

Abstract
PVS-RIPO is a recombinant oncolytic poliovirus designed for clinical application to target CD155 expressing malignant gliomas and other malignant diseases. PVS-RIPO does not replicate in healthy neurons and is therefore non-pathogenic in rodent and non-human primate models of poliomyelitis. A tetrazolium salt dye-based cellular assay was developed and qualified to define the cytotoxicity of virus preparations on susceptible cells and to explore the target cell specificity of PVS-RIPO. In this assay, PVS-RIPO inhibited proliferation of U87-MG astrocytoma cells in a dose-dependent manner. However, HEK293 cells were much less susceptible to cell killing by PVS-RIPO. In contrast, the Sabin type 1 live attenuated poliovirus vaccine strain (PV(1)S) was cytotoxic to both HEK293 and U87-MG cells. The correlation between expression of CD155 and cytotoxicity was also explored using six different cell lines. There was little or no expression of CD155 and PVS-RIPO-induced cytotoxicity in Jurkat and Daudi cells. HEK293 was the only cell line tested that showed CD155 expression and resistance to PVS-RIPO cytotoxicity. The results indicate that differential cytotoxicity measured by the colorimetric assay can be used to evaluate the cytotoxicity and cell-type specificity of recombinant strains of poliovirus and to demonstrate lot to lot consistency during the manufacture of viruses intended for clinical use.
Keywords: Cytopathogenicity, Cytotoxicity, Poliovirus, MTS-based Assay, CD155, Gliomas
1. Introduction
Poliovirus (PV) is a neurovirulent member of the genus Enteroviridae in the family of Picornaviridae. It features a single (+) stranded RNA genome of approximately 7,400 nucleotides enclosed by a compact icosahedral capsid. The unusual restricted specificity of poliovirus for spinal motor neurons in the human central nervous system is responsible for the distinct pathology and clinical manifestations of paralytic poliomyelitis (Thompson and Tebbens, 2006). Poliovirus and its affinity for its cellular receptor CD155, an immunoglobulin superfamily molecule, provide a unique opportunity to direct specific viral target tropism against cancerous cells such as glioblastoma multiforme (Gromeier et al., 2000; Ochiai et al., 2006). CD155 has been identified recently as a key determinant of tumor cell migration, invasion, and metastasis (Sloan et al., 2005), and transcriptional activation of the CD155 gene has been linked to signaling pathways deregulated commonly in malignancy, including pathways regulated by sonic hedgehog (shh)/gli (Solecki et al., 2002), fibroblastic growth factor, and oncogenic ras (Hirota et al., 2005). Interaction with the CD155 receptor induces irreversible morphological changes of viral particles, resulting in penetration of the cytoplasmic membrane and release of the viral genomes into the host cell cytoplasm (Gromeier et al., 2000).
The poliovirus plus-strand RNA genome initiates translation in a cap-independent manner via an internal ribosome entry site (IRES) in the 5′ untranslated region. Viral translation is co-determined by cellular IRES trans-acting factors which can influence viral propagation in a cell-type-specific manner (Merrill and Gromeier, 2006). A poliovirus recombinant PV1-RIPO, in which the cognate poliovirus IRES is replaced with its counterpart from human rhinovirus type 2 (HRV2), was constructed in a poliovirus type 1 (Mahoney) [PV1 (M)] backbone (Gromeier et al., 1996). PVS-RIPO was generated by exchange of the wild-type PV1(M) open reading frame (ORF) and 3′UTR with its counterparts from type 1 live attenuated Sabin (PV(1)S) vaccine strain (Ochiai et al., 2004). Insertion of the HRV2 IRES into the poliovirus genome eliminated poliovirus propagation in cells of neuronal derivation and abolished neuropathogenicity in mice transgenic for the human poliovirus receptor CD155 and in nonhuman primates (Gromeier et al., 1999). Despite severely repressed viral propagation in neurons, the chimeric viruses replicate with wild-type kinetics in malignant glioma cells (Gromeier et al., 2000). Cell type-specific dysfunction of the HRV2 IRES in neuronal cells has been attributed to the presence of a brain-specific translational repressor complex present in the CNS but absent from glioblastoma cells (Merrill et al., 2006; Merrill and Gromeier 2006).
PVS-RIPO/HRV2 IRES chimeras exhibit a striking cell type-specific replication deficit in various cell lines of neuronal lineage [i.e. Sk-N-Mc neuroblastoma (Gromeier et al., 1996; 1999) and HEK293 cells (Campbell et al., 2005)]. HEK293 cells, derived originally from primary human embryonic kidney cultures containing cells of neuronal lineage (Shaw et al., 2002), have been established as a cell culture model for evaluation of poliovirus phenotypes (Campbell et al., 2005). Merrill et al. (2006) used HEK293 cells to conduct PVS-RIPO propagation assays in neuron:glioblastoma heterokaryons. Interestingly, PVS-RIPO propagation in HTB-14 (U87-MG) glioblastoma cells and HEK293 fused cells is repressed profoundly, indicating that neuron-specific trans dominant inhibitors of HRV2 IRES are present in HEK293 cells, thus validating the relevance of this cell-line as a model for neuronal cell function in regard to the cell-type specific repression of PVS-RIPO propagation (Merrill and Gromeier, 2006).
Based on the differential growth properties of PVS-RIPO in malignant cells vs. normal neurons, PVS-RIPO has been evaluated in preclinical research for its potential application for treatment of glioblastoma and other malignancies (Ochiai et al., 2004). Cellular specificity of virus replication and cytopathogenicity are critical elements in defining the anti-tumor potency of this recombinant virus. Hence, identification of cellular systems susceptible and resistant to PVS-RIPO mediated cytopathic effects and development of quantitative cytopathic assays are essential for clinical development. HEK293 cells have certain advantages over neuronal cell lines as a tissue culture model for evaluating neuronal virus growth phenotypes. Campbell et al. (2005) reported that HEK293 cells recapitulate faithfully the neurovirulence of poliovirus observed in experimental animals and do not convert spontaneously from neuron-like to fibroblast-like phenotypes, a common property of many neuroblastoma cell lines.
Viral replication is determined by measuring virus-induced cytopathic effect (CPE) in infected cells. The methods used most widely are the plaque assay and endpoint titration assay (Dulbecco and Vogt, 1954; Hierholzer and Killington, 1996). Values determined by these separate methods are expressed as plaque forming unit (pfu) and tissue culture infectious dose 50% (TCID50), respectively. A disadvantage of these methods in general is that they are time consuming and labor intense and require daily attention for approximately 5–10 days. Different methods of cell viability determination such as the Trypan blue dye exclusion method, thymidine incorporation assay, assays using MTT (3-(4,5-dimethylthiazol-2yl)-2,5 diphenyl tetrazolium bromide) (Mosmann,1983) and its derivatives, MTS (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt) or CellTiter 96 Aqueous One Solution containing MTS and an electron coupling reagent phenazine ethosulfate, and several fluorescent and chemiluminescence-based cell proliferation or cytotoxicity assays have been used to assess viral cytopathic effects (Malecki et al., 2000; Heese et al., 2000; Gauduchon et al., 2005; Zhang et al., 2002; Giridharan et al., 2002; Tonello et al., 2002). In addition, cell viability assessment has been used in several cases as an alternate method for determination of virus infectivity (Heldt et al., 2006).
The MTT-based colorimetric method has been used to quantify cytolytic replication for a number of picornaviruses in cell culture (Andersson et al., 2005) and for measuring the infectious titer for a number of parvoviruses (Heldt et al., 2006). The MTS-based cytotoxicity assay could be used as an alternate method for defining the infectious titer of poliovirus in MTS50 units under defined conditions analogous to those described earlier for other piconaviruses. Ochiai et al., (2004) used a one-step growth assay to demonstrate the differential susceptibility of PVS-RIPO toward MCF-7/HER-18 tumor cells and HEK293 cells. In order to establish a more efficient, quantitative, and reproducible assay, a MTS-based assay was developed to evaluate PVS-RIPO induced differential cell killing of U87-MG glioblastoma cells and HEK293 cells. The utility of this assay for monitoring viral cytotoxic effects during the manufacture was also explored and it was confirmed that expression of the CD155 receptor as analyzed by FACS and antigen-captured ELISA is not the determinate of this specificity.
