemicon), IL-6 (10 ng/mL, Calbiochem), IL-10 (10 ng/mL, Chemicon), IL-2 (5ng/mL, Calbiochem), IL-7 (20 ng/mL), Flt3 ligand (20ng/mL), stem cell derived factor (20 ng/mL, Chemicon), IL-3 (10 ng/mL) and agonist anti-CD40 mAb (5 μg/mL, clone HM40-3; BD Biosciences).
Cultures were exposed to 3.5 μg/mL lipopolysaccharide (LPS) (Sigma-Aldrich) to activate B lymphocytes and induce plasma cell formation. Secreted IgM was quantitated for all B cell cultures using a total human IgM ELISA assay (Diagnostic Automaton, Inc.) as described by the manufacturer. Supernatant fluids were removed from 3D and 2D cultures and levels of secreted IgM were determined. All samples were run in triplicate and averaged optical densities (OD) at 450 nm were compared to a standard curve using optical densities obtained for each of the standards.
2.7. Stimulating mature B Cells in vitro
To expand B cell numbers, cells were stimulated by IgM crosslinking using 5 μg of anti-IgM antibody prior to antigen specific stimulation of cultures. Prior to antigen exposure, 2 × 106 paraformaldehyde fixed cord blood donor matched CD4+ T cells were added to the bioreactor chamber to facilitate the B cell maturation. For antigen specific exposures, heat killed whole Influenza strain A New Caledonia/99/ H1N1 virus was grown in allantoic cavities of 10-day old embryonated hen’s eggs as previously described [35]. The allantoic fluid was pooled after collection and frozen at -70°C until titered to 107 or 108 when assayed in Madin-Darby Canine Kidney (MDCK) cells (ATCC). Heat killed virus was prepared by incubating pooled virus at 56°C for one hour with constant agitation. Virus titers were checked from heat killed stocks and were always 0. For sham-exposures, allantoic fluid from uninfected eggs was collected, pooled and frozen at -70°C until used.
Bioreactor chamber supernatants were tested for neutralizing antibody production against influenza virus using a micro-neutralization assay [36] using infectious influenza A/New Caledonia/99 virus. Each sample was tested in at least two replicate experiments beginning at a dilution of 1:2, with the mean titer of replicate assays reported. The hemaglutinin inhibition (HAI) test was done to evaluate the presence of anti-influenza A antibody in the culture supernatants. The ELISA for nucleoprotein (NP) (Immgenex) was done following the manufacturer’s directions. The ELISA for anti-hemagglutinin (HA) antibodies was done as follows. In brief, polystyrene 96-well ELISA plates were coated with 100 ng/well of A/New Caledonia HA prepared as previously described [37]. Supernatants were tested in triplicate, and the OD value reported after detection using an alkaline phosphatase-conjugated goat anti-human IgG antibody (BioSource International, Camarillo, CA). For the anti-HA ELISA, culture supernatants were added to wells in serial two-fold dilutions beginning at 1:2. A positive control sample consisting of a pool of four post-vaccination sera with a neutralizing titer of 1:100 was run on each plate as a positive control sample. Results were reported as units of ELISA activity, and although a four-fold or greater increase in units is considered evidence of a significant response to vaccination in vivo we determined a positive response for these in vivo cultures to be at least a consistent two-fold increase in at least three of the assays used. A serum sample was considered positive for anti-HA IgG antibody if a titer of 1:8 was obtained in at least two independent assays. Cultures were considered to have made a response to the antigen if one or more samples met the criteria above and demonstrated a four-fold or greater rise in antibodies from the starting pre-immunized cultures which always were 0 for all assays.
2.8. In vivo experiment
For SCID mouse implantations of ICC matrix constructs HSCs and stromal cells were seeded onto matrices as described above and cultured in differentiation media for three days. A single scaffold of approximately 0.5 cm in diameter was implanted on the back of each of eight male SCID mice and was allowed to remain for two weeks. The bone marrow construct, peripheral blood and spleen cells were collected and were analyzed for the cell phenotypes produced. 0.5 × 106 cells were stained as described by the manufacturer, BD Biosciences (San Jose, CA), for expression of human HSC markers CD34, CD150, CD1a, CD7, CD10, CD117, CD90, CD133 and CD110 and mature leukocyte lineage markers HLA-class-I, CD45, CD3, CD19, CD16+56, CD14, IgM and IgD. 7 μm frozen sections of the ICC matrices were stained for expression of CD34, CD150, CD133 as well as human HLA-class I.
3. Results and discussion
3.1. ICC Scaffolds
The basic structure put in the foundation of scaffold design utilized cell growth matrixes with ICC geometry, which were recently introduced [33, 38]. Primary colloidal crystals are hexagonally packed lattices of spheres (Fig. 1A), with a wide range of diameters from nanometers to micrometers. ICCs are similarly organized structures where the spheres are replaced with cavities, while the interstitial spaces are filled (Fig. 1B-E). When ICC cavities exceed the diameter of cells, they can be used as 3D cell scaffolds. The open geometry of the ICC lattice, high porosity (74% free space), full interconnectivity, and large surface area make ICC an attractive structure for studies of 3D effects in cell cultures. It is also apparent that the ICC structure is geometrically similar to the 3D morphology of supporting bone marrow tissue in a trabecular bone, which is important for creation of the microenvironmental niches that maintain stem cell survival and promote maturation [39-41]. In addition, ICC topology is convenient because it affords a simple method of control over cellular interactions and migration by varying the sphere diameter. Lastly, dynamic culture within a rotary cell culture vessel was beneficial not only by creating more realistic physiological environment but also by maximizing the role of specifically designed ICC geometry by generating continuous convective media flow into ICC pores (Fig. 1E).
Fig. 1. Cellular scaffolds with ICC topography.

Scanning electron microscopy image of (A) colloidal crystals made from 110 μm polystyrene beads, and (B) ICC scaffolds from silicate gel. (C) ICC scaffolds after 5 days in culture with HS-5 bone marrow stromal cells. (D) 3D reconstructed poly (acrylamide) hydrogel ICC scaffold after 5 bilayers of FITC-albumin and PDDA coating. The diameter of the spherical cavities is 110 μm, interconnecting pores is ca. 15-25μm. (E) 3D schematics of ICC topology and cell contacts within. Floating HSCs enter into a pore through interconnected channels that have diameters 2-3 times larger than that of a single cell. Temporarily entrapped HSCs undergo intense contacts with stroma cells residing on the pore surface.
Manufacturing of the ICC scaffolds was described elsewhere [32,33, 38]. Throughout this study poly(styrene) beads with a diameter of 110 μm were used, producing the cavities of the same size connected by 10-20 μm channels (Fig. 1B-D). As the beads provide a template for the scaffold cavities, the diameter of the beads is directly proportional to the diameter of the resulting cavities. Similarly, the size of channels connecting the cavities can be tuned by the bead diameter, as the degree of contact upon annealing is greater for larger beads, and also by annealing temperature and time [42]. This bead diameter was chosen because it provides efficient contacts between adhesion and dispersion cells [43], and allows for natural cell migration through the channels between the cavities, which is imperative for replication of hematopoietic tissues (Fig. 1C). The materials of a typical ICC scaffolds used here can be either silicate or poly(acrylamide) hydrogel, both of which were demonstrated to be compatible with human cell cultures [32,33, 38]. To provide adequate adhesion of bone marrow support cells, hydrogel matrixes were coated by clay and poly(diallyldimethyl ammonium chloride) multilayer following the layer-by-layer technology [44], which results in a thin layer of nanocomposite on the walls of the scaffold [32] (Fig. 1D). Such coatings facilitate cell adhesion, which is typically quite poor on native unmodified hydrogels [45]. Moreover, the nanoscale nature of the coating is essential for several reasons. (i) The hybrid organic-inorganic composite is mechanically compatible with the hydrogel and does not delaminate. (ii) It provides high Young’s modulus [44] necessary for successful stromal cell adhesion [46,47]. (iii) Nanocomposites have minimal light scattering because the characteristic diameter of the inorganic component is smaller than the wavelength of light, which is quite relevant for optical interrogation of biological processes. These features markedly differentiate ICC hydrogel scaffolds from those previously used for bone marrow cultures. In fact, the nanocomposite bilayers add another layer of tunability to the construct related to cell adhesion in that the surface stiffness of the scaffold can be adjusted with the number of bilayers coated on its surface. For example, freestanding films of the same materials were shown to have increased Young’s modulus with increasing numbers of bilayers. [44] The ability to tune mechanical properties and increase surface stiffness by addition of nanocomposite bilayers is important because substrate mechanics have been shown to influence cell adhesion [48-50]. Additionally, the bilayers provide a surface charge necessary for cell adhesion. Lastly, multilayer films have also been shown to guide cell differentiation [51, 52]. In the framework of this study, no difference in silicate and hydrogel ICC scaffold performance in terms of bone marrow functionality was observed, although hydrogel scaffolds are preferred due to much higher transparency and ease of confocal microscopy examination.
3.2. Bone marrow construct
Self-renewal and maintainance of an undifferentiated population of CD34+ HSCs and production of fully functional immune cells of specific leukocyte lineages are the two basic functions of bone marrow most essential for applications mentioned above. Bone marrow stroma is comprised of a complex reticulum containing hematopoietic precursors, as well as non-hematopoietic cells such as fibroblasts, epithelial cells, nerve cells, reticular cells, adipocytes and osteoid cells [2-5, 9, 53]. It is unknown how many or all of these cell types may be necessary to support the development of fully functional leukocytes.
To mimic bone marrow stroma tissue function, human bone marrow stromal cells were seeded on scaffolds and cultured for 3 days to allow the formation of a support cell layer on the scaffold surface prior to the addition of CD34+ HSCs. Bone marrow stromal cells were stained for expression of the cell marker CD105 (green) to allow for visualization of the stromal cell network that was formed by day 3 (Fig. 2A). On day 3 CD34+ were then seeded onto stromal cell-ICC constructs (Fig. 2B). CD34+ HSCs were chosen because they have been shown to provide long term multi-lineage engraftment capability [54, 55]. CD34+ HSCs were isolated from human peripheral blood (PB), umbilical cord blood (CB) or bone marrow (BM). All cells were positive for CD34 and were lineage-1 (lin-1) negative when seeded onto the scaffolds. A small portion (1-2%) of CD34-expressing cells was positive for CD150, a cell marker also associated with long term multi-cell lineage reconstitution in irradiated mice [8]. Analogous cultures were also made on 2D plates to establish the importance of the 3D geometry in ICC scaffolds. Examination of ICC cultures on day 28 showed the continued presence of CD34+ HSCs (red) (Fig. 2C); CD34+ antibody-labeled cells (red) are shown interacting within the scaffold with CD105 positive stromal cells (green). Cells in the ICC cultures formed numerous actin-rich cell processes (Fig. 2D), which were absent in cell cultures on flat substrates. In these sections CD34 positive cells (green) were stained for actin using rhodamine phalloidin (red). Similarly, maintenance of a population of CD150+ cells (Fig. 2E) was seen in ICC matrices but not in donor matched 2D cultures after 28 days. Data from flow cytometry show that there were significantly higher percentages of CD34+ cells in ICC cultures after 28 days, regardless of the original cell source, when compared to 2D plate cultures (Fig. 2F-K, L). Figs. 2F, 2H, and 2J correspond to 3D ICC cultures, while 2G, 2I, and 2K correspond to 2D plate cultures. Cells in cultures 2F and 2G, 2H and 2I, and 2J and 2K were isolated from bone marrow, cord blood, and peripheral blood, respectively. One can see that a significantly greater percentage of cells in ICC scaffolds expressed CD34 (red) over the isotype control (green line). These data are summarized in Figure 2L. This proves that an undifferentiated population of CD34+ cells was maintained over time and demonstrates the importance of the chosen 3D ICC organization of the cell cultures for replication of reproductive functionality of bone marrow. Higher numbers of of mitotic figures were seen in 3D cultures than donor matched 2D cultures after 28days (Fig. 2M). This indicated that there was active proliferation of CD34+ HSC. This is also supported by the loss of CFSE fluorescence intensity (Fig. 2N-O). In Fig. 2N, a large population of cells grown in 3D ICC cultures (85.6%) displayed decreased intensity of CFSE fluorescence after 7 days (green); the decrease in intensity was much less (14.6%) for 2D cultures, indicating less reproduction in 2D. This is summarized for different HSC from different cell sources in Fig 2O. ICC scaffolds demonstrate substantially more CD34+ proliferation than plate cultures.