2. Materials and Methods
2.1. Cells and cell culture
U87-MG, an astrocytoma cell line, was obtained from Dr. John H. Sampson (Duke University, Durham, North Carolina) and cultured in DMEM (Dulbecco's Modified Eagle's Medium) with L-Glutamine, supplemented with 10% fetal bovine serum (FBS) and 1% non-essential amino acids solution. HEK293 cell line was obtained from Magenta Corporation and cultured in DMEM with L-Glutamine, supplemented with 10% FBS. M21/P6, a human malignant melanoma cell line, was obtained from Dr. Jean Surfus, (University of Wisconsin, Madison, Wisconsin) and cultured in RPMI-1640 medium with 10% FBS, 2 mM L-glutamine, 10 mM HEPES, and penicillin (100 units/mL) streptomycin (100 μg/mL). H226, an adherent non-small cell lung carcinoma cell line (Batra et al., 1998), was cultured in DMEM with 2 mM L-Glutamine and 10% FBS. The Daudi cell line was obtained from Dr. Ira Pastan (National Cancer Institute, Bethesda, MD) and cultured in RPMI-1640 medium with 20% FBS, 1 mM Sodium pyruvate, 2 mM L-Glutamine, Penicillin (100 units/mL)/ Streptomycin (100 μg/mL). The Jurkat cell line was obtained from ATCC and cultured in RPMI-1640 medium with 10% FBS, 1 mM Sodium pyruvate, 2 mM L-Glutamine, Penicillin (100 units/mL)/ Streptomycin (100 μg/mL).
2.2. Virus
The construction of chimeric PVS-RIPO was reported previously (Ochiai et al., 2004). PVS-RIPO production was performed in the Biopharmaceutical Development Program, SAIC Frederick, Inc., Biological Resource Branch of National Cancer Institute, Frederick, MD. Poliovirus type 1 Sabin (PV(1)S) vaccine strain was obtained from National Institute for Biological Standards and Control (Potters Bar, Hertfordshire, United Kingdom). The PVS-RIPO virus seed obtained from Dr. Gromeier's laboratory is used as the Reference Standard for the assay development and comparison of activity of different virus preparations.
2.3. Antibodies
Affinity-purified primary polyclonal rabbit anti-CD155 antibody D480 was generated as described previously (Merrill et al., 2004) and anti-CD155 monoclonal antibody D171 was kindly provided by E. Wimmer (Stony Brook University, Stony Brook, New York). Biotinylated Goat anti-Rabbit IgG was obtained from Vector Laboratories (Burlingame, CA). Streptavidin-HRP was obtained from R&D systems (Minneapolis, MN). FITC-labeled anti-CD155 antibody D171 was obtained from MBL International (Woburn, MA). Phycoerythrin (PE)-anti-poliovirus receptor related 2 protein (αPRR2), was obtained from BD BioSciences Pharmingen (San Diego, CA).
2.4. Differential cell killing assay
Unless specified otherwise U87-MG or HEK293 cells were plated in a 96-well plate at (0.2- 4) × 104 cells/well [(0.2-4) × 105 cells/mL, 0.1 mL/well]. Test articles were diluted to an initial titer of 2 × 107 pfu/mL (based on the titer of PVS-RIPO determined on Vero cells) followed by serial 3-4 fold dilutions. The diluted virus samples (100 μL/well) were transferred into the plates containing the cells within 2 h. The final multiplicity of infection (MOI) of test articles ranged from 50 to 0.0001 pfu/cell (at the time of seeding or infection). The plates were incubated at 37°C in a 5% CO2 and 80% humidity for 48 h. After 48 h incubation, CellTiter96® AQueous One Solution was added (20 μL/well) and incubated for another 4 h at 37°C in 5% CO2 and 80% humidity. SDS (25 μL/well of 10% solution) was added to the wells. The plates were read within 5 minutes at 490 nm on a plate reader. The background readings in the wells with medium (no cells) were subtracted from the sample well readings. In experiments where infection was carried out 20-24 h after cell seeding, the MOI was calculated based on 2× the cell seed density as the doubling time of the cells was ∼24 h. When infection was carried out immediately after cell seeding, the MOI was calculated based on actual cell seed density.
Data analysis was performed using a non-linear 4-parameter curve fit (SoftMax Pro Software program from Molecular Device). The 4-parameter curve fit equation applied for analysis is shown as y= {(A-D)/ [1 + (x/C) ˆ B]} +D. Parameter A refers to the asymptote (OD read out) at the low end of the x-axis (corresponding to no virus), D refers to the asymptote of the four parameter curve at the high virus infectious titer. Coefficient C corresponds to the x value (concentration) at the mid-point between A and D. The C value thus corresponds to the virus infectious titer (MOI or pfu/cell) corresponding to half maximal cell killing (IC50). The coefficient B describes how rapidly the curve makes its transition from the asymptotes in the center of the curve (Nix and Wild, 2001). The % cell survival at different MOIs was calculated using the formula: % cell survival = [absorbance of the infected well ÷ absorbance of the control well (uninfected cells) ×100. ED50 is used to denote the virus infectious titer that gives 50% cell killing. This is determined by plotting the % cell survival against MOI.
2.5. Titration of viruses and titer calculations in U87-MG
The MTS assay was used to calculate the virus titer in terms of MTS50 (50% virus infectious titer) (Heldt et al., 2006). The average optical density (OD) of uninfected control cells was set as 0% cytotoxicity. The % cytotoxicity at different dilutions was calculated using the formula: % cytotoxicity = (average OD of uninfected control cells -average OD of infected cells) / average OD of uninfected control cells × 100. From the plot of % cytotoxicity versus dilution factor, the 50% cytotoxicity was determined to be at a certain dilution factor. This value was converted to units/mL and stated as the MTS50 titer (Heldt et al., 2006). For example, if the 50% cytotoxicity was found to be at 103 dilution factor and 50 μL had been initially added to the first well for infection, then the MTS50 titer would be: (103) /0.050 mL = 2 × 104 MTS50/mL = 4.3 log10(MTS50/mL).
For comparison of the MTS assay with the TCID50, a TCID50 assay (Dulbecco, 1988; Hierholzer and Killington, 1996) was performed using equivalent plates to those used for the MTS assay under identical conditions including cell seeding, culture, virus infection, and post-infection culture time. At the defined time point, the plates were read by visual observation under a microscope to determine the number of wells showing CPE, and were scored. The TCID50/mL value was calculated by the Spearman Kärber method (Hierholzer and Killington, 1996).
2.6. Solid-phase antigen capture/ELISA assay
CD155 expression levels were determined by using a combined solid-phase antigen capture/ ELISA assay. The antigen capture in 96-well plate was performed following the procedure reported earlier (Merrill et al., 2004). Cells were solubilized in cold phosphate buffered saline (PBS) containing 0.5% NP-40 (Sigma) and 1% protease inhibitor cocktail. Total protein concentrations of cell lysate were measured by using bicinchoninic acid (BCA) protein assay (Smith, et al 1985). Equivalent amounts of total solubilized protein were used for antigen capture. Individual wells in ELISA plates were treated with 50 μL of 0.2 M Sodium Carbonate-bicarbonate Buffer (pH 9.4) containing 7.5 μg of anti-CD155 antibody D171 and incubated overnight at 4°C. On the second day, the plate was washed 3 times with PBS-Tween (PBST) and blocked with 3% non-fat dry milk-PBST for 30 min at 37°C. The cell homogenate was added to the D171 coated wells and incubated at 37°C for 1 h. The unbound homogenate was removed by washing three times with PBS. The wells containing bound antigen were incubated with affinity purified-primary polyclonal anti-CD155 antibody D480 (diluted 1:500 in 3% BSA-PBST, 200 μL/well) for 1 h at room temperature. After washing with PBST, the plate was incubated with biotinylated anti-rabbit IgG (diluted 1:250, 200 μL/well) for 2 h at room temperature followed by incubation with Streptavidin-HRP (R&D systems, diluted 1:200, 200 μL/well) for 30 min. At the end of the incubation period, the plate wells were emptied and washed three times in PBST. Plate wells were then incubated with 50 μL of the TMB substrate solution at room temperature. The reaction was stopped by adding 50 μL/well of 2 N sulfuric acid, and the absorbance at 450 nm was recorded using a microplate reader.