Fig. 2. Examination of bone marrow constructs.
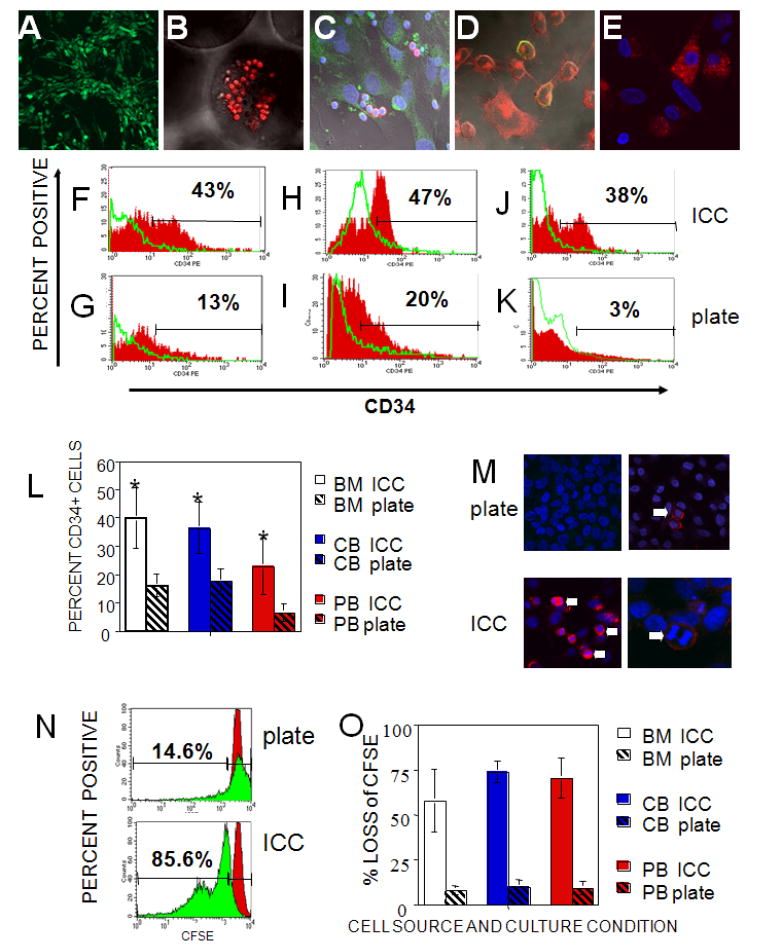
(A-E) Confocal microscopy images of 7 μm frozen sections of hydrogel ICC scaffolds. (A) Stromal cells cultured for 3 days were stained with a CD105 stromal cell marker for visualization of the developing stromal cell network (green), 200X. (B) CD34+ HSCs (red) were seeded onto the ICC scaffold and imaged after 1 day of stromal cell culture, 400X. (C) Examination of the ICC scaffold cultures on day 28 showing stromal cells CD105 (green) and CD34+ HSC (red), 630X. All cell nuclei were stained with DAPI (blue). (D) Sections of 28 day ICC/HSCs cultures stained for actin (red) and CD34 (green), 630X. (E) Same cultures stained for CD150 (red). DAPI nuclear stain (blue) was used. Flow cytometry data for donor matched 3D ICC scaffold (upper row) and 2D plate (lower row) cultures for cells isolated from (F,G) bone marrow, (H,I) cord blood, and (J,K) peripheral blood. 10,000 cytometry events were collected for all samples. Solid red histograms show CD34 levels with isotype controls for each sample shown as the green line histogram overlay. (L) Comparative evaluation of CD34+ cells on day 28. Significantly more CD34+ cells were seen in ICC cultures for bone marrow (BM) (P=0.01), cord blood (CB) (P=0.004), or peripheral blood (PB) (P=0.03) than for donor matched 2D plate culture in a total of 6 experiments. (M) Confocal images of 28 day 2D plate culture (M, top left and right, 400x) and 3D ICC cultured cells (M, bottom, 400x left, 630x right) stromal cell peripheral blood derived CD34+ cell seeded cultures. Plate and ICC cultures were stained for cell surface expression of CD34 (red) and DAPI (blue) nuclear stain. There are few mitotic figures seen in CD34+ cells in plate (top micrographs) compared to the ICC (bottom micrographs) cultures. Numerous mitotic figures indicate proliferation of CD34+ cells (white arrows) were always seen in 28 day ICC but not in plate cultures. (N) Flow cytometric analysis of CFSE levels in cord blood derived CD34+ HSCs for ICC scaffold (N, bottom) and donor matched 2D plate cultures (N, top). Solid red histograms show CFSE level of CD 34+ cells on the same day as ICC scaffolds were seeded. Solid green histograms show CFSE levels on day 7 of culture. 10,000 cytometry events were collected for all samples. (O) Comparison of 2D versus 3D cell cultures in ICC scaffolds by HSC proliferation analysis using CFSE loss for CD34+-derived from bone marrow (BM), cord blood (CB) or peripheral blood (PB) (averages for 5 experiments).
3.3. Stem cell differentiation
Bone marrow is also the site of long term antibody production after viral infection [53] and bone marrow stroma has been shown to play a role in plasma cell life cycle [40]. Similar to the maintenance of HSCs populations, the production of B cells is an essential component for the development of ex vivo bone marrow, immune system studies, the development of human monoclonal antibodies, drug evaluation, and disease treatment. B cell development involves a series of stages where close 3D contact between bone marrow stroma and the developing B cell is critical and hard to realize in 2D plate cultures.