2.7. Fluorescence-activated cell sorter (FACS) analysis
For immuno-staining, cells were harvested and suspended in phosphate buffered saline. Fixed or non-fixed cells (106/mL) were incubated with fluorescence-labeled antibodies for 45 min at 2-8°C and then centrifuged for 5 min at 1000 rpm. The cell pellet was washed in PBS and re-suspended in 1 mL PBS for FACS analysis. FACS analysis was performed using FACSCalibur™ from BD Biosciences.
2.8. Morphological analysis
U87-MG cells were seeded at 104/well and cultured at 37°C and 5% CO2 for 24 h. The cells were infected with different multiplicities of infection (MOI). After incubating while gently rocking for 30 min at room temperature to allow for virus attachment, the cultures were thoroughly rinsed 3 times with growth medium to remove unbound virus particles. Fresh growth medium was added to the washed cells. The plate was incubated in a CO2 incubator maintained at 37°C. After incubation for 24 h, cell morphology was analyzed by light microscopy.
3. Results
3.1. Cytopathogenicity of PVS-RIPO in the human astrocytoma cell line U87-MG
To analyze the oncolytic potential of PVS-RIPO in glioma cells in culture, the astrocytoma cell line U87-MG was infected with different multiplicities of infection (MOIs) of PVS-RIPO. There was no evidence of infection in mock-treated cultures (MOI = 0) while classical CPE was observed with virus treated cells (Figure 1A). MTS-based assays also indicated that cytotoxicity diminished as the dilution factor of the virus inoculums increased (Figure 1B). The MTS50 titer was determined to be 4.36 log10 MTS50/mL. The TCID50 assay was performed using equivalent plates to those used for the MTS assay under identical conditions. A similar dose-dependent cytopathogenicity was observed with a TCID50 titer of 5.68 log10 TCID50/mL. The data are consistent with the earlier report that the MTS dye-based assay is more sensitive compared to visual CPE observation (Andersson, et al., 2005). These results indicate that the effects of PVS-RIPO infection on U87-MG astrocytoma cells are consistent with previous reports of CPE in PVS-RIPO infected primary glioma cultures (Merrill, et al., 2004) and other established glioma cell lines measured by plaque assay (Gromeier, et al., 2000).
Figure 1. Cytopathogenicity of PVS-RIPO on the human astrocytoma cell line U87-MG.
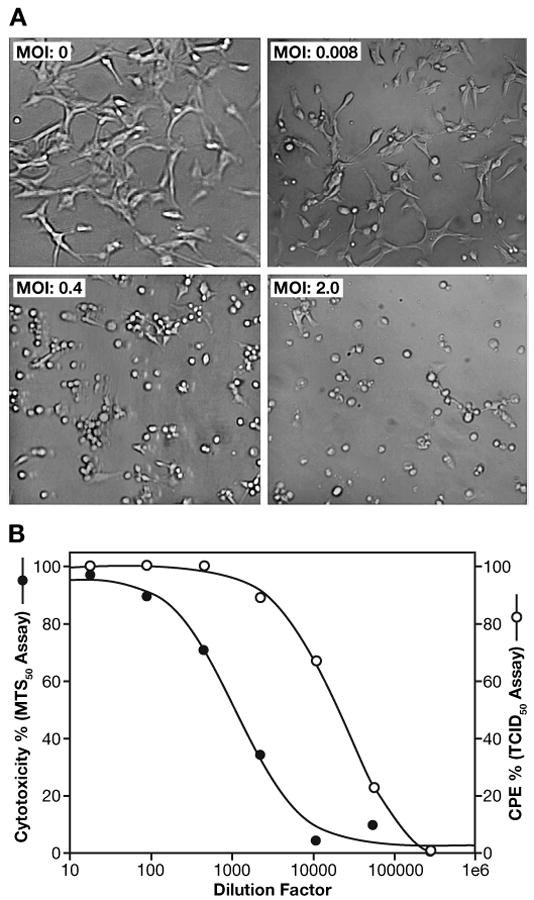
A. Morphology of U87-MG cells without virus infection (MOI: 0) and infected with PVS-RIPO at different MOIs (0.008, 0.4 and 2.0). Details as described in Materials and Methods. (Magnification: 10X).
B. Comparison of cytopathogenicity of PVS-RIPO determined by MTS-based assay and TCID50 assay. U87-MG cells were seeded at 5 × 103 cells/well in two flat bottomed, 96-well cell culture plates and incubated at 37°C in a humidified 5% CO2 incubator for 24 h. On the second day, the virus was diluted in five-fold serial dilutions with culture medium in a separate 96-well plate starting at a titer of the indicated dilution factor in quadruplicate wells. After carefully removing the media from the plate containing the cells, the cells were infected with 50 μL/well of virus sample to be titrated in quadruplicate. After 1 h 150 μL/well of culture medium was added. The infected cells were then maintained at 37°C in a humidified 5% CO2 environment. At the defined time point (48 h post-infection), one plate was observed by visual CPE account (TCID50 assay) and another was assayed by MTS-dye. TCID50 and MTS50 value were calculated from the plot of % CPE versus dilution factor, or the plot of %cytotoxicity versus dilution factor, respectively.
3.2. Effect of incubation time on PVS-RIPO induced cytotoxicity
To investigate the effect of incubation time on PVS-RIPO induced cytotoxicity, U87-MG cells were infected with PVS-RIPO at different MOIs and incubated for 24, 48, or 72 h. PVS-RIPO induced effect was evaluated by monitoring inhibition of cell proliferation using the MTS assay. The results (Figure 2A) demonstrate that cytotoxicity increases with an increase in incubation time. Uninfected U87-MG control cells showed a time-dependent increase in cell viability as expected indicating that the cells are in growth phase from the time of infection to day 3 post-infection. The absorbance value at all MOIs is lower than that of the control indicating cytotoxicity. As virus infectious titers (MOI) increase, there is enhanced cytotoxicity. At a MOI of 0.003, the lowest virus titer tested, ∼20% loss of viability was noted on day 1, ∼70% loss on day 2, and more than 85% loss of viability was observed on day 3 compared to the uninfected control cells on the respective days. Enhanced cytotoxicity with increasing incubation time is indicative of virus replication and shedding resulting in new infection of previously uninfected cells in culture. Figure 2B shows the % cell survival as a function of virus MOI on different days post-infection. There were only minor differences in the absorbance readings for the control and infected wells on day 1 post infection, and this resulted in a larger %error as reflected in the observed fluctuations of the dose response curve on day 1.
Figure 2. Optimization of MTS based cell proliferation assay conditions.

A-B. Effect of post-infection culture time on cytopathogenicity of PVS-RIPO in U87-MG. U87-MG cells were seeded into a 96-well plate and cultured for 24 h. After removing the medium, 50 μL/well of PVS-RIPO at different MOIs were added. Additional 150 μL/well of culture medium were added after 1 h incubation. After additional 24, 48, or 72 hours, MTS was added (20 μL/well) and incubated for 4 h. Twenty-five micro liters of 10% SDS were added and the OD at 490nm was read after 10 minutes. (A): Absorbance at 490 nm, which is an index of cell counts or cell viability, is plotted as a function of post-infection culture time for the control (non-infected) and cells infected with different doses of PVS-RIPO. (B): % cell survival [(OD 490 nm of infected cells ÷ OD 490 nm of Control cells (uninfected) × 100)] is plotted as a function of virus titer (MOI).
C. Effect of cell seeding density on cytopathogenicity of PVS-RIPO in U87-MG cells. U87-MG cells were seeded at different cell densities (5×103/well or 1 × 104/well) into two 96-well plates and cultured for 24 h. Two different PVS-RIPO lots were diluted and added at different MOIs into plates containing different cell densities. After an additional 48 h, MTS was added (20 μL/well) and incubated for 4 h. Twenty-five micro liters of 10%SDS were added and after 10 minutes the OD at 490nm was read.
D. Cell culture related variations of PVS-RIPO induced cytotoxicity. Eight experiments were performed over 1 year using U87-MG cells at different post-thaw cell passage infected with the same lot of PVS-RIPO. MTS50 value obtained from each assay is plotted as a function of cell passage number.
3.3. Effect of cell seeding density on PVS-RIPO induced cytotoxicity
The cytotoxic effect of two different virus preparations was determined at two different cell seeding densities (5×103 and 1×104 cells/well). The cells were infected with different doses of virus in 5 fold serial dilutions starting with 106 pfu/mL. Two days post-infection, dose-dependent cell-killing effects were measured using the MTS assay. Initially it appeared that the virus concentration for half-maximal cell killing was higher at the high cell seed density (data not shown). However, when the data were normalized for MOI (pfu/cell), the virus infectious titer (MOI) for half maximal cell killing was almost the same at all cell seeding densities (Figure 2C).
3.4. Effects of other cell culture related factors on PVS-RIPO induced cytotoxicity
Different post-thaw cell passage numbers of U87-MG cells were examined to determine cell culture related variations in the cytotoxicity of PVS-RIPO on U87-MG cells. Figure 2D shows the variations in the MTS50 values obtained from eight experiments performed over 1 year using U87-MG cells at different post-thaw cell passage infected with the same lot of PVS-RIPO. Although there was a variation in MTS50 ranging from 4.07 to 5.17 log10Unit, no direct correlation between MTS50 and cell passage number or storage time (stability) of the virus was observed in PVS-RIPO induced cytotoxicity of U87-MG.
3.5. Effect of virus propagation in Vero cells on cytotoxicity of PVS-RIPO
Since PVS-RIPO is produced in Vero cells, the MTS-based U87-MG cell cytotoxicity assay was used to evaluate the effect of PVS-RIPO propagation in Vero cells on cytopathogenicity of PVS-RIPO. Vero cells were transfected with PVS-RIPO RNA by an electroporation process. The virus shed from the transfected cells was isolated and labeled propagation cycle zero (C0). Subsequent serial infections (C1, C2, C3 etc.) were performed using the virus isolated from the previous propagation cycle. The cytotoxicity of viruses isolated at different passages through Vero cells was compared with a PVS-RIPO reference standard, and also with PV(1)S. Dose-dependent inhibition of U87-MG cell growth was observed for PVS-RIPO isolated from Vero cells at different propagation cycles. IC50 values for each preparation are shown in Table 1. The virus preparation (C3) after three cycles of propagation through Vero cells displayed slightly lower cytotoxicity compared to the virus preparation from earlier passages (C0 to C2). The PVS-RIPO reference standard, as well as PV(1)S showed comparable activity. Even though the cytotoxicity of virus obtained from propagation cycle 3 was slightly lower (Table 1), the variation was within the anticipated variation limit of the plaque assay used for virus infectivity titer determination, indicating that propagation in Vero cells does not affect the cytotoxicity of PVS-RIPO.
Table 1. Effect of virus propagation in Vero cells on cytopathogenicity of PVS-RIPO by MTS-based assay of U87-MG cells.
| Virus | Propagation Cycles* | IC50 (pfu/mL) | IC50 (MOI**) |
|---|---|---|---|
| PVS-RIPO | C0 | 6245 | 0.062 |
| Preparations | C1 | 2882 | 0.029 |
| From Vero | C2 | 3459 | 0.035 |
| cells | C3 | 12497 | 0.124 |
| PVS-RIPO Control | 8700 | 0.087 | |
| PV(1)S | 5300 | 0.053 | |
C0-C3 represent the propagation cycles in Vero cells after infection (0 to 3 respectively)
MOI is multiplicity of infection (pfu/cell)
3.6. Cell type specificity of PV(1)S and PVS-RIPO
The basis for potential therapeutic application of PVS-RIPO is the loss of viral replication in normal neuronal cells, while retaining oncolytic activity in tumor cells. To evaluate the differential effect of PVS-RIPO infection on neuronal cells in comparison to glioblastoma cells, the cytotoxic effects of PVS-RIPO on HEK293 and U87-MG cells were compared using the MTS colorimetric assay. Ideally, the conditional replication phenotype of PVS-RIPO in glioblastoma would be evaluated in parallel with indiscriminate replication in neuronal or glioblastoma cells of fully neurovirulent poliovirus strains, e.g. wild-type serotype 1 (Mahoney) (Gromeier et al., 1996). However, due to regulatory restriction of the use of wild-type polioviruses in the facility where this work was conducted, PVS-RIPO was compared with its backbone, PV(1)S. PV(1)S itself displays reduced replication potential in HEK293 cells when compared to its wild-type progenitor (Campbell et al., 2005), reflecting its attenuated neurovirulence phenotype. However, all cell culture-based assays suggest that the ‘neuronal incompetence’ of PV(1)S is far less pronounced than with PVS-RIPO (Campbell et al., 2005). Since PVS-RIPO is derived from PV(1)S) by insertion of a heterologous HRV2 IRES, the assays described below essentially determine the effect of the HRV2 IRES on cell type-specific replication of PVS-RIPO.
In HEK293 cells, PV(1)S displayed significant cytotoxicity in a dose-dependent manner (Figure 3A), suggesting that HEK293 cells meet all the requirements for viral entry, replication and oncolytic or cytolytic activity of polioviruses. The MTS50 value for PV(1)S in HEK293 cell was 5.94 log10 (MTS50/ml) compared to 6.87 log10 (MTS50/ml) in U87-MG cells. Growth of PV(1)S in HEK293 cells, albeit at moderate levels compared to wild type serotype 1 strains, was reported before (Campbell et al., 2005). In contrast, no detectable cytolysis of HEK293 cells was observed with PVS-RIPO up to an MOI of 1 pfu/cell although some cell death was observed at an MOI >1 pfu/cell (Figure 3A). In U87-MG cells, both PV(1)S and PVS-RIPO showed dose-dependent cytolysis (Figure 3B). IC50 values were about 10-4 pfu/cell for PV(1)S or 10-2∼10-3 pfu/cell for PVS-RIPO, respectively. With an MOI of >1 pfu/cell, almost complete cytolysis was observed with either virus. Under identical conditions, PVS-RIPO had a pronounced oncolytic effect on U87-MG indicating differential cell killing of U87-MG and HEK293 cells. The results are consistent with the earlier report (Campbell et al., 2005), and support the use of HEK293 cells as a cell culture model for evaluation of neurovirulent phenotypes of poliovirus.
Figure 3. Cytotoxicity comparison of PV(1)S and PVS-RIPO on U87-MG and HEK293 cells.

A. Cytotoxicity of PV(1)S and PVS-RIPO on HEK293 cells. HEK293 cells were seeded at 2 ×104 cells/well followed by incubation with PV(1)S or PVS-RIPO at different virus MOIs (pfu/cell). After 48 h incubation, Aqueous One cell titer reagent was added and incubated for 4 h. SDS was added to a final concentration of 1% and after 10 min the OD 490 nm was recorded using a 96-well plate reader.