To assess the ability of the artificial bone marrow constructs to produce functional immune cells, we focused on B-lymphocyte production since B cells normally undergo the process of differentiation (as well as negative and positive selection) in the bone marrow [4, 5, 54, 56]. After 3 days of culture, ICC/stromal cell constructs containing growth factors to drive the B cell differentiation were seeded with CD34+ HSCs isolated from unbilical cord blood. Cell cultures were examined for stage-specific markers of development and functionality on days 1, 7, 14, 28 and 40. ICC cultures showed nuclear specific expression of recombination activating gene-1 protein (RAG-1) by day 7 (Fig. 3A). Recombination activating gene-1(RAG-1) is a nuclear protein involved in lymphocyte development which is expressed by immature B- and T-lymphocytes. By day 14 there was measurable RAG-1 expression (red) by IgM+ cells (green). Also seen by day 14 (Fig. 3B) was cell surface expression of IgM (green) and CD19 (red), indicating differentiation of the CD34+ HSC into B-cells. Co-expression of IgM (green) with IgD (red) was seen by day 28 (Fig. 3C) confirming differentiation of CD34+ into mature antigen-naive B lymphocytes. Figs D and E show corresponding quantitative flow cytometry histograms from one representative experiment to evaluate expression of IgM and IgD on cells from ICC scaffolds after 28 days. Averaged data for cells from six different donors evaluating expression of CD40, IgD, IgM as well as coexpression of IgD and IgM is displayed in Fig 3F and shows the comparison between 3D and 2D cultures. Expression of CD40 (P=0.0002) and IgM/IgD co-expression (P= 0.021) was significantly higher in donor matched ICC cultures than in 2D cultures (Fig. 3F).
Fig. 3. Results of B cell differentiation.
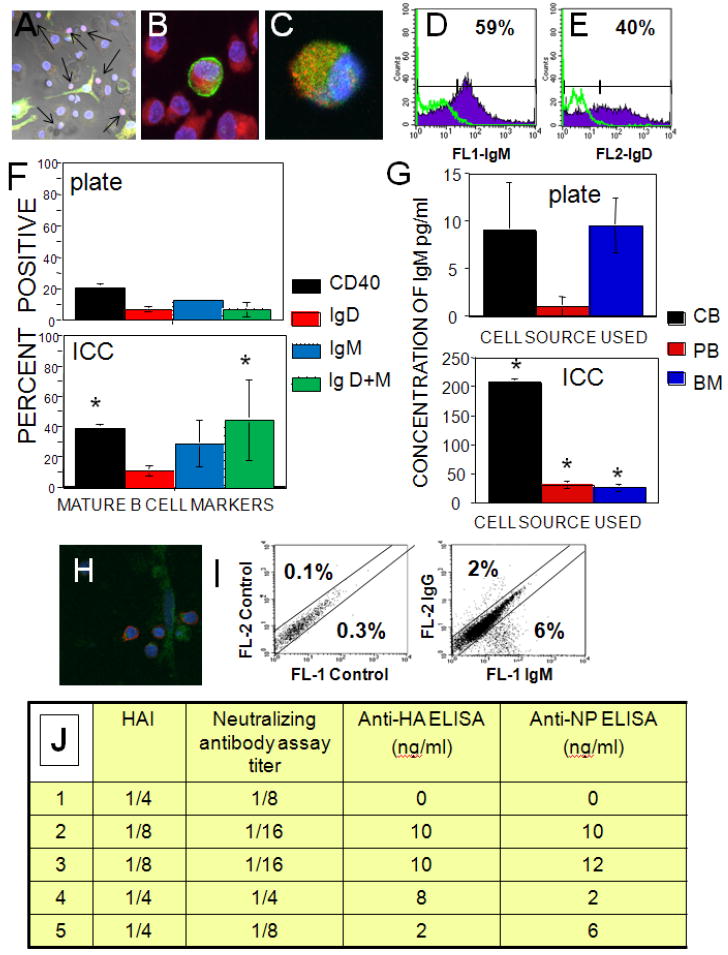
(A-C) Confocal microscopy images of 7μm sections of hydrogel scaffolds supporting CD34+ HSCs from cord blood. DAPI nuclear stain is blue for all images. (A) Nuclear RAG-1 (red) expression and surface expression of IgM (green), day 7, 200X. Arrows point to RAG-1 positive nuclei. (B) Cell surface co-expression of CD19 (red) and IgM (green), day 14, 630X. (C) Co-expression of cell surface IgM (green) and IgD (red) day 28, 630X. (D-E) Flow cytometric evaluation of cell surface expression of immunoglobulin M or D from one representative experiment for (D) IgM and (E) IgD. 10,000 events were collected for all samples, isotype controls shown as green overlays. (F) The average expression of CD40, IgM, IgD and IgM+IgD co-expression for plate and ICC cultures using CD34+ from cord blood (6 independent experiments). (G) Comparison of IgM production for LPS stimulated plate and ICC cultures from cord blood (CB), peripheral blood (PB) or bone marrow (BM) CD34+ HSCs. (H) Confocal micrograph of 7 μm section of ICC culture stained for expression of IgG (red) and CD105 (stromal cell marker, green). (I) Flow cytometry data for IgG versus IgM expression from one representative experiment showing class switch for cord blood CD34+ HSC cells after exposure to heat killed-influenza virus. 10,000 events each, isotype control data are on the left plot. (J) Evaluation of specific influenza antibody production by HAI assay, neutralizing antibody assay titer, anti-HA IgG antibody ELISA, and anti-nuclear protein (NP) ELISA (5 experiments each). Results are arranged according to the lowest dilution of culture supernatant exhibiting inhibition in case of HAI and neutralizing antibody or ng/ml concentrations of corresponding proteins in case of anti-HA and NP ELISAs.
These results show the expression of phenotypic cell surface markers of B-cells, which is an important step in the development of bone marrow replicas; however, this fact does not necessarily prove the functionality of the ex vivo generated B lymphocytes. To evaluate the ability of these B lymphocytes to respond to mitogenic or antigenic stimulation and fully mature into antibody producing cells, B lymphocytes isolated from 28 day ICC scaffold constructs and donor matched plate cultures were exposed to bacterial LPS, a major structural component of the outer wall of gram-negative bacteria and initiator of immune response to bacterial infection. Secreted IgM was quantified using a human IgM ELISA for all B cell cultures differentiated from CB-derived, PB-derived and BM-derived CD34+ cells. Significantly higher levels of IgM were produced from B lymphocytes generated in the ICC scaffold regardless of the initial source of the CD34+ cells (Fig. 3G, notice difference in scales).