B. Cytotoxicity of PV(1)S and PVS-RIPO on U87-MG cells. U87-MG cells were seeded at 2 ×104 cells/well followed by incubation with PV(1)S or PVS-RIPO at different virus MOIs (pfu/cell). After 48 h incubation, Aqueous One cell titer reagent was added and incubated for 4 h. SDS was added to a final concentration of 1% and after 10 minutes the OD 490 nm was recorded using a 96-well plate reader.
3.7. PV Receptor (CD155) and cytopathogenicity
The dependence of poliovirus cell killing on CD155 expression is well established (Mendelsohn et al., 1989). Since the poliovirus capsid is unaltered in PVS-RIPO, receptor interaction and host cell entry should be identical for both PVS-RIPO and PV(1)S (Gromeier et al.,1996). To elucidate the specific cytotoxic effects of PVS-RIPO in relation to receptor expression, a number of cell lines maintained routinely in our laboratory were tested for cytotoxicity and CD155 levels. Efficient killing of HEK293 cells by neurovirulent wild-type polioviruses and moderate cytopathogenicity of PV(1)S suggest that CD155 expression is not a factor in determining susceptibility of these cells to PVS-RIPO cell killing. CD155 expression on HEK293 cells was specifically evaluated to confirm that the absence of PVS-RIPO cytotoxicity in these cells was not related to a block at the level of cell entry. Results are shown in Figure 4(A–D) and Table 2.
Figure 4. Relationship between expression of PV receptor (CD155) and cytopathogenicity.
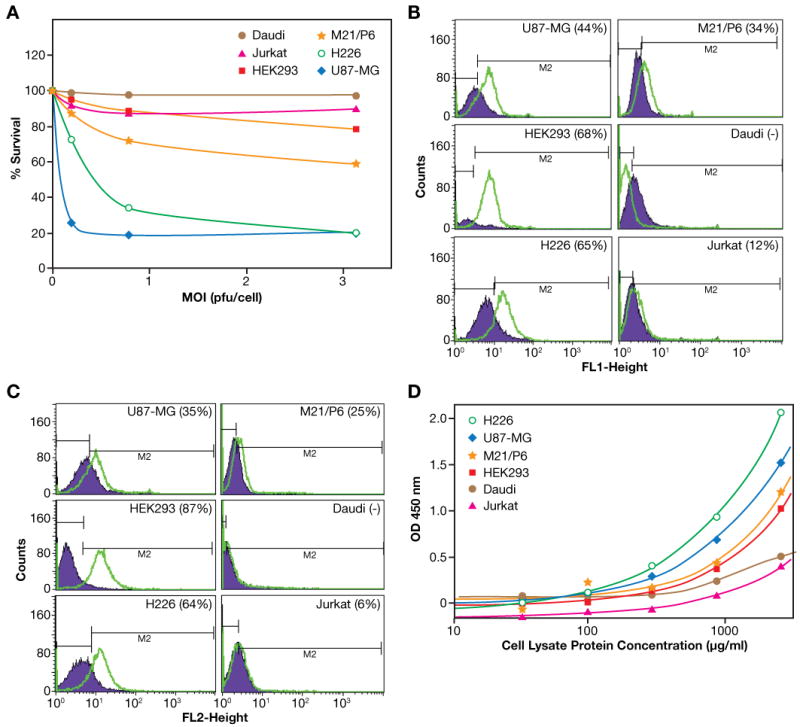
A. Comparison of the cytotoxicity of PVS-RIPO on different cell lines. Cells were seeded (4 ×104 in 100 μL/well) in a 96-well plate, added 100 μL PVS-RIPO to achieve the indicated MOI, and continued the incubation for another 48 h. MTS (25 μL) was added and incubated for additional 4 h. Twenty-five micro liters of 10% SDS were added and after 10 minutes the OD at 490 nm was read.
B. FACS staining with FITC-αCD155. Cells (106/100μL) were incubated with FITC-αCD155 or an isotype control FITC- IgG1κ for 10 min. After washing, cells were analyzed on FACS. Figure label at the top denotes the cell.
C. FACS staining with PE-α PRR2 on different cell lines. Cells (106/100μL) were incubated with PE-αPRR2 or an isotype control PE- IgG1κ for 10 min. After washing, cells were analyzed on FACS. Figure label at the top denotes the cell line.
D. Comparison of CD155 expression on different cell lines determined by a solid-phase antigen capture-ELISA. Details as described in Materials and Methods.
Table 2. Comparison of CD155 FACS staining, CD155 capture ELISA and cytopathogenicity of PVS-RIPO on different cell lines.
| Cell Type | Positive% (anti-CD155) | Positive% (anti-PRR) | Capture ELISA using anti-CD155 OD Read Out at 2.6 μg/mL cell lysate protein | Cytolytic activity of PVS-RIPO |
|---|---|---|---|---|
| U87-MG | 44% | 35% | 1.48 | Effective killing (ED50 <0.1 MOI) |
| HEK293 | 68% | 87% | 1.0 | Less susceptible to cytolysis (no killing up to MOI 1.0) |
| H226 | 65% | 64% | 2.02 | Effective killing |
| M21/P6 | 34% | 25% | 1.17 | Moderate killing |
| Jurkat | 12% | 6% | 0.36* | Not susceptible to cytolysis |
| Daudi | Negative | Negative | 0.47* | Not susceptible to cytolysis |
The read out are marginally above background readout as indicated by the lower plateau read out
Daudi B cells and Jurkat T cells show little or no susceptibility to PVS-RIPO cell killing while H226 and M21/P6 tumor cells showed various degrees of killing and U87-MG cells were killed very effectively (Figure 4A). Figure 4B shows the surface staining of fixed cells using FITC-αCD155 (D171). As expected, there was little or no evidence of CD155 expression on either the Daudi or Jurkat cells. The other cell lines were stained effectively. Similar results (Figure 4C) were obtained with PE-αPRR2, an antibody to a cell surface molecule related to the poliovirus receptor. Figure 4D shows CD155 expression determined by the immuno-capture ELISA using the antibody combination D171 and D480. The expression of CD155 as determined by the immuno-capture ELISA and FACS staining with FITC-αCD155 or PE-αPRR2, and cytotoxicity on the various cells tested are summarized in Table 2. These results confirm that Jurkat and Daudi cell lines, which showed little or no PVS-RIPO- or PV1(S)-induced cytotoxicity, exhibited very little or no expression of CD155, whereas the three tumor cell lines that were effectively killed by PVS-RIPO showed high CD155 expression by both FITC-αCD155 staining and an antigen-capture ELISA. In contrast, HEK293 cells, which were resistant to PVS-RIPO cell killing, also expressed high levels of CD155. These results confirm that the lower sensitivity of HEK293 cells to PVS-RIPO cell-killing is not related to the level of CD155 expression.
3.8. Assay variability
Preliminary assay optimization and qualification studies were performed to assess the feasibility of developing an MTS-based assay for testing lot to lot consistency and stability. Results shown in Figure 2 indicated that cell seed density, incubation time, cell passage number and/or other unidentified cellular factors may influence the MTS based cytotoxicity assay. In order for the assay to be quantitatively relevant the cell seed density and culture time should be controlled to obtain ideal linear growth kinetics. Cell growth kinetics for U87-MG was followed at three different cell seed densities (2.5, 5.0 and 10 × 103 cells/well in 100 μL seed) (data not shown). A satisfactory linear cell density response, as measured by the MTS based colorimetric cell proliferation assay, was obtained for five days post-seeding for cell seeding densities of 2.5 × 103 and 5.0 × 103 cells/well. In the case of cell seeding density of 104 cells/well, there was a linear response for up to three days post-seeding. The absorbance reading reached a plateau on day 4. Cells seeded at 5.0 × 103 cells/well cultured for 24 h before infection were used in the assay optimization pre-qualification studies. The intra-variation (intra-plate or well-well) in cytotoxicity of PVS-RIPO on U87-MG cells was determined by performing the assay in a 96-well plate and comparing three standard titration curves generated from different wells (Figure 5A). The plate-to-plate variability of the assay was assessed by looking at the 4-parameter curve fit analysis of the proliferation inhibition curves of samples run in different plates on the same day. MTS assays using three plates were performed on the same day under identical conditions by the same technician. There is an excellent plate-to-plate consistency as demonstrated in Figure 5B.
Figure 5. Assay variation.

A. Intra-plate variation of PVS-RIPO-induced cytopathogenicity. U87-MG cells were seeded into a 96-well plate at the 5 × 103/well and cultured for 24 h. One hundred micro liters of PVS-RIPO were added at different MOIs. After 48 h incubation, MTS was added (20 μ L/well) and incubated for 4 h. Twenty-five micro liters of 10% SDS were added and after 10 minutes the OD at 490 nm was read. Each curve represents the mean OD value ± SD of triplicate wells in different series within the same plate.
B. Inter-plate variability of PVS-RIPO-induced cytopathogenicity. U87-MG cells were seeded into three 96-well plates at 5 × 103/well and cultured for 24 h. The PVS-RIPO was added at different MOIs. After an additional 48 h, MTS was added (20 μL/well) and incubated for 4 h. Twenty-five micro liters of 10% SDS were added and after 10 minutes the OD at 490 nm was read. Each curve represents the mean read out for triplicate wells in different plates.
MTS based cell assays for several cytotoxic viral agents have been performed in 96-well plates by seeding cells into the wells and pre-incubating 16-20 h before infection to allow entry into growth phase (Heldt et al., 2006, Andersson, et al., 2005). However, the cell number for calculation of the MOI is not precisely known at the time of infection. In order to define the ED50 in terms of MOI (pfu/cell) and also to compare cytotoxicity of PVS-RIPO in the susceptible astrocytoma and non-responsive HEK293 neuronal cell line, we tested the cytotoxicity of the two cells lines following infection within 2 h after cell seeding in the 96 well plates. Cells were detached from the flasks and washed in culture medium and seeded at a cell density of 2-4 ×104/well in 100 μL followed by addition of 100 μL of virus within 2 h. Cells were incubated for 24 or 48 h after the addition of virus. Incubation for 24 h post-infection did not provide satisfactory cytotoxicity curves whereas satisfactory results were obtained at 48 h post-infection (data not shown). The differential cell killing assays using U87-MG and HEK293 cells under the defined conditions were repeated 8 times over a one year period using the same virus lot. The results are shown in Table 3. In all cases the U87-MG cells were effectively killed with a half maximal killing at <0.1 MOI. Estimation of ED50 or MTS50 values for PVS-RIPO in HEK293 cells was not feasible due to lack of curve-fit correlation up to an MOI of 100 pfu/cell. Also at high virus titers (> 1 MOI), significant fluctuations in the absorbance readings were detected with frequent zig-zag type dose response. Obviously, there is inherent assay variability attributable to both the cell growth assay and factors associated with viral infection and cytotoxicity. Under the defined conditions, the half maximal cell killing dose for U87-MG cell (C value) showed an inter-assay variation with an average of 0.022 ± 0.014 MOI (n=8). The ED50 values corresponding to virus infectivity titer causing 50% cytotoxicity was 0.056 ± 0.033 MOI (n = 8). MTS50 values averaged 4.49 ± 0.32 log10 unit with 7.2%CV. These results clearly demonstrate that an MTS assay using U87-MG and HEK293 cells can be used to evaluate cell-type specific killing by PVS-RIPO and could potentially be used to monitor lot to lot variation and stability of different virus preparations following manufacturing of clinical lots.
Table 3. Inter-day variations in PVS-RIPO-mediated CPE on U87-MG and HEK293 cells.
| Exp# | U87-MG | HEK293 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cell Passage # | A* | B | C (MOI) |
D | R2 | ED50 (MOI) |
MTS50 | Effect | Cell Passage # | |
| 1 | 3 | 1.546 | 0.587 | 0.031 | 0.434 | 0.991 | 0.117 | 4.07 | no effect <1MOI | 3 |
| 2 | 4 | 1.453 | 0.670 | 0.038 | 0.255 | 0.996 | 0.070 | 4.30 | no effect <1MOI | 4 |
| 3 | 31 | 1.760 | 0.848 | 0.047 | 0.073 | 0.990 | 0.054 | 4.42 | no effect <1MOI | 14 |
| 4 | 7 | 1.486 | 0.761 | 0.014 | 0.413 | 0.993 | 0.042 | 4.50 | no effect <1MOI | 7 |
| 5 | 10 | 1.237 | 0.919 | 0.022 | 0.470 | 0.987 | 0.094 | 4.17 | no effect <1MOI | 10 |
| 6 | 5 | 1.515 | 0.478 | 0.009 | 0.386 | 0.994 | 0.037 | 4.54 | no effect <1MOI | 4 |
| 7 | 5 | 1.334 | 0.718 | 0.015 | 0.240 | 0.996 | 0.027 | 4.72 | no effect <1MOI | 4 |
| 8 | 3 | 2.225 | 0.510 | 0.003 | 0.562 | 0.986 | 0.010 | 5.17 | no effect <1MOI | 3 |
| Average | 1.570 | 0.686 | 0.022 | 0.354 | 0.992 | 0.056 | 4.49 | no effect <1MOI | ||
| Std. Dev. | 0.286 | 0.146 | 0.014 | 0.146 | 0.004 | 0.033 | 0.32 | - | ||
| CV% | 18.25 | 21.33 | 63.28 | 41.09 | 0.36 | 59.09 | 7.21 | - | ||
A, B, C and D are the curve-fit parameters, R2 is the curve fit correlation.
ED50 refers to the virus infectious dose (MOI) that causes 50% cell killing.
C value refers to the virus infectious dose (MOI) that causes half maximal cell killing
MTS50 refers to the 50% infectious dose (log10 dilution factor/mL) at which the average infected well absorbance is 50% of the average uninfected cell absorbance
4. Discussion
A variety of naturally occurring or genetically engineered viruses are being tested for their potential as anti-cancer, or oncolytic agents. The basic overriding principle shared by these agents is selective virus replication and cytotoxicity in the intended target and resistance of normal bystander tissues. Therefore, it is imperative to investigate rigorously the factors dictating host cell attachment/entry, viral translation and genome replication in target cancerous cells and surrounding normal cells. Naturally, these analyses rely primarily on representative cell culture models. Development of a cell culture-based selective cytotoxicity assay for PVS-RIPO, a recombinant oncolytic poliovirus under consideration for clinical application against glioblastoma multiforme, is described in this paper.
PVS-RIPO, in which the cognate PV IRES is replaced with its counterpart from HRV2 (Gromeier et al., 1996), grows with wild-type kinetics in HeLa and malignant glioma cells (Gromeier et al., 2000) but is severely attenuated for growth in neuron-like cell lines commonly used to assess poliovirus attenuation phenotypes (Agol et al., 1989; La Monica and Racaniello, 1989; Gromeier et al., 1996; Merrill and Gromeier 2006). Growth deficiency in neuronal cell lines also correlates with elimination of pathogenicity in mice transgenic for the poliovirus receptor CD155 (Gromeier et al., 1996) and in nonhuman primates (Gromeier et al., 1999).
The main purpose of this work was to develop a reliable assay to measure cell type-specific replication deficits of PVS-RIPO in cells of neuronal derivation. Due to regulatory constraints prohibiting the use of wild type neurovirulent polioviruses, these studies are based on comparing PVS-RIPO to PV(1)S, itself an attenuated poliovirus strain. HEK293 cells, isolated from embryonic kidney cells transformed by type 5 adenovirus DNA (Graham et al., 1977) were recently reported to exhibit neuronal characteristics suggesting preferential transformation of neuroblastic precursors in human embryonic kidney (Shaw et al., 2002). These observations establish HEK293 cells as a tissue culture model superior to unstable neuroblastoma cell lines for the evaluation of poliovirus neurovirulent phenotypes. The attenuation phenotype of PVS-RIPO as determined by differential cytotoxicity in U87-MG versus HEK293 cells relative to PV1(S) was substantial. This assay using U87-MG astrocytoma and HEK293 cells could be adapted as a specific test for demonstration of cellular target specificity of PVS-RIPO preparations for pre-clinical and clinical studies.