In a subset of experiments artificial bone marrow constructs were prepared as described above and were seeded with human cord blood derived CD34+ HSCs. Cultures were primed to proliferate using anti-IgM cross-linking of B cells in the bioreactor chamber and within the construct on day 14 of culture. On day 14 paraformaldehyde-fixed donor matched CD4+ T cells were added to the bioreactor chamber and cultures were then exposed to heat-killed whole influenza A virus with multiplicity of infection (MOI) of 10 on days 28-30 of culture. Cultures yielded mature IgG expressing cells after 40 days of culture as analyzed by confocal microscopy (Fig. 3H) or flow cytometry (Fig. 3I) with an average production of 13.5 ± 9.4% IgM expressing (with no expression of IgD) and 3.1 ± 1.9 % IgG expressing cells, while isotype controls for all experiments were ≤ 0.2%. Examination of influenza A antigen specific antibody production by hemagglutinin inhibition (HAI) assay, neutralizing antibody assay titer, anti-HA IgG antibody ELISA, and anti-nuclear protein (NP) ELISA showed consistent production of specific antibody in all ICC scaffold cultures but never in donor matched 2D cultures receiving the same treatments (Fig. 3J).
ICC scaffold/stromal cell constructs seeded with CFSE-labeled cord blood derived CD34+ HSCs were cultured in vitro for 3 days and then implanted on the backs of eight SCID mice to test their in vivo functionality. The animals were sacrificed after 2 weeks and then the implanted bone marrow construct (Fig. 4A), peripheral blood and spleens of animals were collected for leukocyte subset phenotyping. The regions near the construct were highly vascularized (Fig. 4A) although few red blood cells were seen in the construct itself. Flow cytometric evaluation of cells obtained from the construct, peripheral blood or spleens and confocal evaluation of the bone marrow construct itself showed that the majority of cells in the ICC construct were human major histocompatibility complex (MHC) class I+ (Fig. 4B, H) and subsets of HSCs including CD34+ (Fig. 4C, H), CD150+ (Fig. 4D, H) and CD133+ (Fig. 4E, H) were maintained. CD133+ and CD150+ are HSCs cell types that have been shown along with CD34+ cells to function in the repopulation of leukocytes [57,58] in vivo. High levels of CD19+ B lymphocytes (Fig. 4F, H) and IgM expressing cells (Fig. 4G, H) as well as B cell precursors (Fig. 4H) were also seen. Examination of the cells populating in the spleen and peripheral blood showed that the majority of leukocytes were of human origin as indicated by MHC class I staining (Fig. 4H) and that the predominant cell type found was CD19+ positive (Fig. 4H). In the spleens of mice receiving implants most of the CD19+ cells were shown to co-express IgM and IgD (Fig. 4H). Median engraftment time for bone marrow reconstitution by transplant of bone marrow or cord blood is typically 18-26 days. One can suggest that engraftment of HSCs in the artificial bone marrow construct with continued production of CD34+ cells is similar to that seen in human or murine reconstitution of irradiated marrow [54, 55]. The level of cells was surprisingly high after engraftment of the bone marrow surrogate possibly because it is a better system overall than some used before and supports not only production of mature cells types but production of immature precursor cells at levels closer to that found in normal bone marrow. As seen before, there were high levels of CD34+ HSC replication, which could be maintained for an extended period of time.
Fig. 4. Animal testing of ICC scaffolds.
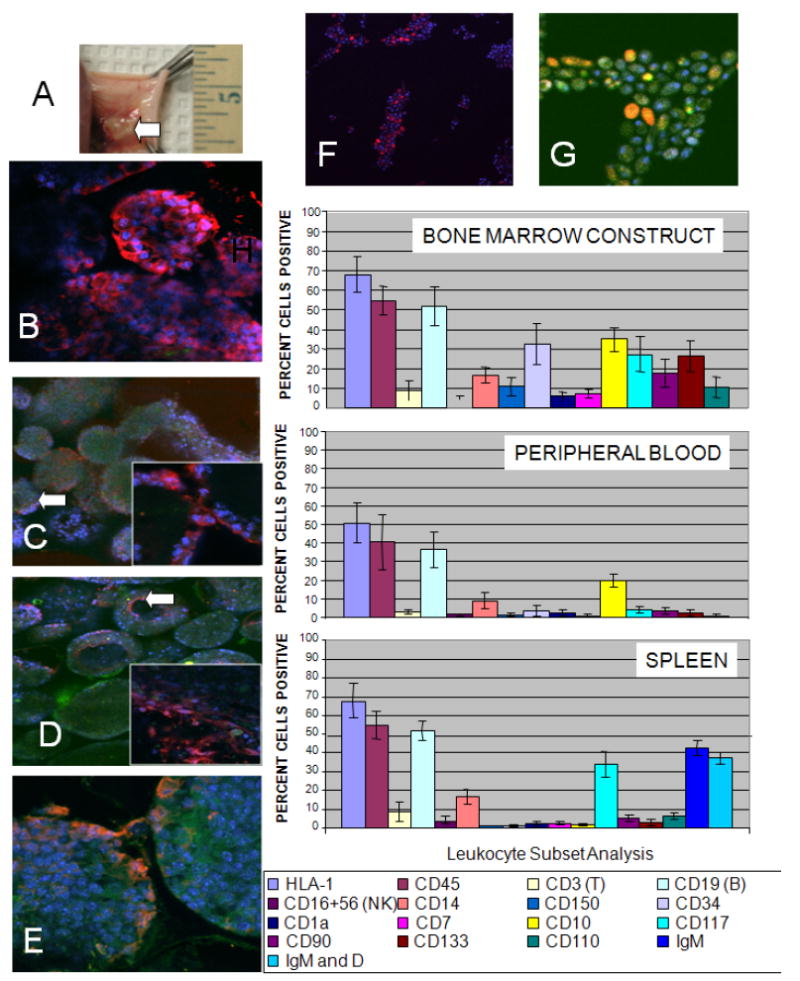
Evaluation of bone marrow construct, bone marrow derived cells, peripheral blood and spleen cells after 2 weeks implantation of hydrogel ICC scaffolds on the backs of eight SCID mice. The scaffolds were seeded with CFSE labeled cord blood derived CD34+ HSCs and cultured for 3 days before implantation. (A) A high degree of vascularization is seen in the regions near the site of the implanted ICC construct. (B-G) Confocal microscopy images of 7 μm frozen sections of the ICC-bone marrow construct. DAPI (blue) and CFSE (green). (B) Human MHC-Class I (red), 400X (green channel was not overlaid for clarity). HSC markers: (C) CD34 (red), insert 630X (D) CD150 (red), insert, 630X and (E) CD133 (red), (F) CD19 (red) (green channel not overlaid for clarity) and (G) IgM expression (red), magnification 630X. (H) Flow cytometry evaluation of cell phenotypes found in the bone marrow construct, peripheral blood and spleens of SCID mice receiving constructs. For all flow cytometry data 10,000 events were collected for each sample. Isotype control staining was less than 2% cells positive for all antibodies used.