The results presented here suggest that under optimized assay conditions the U87-MG cell line can be used to quantitatively measure cytotoxicity following infection with various strains of poliovirus including the recombinant PVS-RIPO. Earlier studies evaluating different susceptible tumor lines as well as HeLa cells in one-step cell growth assays demonstrated that at a MOI of 10 pfu/cell, infection as measured by viral productivity, peaks at 8-12 h (Dobrikova et al., 2003). In the MTS-based assay, significant cytotoxicity was detected at a MOI of <0.1 pfu/cell within 24 h after infection and cytotoxicity increased with incubation time. Under these experimental conditions, complete inhibition of cell growth was not achieved at a MOI of 5-10 pfu/cell at 24 h post infection whereas almost complete inhibition was achieved at a MOI <0.2 pfu/cell after 48 h post-infection. These results also suggest that cell growth characteristics at the time of infection, as well as during post-infection culture influence cytotoxicity.
With a defined and characterized reference lot of the virus, inter-lot variation could be monitored. It should be noted that lot to lot variation is subject to the variation limits of the viral infectivity assay used to define the virus titer (plaque or TCID50 assays) where the assay variations are much wider. The MTS-based assay described here can be further optimized and standardized to give an MTS50 index comparable to that defined for picornavirus by an earlier MTT-based assay (Andersson et al., 2005). The MTS50 assay in turn could be used as an alternate method for viral titer determination, which is usually determined by classic plaque assay or TCID50 assay. Cytotoxicity as determined by the MTS assay may be influenced by factors such as cell passage number, cell seeding density, time of post-infection period and other cellular factors that are not well defined. When cells from three different vials thawed at different times were passaged for different passage cycles and assayed on the same day it appeared that the passage number may influence the cytotoxicity. The longer passaged cells (passage 31) were more sensitive to cytotoxicity compared cells at passage 3 and 10. However, careful examination of all the data generated suggest that the observed difference could not be ascertained to passage number (Figure 2D). Inherent variations in bioassays based on cell proliferation or cell proliferation inhibition are well documented (Mire-Sluis et al., 1995; Meager, 2005). Such variations are more pronounced when activity is expressed in absolute units (C values of 4-parameter curve and ED50 values). For the cell proliferation based cytotoxicity assays, the relative activity compared to a well defined reference standard is preferred over absolute units for lot to lot comparison and stability monitoring. Apart from the variation anticipated for cell-based assay, the use of virus titer based on plaque assay in Vero cells adds a major variability factor associated with the inherent variation in the plaque assay. The cytotoxicity or other cell based assays for virus infectivity is more complex and the virus replication and cell growth related factors will contribute to the inherent assay variation when activity is expressed in absolute units such as half maximal dose response titer or ED50 values. Expression of cytotoxicity as MTS50 units is independent of virus infectivity titer based on plaque assay because MTS50 is defined in terms of virus dilution.
Assay optimization could be extended over the early clinical development phase to explore the effect on neuroblastoma cell lines and to explore the availability of other neuronal model cell lines. The optimization studies could be continued to further define the optimum cell passage number, infectious titer and post infection culture time with U87-MG and HEK293 cells. With such optimization studies, a cell culture based cytotoxicity assay using the MTS dye could be qualified and validated for use as a quantitative product release assay for measurement of virus titer and specificity for later stage clinical trials and commercial applications.
PVS-RIPO induced cytotoxicity as defined by C value, ED50 or MTS50 could potentially be developed to meet the requirements for a release test defining virus infectivity and as a tool for lot to lot comparison. Under identical conditions, the standard deviation of MTS50 value obtained from 8 runs using a reference lot over one year was less than 0.5 log10 Unit with 7.2%CV. Expression of cytotoxicity in MTS50 units have the same variation as ED50 though the standard deviation for inter-day estimates may appear lower because the values are expressed in log units. Half maximal cytotoxicity dose (C value of the four parameter curve) and 50% cytotoxicity dose (ED50) are expressed in virus infectivity titer determined by Vero cell infectivity. MTS50 on the other hand is a measure of the log dilution factor of the stock virus corresponding to 50% cytotoxicity. For a given Lot, the trend for ED50 variations are thus opposite of that observed for MTS50. An increase in ED50 results in a decrease in MTS50. However, the ED50 value from day to day assays showed slight differences with C value because PVS-RIPO did not show complete cell killing in some cases. Differential expression of CD155 could be one possibility for this difference.
The correlation between expression of CD155 as determined by FACS staining and Capture ELISA and cytotoxicity following infection with PVS-RIPO was studied in several different cell lines. U87-MG, H226 and M21 cells expressing the CD155 receptor were susceptible to PVS-RIPO cytotoxicity as expected. The T cell Jurkat and B lymphoma Daudi cells showed little or no expression of CD155 and no cytotoxicity was observed following PVS-RIPO infection. HEK293 was the only cell line expressing CD155 that was resistant to PVS-RIPO cytotoxicity. The lack of PVS-RIPO replication in HEK293 cells was therefore not related to a block in viral entry at the level of the receptor. Inhibition of PV-RIPO replication in neurons has been attributed to a translational repressor complex that is present in neuronal cells but is not detected in malignant cells (Merrill et al., 2006; Merrill and Gromeier 2006). The components of this complex have been detected in HEK293 cells (Merrill and Gromeier 2006) indicating that the lack of HEK 293 cell sensitivity to PVS-RIPO cell killing is most likely a function of IRES trans-acting factors that have been previously shown to influence viral propagation in a cell-type-specific manner. The intent of this work is to define the lot to lot consistency and stability of the virus preparations in terms of cytotoxicity in U87-MG cells and differential response on U87-MG and HEK293. The sensitivity of the assay for detection of the presence of neurovirulent virus in the PVS-RIPO preparation could be explored by carefully designed spiking experiments with neurovirulent strains. However, such investigations are difficult at this stage due to restrictions of the use of wild-type neurovirulent polioviruses in the facility.
The observed differences between the half maximal inhibition concentration (C-value of the 4-parameter curve fit) and the ED50 value (concentration required for 50% inhibition of cell proliferation) may indicate possible differences in the susceptibility of the cells to virus induced cytotoxicity. When there is a complete inhibition of cell proliferation at high viral dose, half maximal killing and ED50 values will be comparable. The observed variations could not be attributed to any single factor such as cell passage number or virus stability. Differential expression of CD155 could be a potential cause. The lack of PVS-RIPO induced cytotoxicity on Jurkat and Daudi cells with little or no CD155 expression supports the essential role of CD155. Similar types of variation are observed with PV(1)S induced cytotoxicity in U87-MG cells. Further studies are warranted to understand the cellular factors that contribute to the inter-day variations observed.