4. Conclusions
In conclusion we want to make two points that appear quite important from the fundamental aspects of ex vivo organ regeneration and their practical applications. The data show that (i) proper organization of cells provided by the ICC scaffold has tremendous importance in the functionality of the ex vivo replica, and (ii) the described bone marrow construct replicates two of the key reproductive functions of normal bone marrow. This cell-scaffold system allowed for the growth, differentiation and movement of cells in a 3D environment effectively mimicking the natural in vivo bone marrow environment normally encountered during hematopoiesis and B lymphopoiesis.
Acknowledgments
Technical support of Jordan Kicklighter in the Anesthesiology Manuscript Office and Tom Albrecht and Eugene Knutson of the UTMB Optical Imaging Core was greatly appreciated. Funding from DARPA (J.E.N., N.A.K.) and NSF and NIH (N.A.K.) and assistance of DARPA personnel Robin Sanders and Dr. Al Mc Manus was greatly appreciated.
Footnotes
The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6(2):93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- 2.Kobari L, Pflumio F, Giarratana MC, Li X, Titeux M, Izac B, et al. In vitro and in vivo evidence for the long-term multilineage (myeloid, B, NK, and T) reconstitution capacity of ex vivo expanded human CD34+ cord blood cells. Exp Hematol. 2000;28:1470–1480. doi: 10.1016/s0301-472x(00)00557-9. [DOI] [PubMed] [Google Scholar]
- 3.Barker J, Verfaillie C. A novel in vitro model of early human adult B lymphopoiesis that allows proliferation of pro-B cells and differentiation to mature B lymphocytes. Leukemia. 2000;14:1614–1620. doi: 10.1038/sj.leu.2401869. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, Brandt JS, Ellison FM, Calado RT, Young NS. Defective stromal cell function in a mouse model of infusion-induced bone marrow failure. Exp Hematol. 2005;33(8):901–908. doi: 10.1016/j.exphem.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 5.Punzel M, Moore K, Lemischka I, Verfaillie C. The type of stromal feeder used in limiting dilution assays influences frequency and maintenance assessment of human long-term culture initiating cells. Leukemia. 1999;13:92–97. doi: 10.1038/sj.leu.2401234. [DOI] [PubMed] [Google Scholar]
- 6.Hsiong SX, Mooney DJ. Regeneration of vascularized bone. Periodontol 2000. 2006;41(1):109–122. doi: 10.1111/j.1600-0757.2006.00158.x. [DOI] [PubMed] [Google Scholar]
- 7.Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotech. 2005;23(1):47–55. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 8.Kiel MJ, Yilmaz ÖH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121(7):1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- 9.Islam A, Catovsky D, Goldman JM, Galton DAG. Histomorphological study of cellular interactions between stromal and haemopoietic stem cells in normal and leukaemic bone marrow. Histopathology. 1984;8(2):293–313. doi: 10.1111/j.1365-2559.1984.tb02343.x. [DOI] [PubMed] [Google Scholar]
- 10.Mehta G, Kiel MJ, Lee J, Kotov N, Linderman JJ, Takayama S. Polyelectrolyte-clay-protein layer films on microfluidic PDMS bioreactor surfaces for primary murine bone marrow culture. Adv Funct Mater. 2007;17(15):2701–2709. [Google Scholar]
- 11.Lee J, Cuddihy MJ, Kotov NA. Three-dimensional cell culture matrices: State of the art. Tissue Eng Part B: Rev. 2008;14(1):61–86. doi: 10.1089/teb.2007.0150. [DOI] [PubMed] [Google Scholar]
- 12.Poznansky MC, Evans RH, Foxall RB, Olszak IT, Piascik AH, Hartman KE, et al. Efficient generation of human T cells from a tissue-engineered thymic organoid. Nat Biotech. 2000;18(7):729–734. doi: 10.1038/77288. [DOI] [PubMed] [Google Scholar]
- 13.Nöth U, Steinert AF, Tuan RS. Technology Insight: adult mesenchymal stem cells for osteoarthritis therapy. Nat Clin Pract Rheumatol. 2008;4(7):371–380. doi: 10.1038/ncprheum0816. [DOI] [PubMed] [Google Scholar]
- 14.Chen FH, Rousche KT, Tuan RS. Technology Insight: adult stem cells in cartilage regeneration and tissue engineering. Nat Clin Pract Rheumatol. 2008;2(7):373–382. doi: 10.1038/ncprheum0216. [DOI] [PubMed] [Google Scholar]
- 15.Mironov V, Kasyanov V, Markwald RR. Nanotechnology in vascular tissue engineering: from nanoscaffolding towards rapid vessel biofabrication. Trends Biotechnol. 2008;26(6):338–344. doi: 10.1016/j.tibtech.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 16.Jager M, Feser T, Denck H, Krauspe R. Proliferation and Osteogenic Differentiation of Mesenchymal stem cells cultured onto three different polymers in vitro. Ann Biomed Eng. 2005;33(10):1319–32. doi: 10.1007/s10439-005-5889-2. [DOI] [PubMed] [Google Scholar]
- 17.Kim KS, Lee JH, Ahn HH, Lee JY, Khang G, Lee B, Lee HB, Kim MS. The osteogenic differentiation of rat muscle-derived stem cells in vivo within in situ-forming chitosan scaffolds. Biomat. 2008;29(33):4420–4428. doi: 10.1016/j.biomaterials.2008.08.005. [DOI] [PubMed] [Google Scholar]
- 18.Ma K, Chan CK, Liao S, Hwang WYK, Feng Q, Ramakrishna S. Electrospun nanofiber scaffolds for rapid and rich capture of bone marrow-derived hematopoietic stem cells. Biomat. 2008;29(13):2096–103. doi: 10.1016/j.biomaterials.2008.01.024. [DOI] [PubMed] [Google Scholar]
- 19.Taquvi S, Roy K. Influence of scaffold physical properties and stromal cell coculture on hematopoietic differentiation of mouse embryonic stem cells. Biomat. 2006;27(36):6024–31. doi: 10.1016/j.biomaterials.2006.05.052. [DOI] [PubMed] [Google Scholar]
- 20.Hwang K-C, Kim JY, Chang W, Kim D-S, Lim S, Kang S-M, Song B-W, Ha H-Y, Huh YJ, Choi I-G, Hwang D-Y, Song H, Jang Y, Chung N, Kim S-H, Kim D-W. Chemicals that modulate stem cell differentiation. Proc Natl Acad Sci. 2008;105(21):7467–71. doi: 10.1073/pnas.0802825105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bagley J, Rosenzweig M, Marks DF, Pykett MJ. Extended culture of multipotent hematopoietic progenitors without cytokine augmentation in a novel three-dimensional device. Exp Hematol. 1999:496–504. doi: 10.1016/s0301-472x(98)00053-8. [DOI] [PubMed] [Google Scholar]
- 22.Chua K-N, Chai C, Lee P-C, Ramakrishna S, Leong KW, Mao H-Q. Functional nanofiber scaffolds with different spacers modulate adhesion and expansion of cryopreserved umbilical cord blood hematopoietic stem/progenitor cells. Exp Hematol. 2007;35(5):771–781. doi: 10.1016/j.exphem.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ehring B, Biber K, Upton T, Plosky D, Pykett M, Rosenzweig M. Expansion of HPCs from cord blood in a novel 3D matrix. Cytotherapy. 2003;5:490–499. doi: 10.1080/14653240310003585. [DOI] [PubMed] [Google Scholar]
- 24.Knospe WH, Husseini SG, Fried W. Hematopoiesis on cellulose ester membranes. XI. Induction of new bone and a hematopoietic microenvironment by matrix factors secreted by marrow stromal cells. Blood. 1989;74(1):66–70. [PubMed] [Google Scholar]
- 25.Li Y, Ma T, Kniss DA, Yang S-T, Lasky LC. Human cord cell hematopoiesis in three-dimensional nonwoven fibrous matrices: In vitro simulation of the marrow microenvironment. J Hematother Stem Cell Res. 2001;10(3):355–368. doi: 10.1089/152581601750288966. [DOI] [PubMed] [Google Scholar]
- 26.Liu H, Roy K. Biomimetic three-dimensional cultures significantly increase hematopoietic differentiation efficacy of embryonic stem cells. Tissue Eng. 2005;11(12):319–330. doi: 10.1089/ten.2005.11.319. [DOI] [PubMed] [Google Scholar]
- 27.Oswald J, Steudel C, Salchert K, Joergensen B, Thiede C, Ehninger G, et al. Gene-expression profiling of CD34+ hematopoietic cells expanded in a collagen I matrix. Stem Cells. 2006;24(3):494–500. doi: 10.1634/stemcells.2005-0276. [DOI] [PubMed] [Google Scholar]
- 28.Panoskaltsis N, Mantalaris A, Wu JHD. Engineering a mimicry of bone marrow tissue ex vivo. J Biosci Bioeng. 2005;100(1):28–35. doi: 10.1263/jbb.100.28. [DOI] [PubMed] [Google Scholar]
- 29.Feng Q, Chai C, Jiang X-S, Leong WK, Mao H-Q. Expansion of engrafting human hematopoietic stem/progenitor cells in three-dimensional scaffolds with surface-immobilized fibronectin. J Biomed Mater Res A. 2006;78A(4):781–791. doi: 10.1002/jbm.a.30829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sasaki T, Takagi M, Soma T, Yoshida T. 3D culture of murine hematopoietic cells with spatial development of stromal cells in nonwoven fabrics. Cytotherapy. 2002;4:285–291. doi: 10.1080/146532402320219808. [DOI] [PubMed] [Google Scholar]
- 31.Xiong F, Chen Z, Liu H, Xu Z, Liu X. Ex vivo expansion of human umbilical cord blood hematopoietic progenitor cells in a novel three-dimensional culture system. Biotechnol Lett. 2002;24(17):1421–1426. [Google Scholar]
- 32.Lee J, Shanbhag S, Kotov NA. Inverted colloidal crystals as three-dimensional microenvironments for cellular co-cultures. J Mater Chem. 2006;16(35):3558–3564. [Google Scholar]
- 33.Kotov NA, Liu Y, Wang S, Cumming C, Eghtedari M, Vargas G, et al. Inverted colloidal crystals as three-dimensional cell scaffolds. Langmuir. 2004;20(19):7887–7892. doi: 10.1021/la049958o. [DOI] [PubMed] [Google Scholar]
- 34.Lyons AB, Parish CR. Determination of lymphocyte division by flow cytometry. J Immunol Methods. 1994;171(1):131–137. doi: 10.1016/0022-1759(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 35.Burlington DB, Clements ML, Meiklejohn G, Phelan M, Murphy BR. Hemagglutinin-specific antibody responses in immunoglobulin G, A, and M isotypes as measured by enzyme-lined immunosorbent assay after primary or secondary infection of humans with influenze A virus. Infect Immun. 1983;41(2):540–545. doi: 10.1128/iai.41.2.540-545.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bachmann MF, Ecabert B, Kopf M. Influenza virus: a novel method to assess viral and neutralizing antibody titers in vitro. J Immunol Methods. 1999;225(12):105–111. doi: 10.1016/s0022-1759(99)00034-4. [DOI] [PubMed] [Google Scholar]
- 37.Murphy BR, Phelan MA, Nelson DL, Yarchoan RY, Tierney EL, Alling DW, et al. Hemagglutinin-specific enzyme-linked immunosorbent assay for antibodies to influenza A and B viruses. J Clin Microbiol. 1981;13(3):554–560. doi: 10.1128/jcm.13.3.554-560.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Wang S, Eghtedari M, Motamedi M, Kotov NA. Inverted colloidal crystal hydrogel matrices as three-dimensional cell scaffolds. Adv Funct Mater. 2005;15(5):725–731. [Google Scholar]
- 39.Geiger H, Koehler A, Gunzer M. Stem cells, aging, niche, adhesion and Cdc42. Cell Cycle. 2007;6(8):884–887. doi: 10.4161/cc.6.8.4131. [DOI] [PubMed] [Google Scholar]
- 40.Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. 2003;425(6960):841–846. doi: 10.1038/nature02040. [DOI] [PubMed] [Google Scholar]
- 41.Djouad F, Delorme B, Maurice M, Bony C, Apparailly F, Louis-Plence P, et al. Microenvironmental changes during differentiation of mesenchymal stem cells towards chondrocytes. Arthritis Res Ther. 2007;9(2):R33. doi: 10.1186/ar2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cuddihy MJ, Kotov N. Poly(lactic-co-glycolic acid) Bone Scaffolds with Inverted Colloidal Crystal Geometry. Tissue Eng A. 2008;14(10):1639–1649. doi: 10.1089/ten.tea.2007.0142. [DOI] [PubMed] [Google Scholar]
- 43.Shanbhag S, Lee J, Kotov N. Diffusion in three-dimensionally ordered scaffolds with inverted colloidal crystal geometry. Biomaterials. 2005;26(27):5581–5585. doi: 10.1016/j.biomaterials.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 44.Tang Z, Kotov NA, Magonov S, Ozturk B. Nanostructured artificial nacre. Nat Mater. 2003;2(6):413–418. doi: 10.1038/nmat906. [DOI] [PubMed] [Google Scholar]
- 45.Drury JL, Mooney DJ. Hydrogels for tissue engineering: scaffold design variables and applications. Biomaterials. 2003;24:4337–4351. doi: 10.1016/s0142-9612(03)00340-5. [DOI] [PubMed] [Google Scholar]
- 46.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126(4):677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 47.Thompson MT, Berg MC, Tobias IS, Rubner MF, Van Vliet KJ. Tuning compliance of nanoscale polyelectrolyte multilayers to modulate cell adhesion. Biomaterials. 2005;26(34):6836–6845. doi: 10.1016/j.biomaterials.2005.05.003. [DOI] [PubMed] [Google Scholar]
- 48.Schneider A, Francius G, Obeid R, Schwinté P, Hemmerlé J, Frisch B, Schaaf P, Voegel J-C, Senger B, Picart C. Polyelectrolyte Multilayers with a Tunable Young’s Modulus: Influence of Film Stiffness on Cell Adhesion. Langmuir. 2006;22(3):1193–1200. doi: 10.1021/la0521802. [DOI] [PubMed] [Google Scholar]
- 49.Balaban NQ, Schwarz US, Riveline D, Goichberg P, Tzur G, Sabanay I, Mahalu D, Safran S, Bershadsky A, Addadi L, Geiger B. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat Cell Biol. 2001;3:466–472. doi: 10.1038/35074532. [DOI] [PubMed] [Google Scholar]
- 50.Discher D, Janmey P, Wang Y-L. Tissue Cells Feel and Respond to the Stiffness of Their Substrate. Science. 2005;310(5751):1139–43. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 51.Berthelemy N, Kerdjoudj H, Gaucher C, Schaaf P, Stoltz J-F, Lacolley L, Voegel J-C, Menu P. Polyelectrolyte Films Boost Progenitor Cell Differentiation into Endothelium-like Monolayers. Adv Mater. 2008;20(14):2674–2678. doi: 10.1002/adma.200702418. [DOI] [PubMed] [Google Scholar]
- 52.Dierich A, Le Guen E, Messaddeq N, Stoltz J-F, Netter P, Schaaf P, Voegel J-C, Benkirane-Jessel N. Bone Formation Mediated by Synergy-Acting Growth Factors Embedded in a Polyelectrolyte Multilayer Film. Adv Mater. 2007;19(5):693–697. [Google Scholar]
- 53.Wols HAM, Underhill GH, Kansas GS, Witte PL. The role of bone marrow-derived stromal cells in the maintenance of plasma cell longevity. J Immunol. 2002;169(8):4213–4221. doi: 10.4049/jimmunol.169.8.4213. [DOI] [PubMed] [Google Scholar]
- 54.Civin CI, Trischmann T, Kadan NS, Davis J, Noga S, Cohen K, et al. Highly purified CD34-positive cells reconstitute hematopoiesis. J Clin Oncol. 1996;14(8):2224–2233. doi: 10.1200/JCO.1996.14.8.2224. [DOI] [PubMed] [Google Scholar]
- 55.Spangrude GJ, Brooks DM, Tumas DB. Long-term repopulation of irradiated mice with limiting numbers of purified hematopoietic stem cells: in vivo expansion of stem cell phenotype but not function. Blood. 1995;85(4):1006–1016. [PubMed] [Google Scholar]
- 56.Murti KG, Brown PS, Kumagai M-a, Campana D. Molecular Interactions between human B-cell progenitors and the bone marrow microenvironment. Exp Cell Res. 1996;226(1):47–58. doi: 10.1006/excr.1996.0201. [DOI] [PubMed] [Google Scholar]
- 57.Yilmaz OH, Kiel MJ, Morrison SJ. SLAM family markers are conserved among hematopoietic stem cells from old and reconstituted mice and markedly increase their purity. Blood. 2006;107(3):924–930. doi: 10.1182/blood-2005-05-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kobari L, Giarratana M-C, Pflumio F, Izac B, Coulombel L, Douay L. CD133+ cell selection is an alternative to CD34+ cell selection for ex vivo expansion of hematopoietic stem cells. J Hematother Stem Cell Res. 2001;10(2):273–281. doi: 10.1089/15258160151134980. [DOI] [PubMed] [Google Scholar]