Acknowledgments
This project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400; and also supported [in part] through the NCI-RAID Program of the Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute, National Institutes of Health. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agol VI, Drozdov SG, Ivannikova TA, Kolesnikova MS, Korolev MB, Tolskaya EA. Restricted growth of attenuated poliovirus strains in cultured cells of a human neuroblastoma. J Virol. 1989;63:4034–4038. doi: 10.1128/jvi.63.9.4034-4038.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson P, Alm S, Edman K, Lindberg AM. A novel and rapid method to quantify cytolytic replications of picornavirues in cell culture. J Virol Methods. 2005;130:117–123. doi: 10.1016/j.jviromet.2005.06.016. [DOI] [PubMed] [Google Scholar]
- Batra R, Olsen J, Pickles R, Hoganson S, Boucher R. Transduction of non-small cell lung cancer cells by adenoviral and retroviral vectors. Am J Respir Cell Mol Biol. 1998;18:402–410. doi: 10.1165/ajrcmb.18.3.2784. [DOI] [PubMed] [Google Scholar]
- Campbell SA, Lin J, Dobrikova EY, Gromeier M. Genetic determinants of cell type-specific poliovirus propagation in HEK-293 Cells. J Virol. 2005;79:6281–6290. doi: 10.1128/JVI.79.10.6281-6290.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrikova EY, Florez P, Gromeier M. Structural determinants of insert retention of poliovirus expression vectors with recombinant IRES elements. Virology. 2003;311:2241–253. doi: 10.1016/s0042-6822(03)00191-0. [DOI] [PubMed] [Google Scholar]
- Dulbecco R. Endpoint method-measurement of the infectious titer of a viral sample. In: Dulbecco R, editor. Virology. Second. J.P. Lippincott; Philadelphia: 1988. pp. 22–25. [Google Scholar]
- Dulbecco R, Vogt M. Plaque formation and isolation of pure lines with polymyelitis viruses. J Exp Med. 1954;99:167–182. doi: 10.1084/jem.99.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauduchon J, Gouilleux F, Maillardm S, Marsaud V, Renoir J, Sola B. 4-Hydroxytamooxifen inhibits proliferation of multiple myeloma cells in vitro through down regulation of c-Myc, up-regulation of p27Kip1, and modulation of Bcl-2 family members. Clin Cancer Res. 2005;11:2345–2354. doi: 10.1158/1078-0432.CCR-04-1668. [DOI] [PubMed] [Google Scholar]
- Giridharan P, Somamsundaram ST, Perumal K, Vishwakarma RA, Karthikeyan NP, Velmurugan R, Balakrishnan A. Novel substituted methylenedioxy lignan suppresses proliferation of cancer cells by inhibiting telomerase and activation of c-myc and caspases leading to apoptosis. Br J Cancer. 2002;87:98–105. doi: 10.1038/sj.bjc.6600422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham FL, Smiley J, Russell WC, Nairn R. Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol. 1977;36:59–74. doi: 10.1099/0022-1317-36-1-59. [DOI] [PubMed] [Google Scholar]
- Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci USA. 1996;93:2370–2375. doi: 10.1073/pnas.93.6.2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromeier M, Bossert B, Arita M, Nomoto A, Wimmer E. Dual stem loops within the poliovirus internal ribosomal entry site control neurovirulence. J Virol. 1999;73:958–964. doi: 10.1128/jvi.73.2.958-964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gromeier M, Lachmann S, Rosenfeld M, Gutin PH, Wimmer E. Intergeneric poliovirus recombinants for the treatment of malignant glioma. Proc Natl Acad Sci USA. 2000;97:6803–6808. doi: 10.1073/pnas.97.12.6803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heese K, Nagai Y, Swada T. Induction of rat L-phosphoserine phosphatase by amyloid beta (1-42) is inhibited by interleukin-11. Neurosci Lett. 2000;288:37–40. doi: 10.1016/s0304-3940(00)01197-6. [DOI] [PubMed] [Google Scholar]
- Heldt CL, Hernandez R, Mudiganti U, Gurgel PV, Brown DT, Carbonell RG. A colorimetric assay for viral agents that produce cytopathic effects. J Virol Methods. 2006;135:56–65. doi: 10.1016/j.jviromet.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Hierholzer JC, Killington RA. Virus isolation and quantitation. In: Mahy BWJ, Kangro HO, editors. Virology Methods manual. Academic Press; San Diego: 1996. pp. 25–47. [Google Scholar]
- Hirota T, Irie K, Okamoto R, Ikeda W, Takai Y. Transcriptional activation of the mouse Necl-5/Tage4/PVR/CD155 gene by fibroblast growth factor or oncogenic Ras through the Raf-MEK-ERK-AP-1 pathway. Oncogene. 2005;24:2229–2235. doi: 10.1038/sj.onc.1208409. [DOI] [PubMed] [Google Scholar]
- La Monica N, Racaniello VR. Differences in replication of attenuated and neurovirulent polioviruses in human neuroblastoma cell line SH-SY5Y. J Virol. 1989;63:2357–2360. doi: 10.1128/jvi.63.5.2357-2360.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malecki A, Garrido R, Mattson MP, Henning B, Toborek M. 4-Hydroxynonenal induces oxidative stress and death of cultured spinal cord neurons. J Neurochem. 2000;74:2278–2282. doi: 10.1046/j.1471-4159.2000.0742278.x. [DOI] [PubMed] [Google Scholar]
- Meager A. Measurements of cytokines by bioassays: Theory and applications. Methods. 2005;38:237–252. doi: 10.1016/j.ymeth.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: Molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56:855–865. doi: 10.1016/0092-8674(89)90690-9. [DOI] [PubMed] [Google Scholar]
- Merrill MK, Bernhardt G, Sampson JH, Wikstrand CJ, Bigner DD, Gromeier M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuron-oncology. 2004;10:208–217. doi: 10.1215/S1152851703000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill MK, Dobrikova EY, Gromeier M. Cell-type-specific repression of internal ribosome entry site activity by double-stranded RNA-binding protein 76. J Virol. 2006;80:3147–3156. doi: 10.1128/JVI.80.7.3147-3156.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill MK, Gromeier M. The double-stranded RNA binding protein 76:NF45 heterodimer inhibits translation initiation at the rhinovirus type 2 internal ribosome entry site. J Virol. 2006;80:6936–6942. doi: 10.1128/JVI.00243-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mire-Sluis AR, Page L, Thorpe R. Quantitative cell line based bioassays for human cytokines. J Immunol Methods. 1995;187:191–199. doi: 10.1016/0022-1759(95)00220-1. [DOI] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Nix B, Wild D. Calibration curve-fitting. In: Wild D, editor. The Immunoassay Handbook. Nature Publishing Group; London: 2001. pp. 198–210. [Google Scholar]
- Ochiai H, Moore SA, Archer GE, et al. Treatment of intracerebral neoplasia and neoplastic meningitis with regional delivery of oncolytic recombinant poliovirus. Clin Cancer Res. 2004;10:4831–4838. doi: 10.1158/1078-0432.CCR-03-0694. [DOI] [PubMed] [Google Scholar]
- Ochiai H, Campbell SA, Archer GE, Chewning TA, Dragunsky E, Ivanov A, Gromeier M, Sampson JH. Targeted therapy for glioblastoma multiforme neoplastic meningitis with intrathecal delivery of an oncolytic recombinant poliovirus Clin. Cancer Res. 2006;12:1349–1354. doi: 10.1158/1078-0432.CCR-05-1595. [DOI] [PubMed] [Google Scholar]
- Shaw G, Morse S, Ararat M, Graham FL. Preferential transformation of human neuronal cells by human adenoviruses and the origin of HEK293 cells. FASEB J. 2002;16:869–871. doi: 10.1096/fj.01-0995fje. [DOI] [PubMed] [Google Scholar]
- Sloan KE, Stewart JK, Treloar AF, Matthews RT, Jay DG. CD155/PVR Enhances glioma cell dispersal by regulating adhesion signaling and focal adhesion dynamics. Cancer Res. 2005;65:10930–10937. doi: 10.1158/0008-5472.CAN-05-1890. [DOI] [PubMed] [Google Scholar]
- Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Gromeier M, Mueller S, Bernhardt G, Wimmer E. Expression of the human poliovirus receptor/CD155 gene is activated by sonic hedgehog. J Biol Chem. 2002;277:25697–25702. doi: 10.1074/jbc.M201378200. [DOI] [PubMed] [Google Scholar]
- Thompson KM, Tebbens RJ. Retrospective cost-effectiveness analyses for polio vaccination in the United States. Risk Anal. 2006;26:1423–1440. doi: 10.1111/j.1539-6924.2006.00831.x. [DOI] [PubMed] [Google Scholar]
- Tonello F, Seweso M, Marin O, Mock M, Montecuccp C. Screening inhibitors of anthrax lethal factor. Nature. 2002;418:386. doi: 10.1038/418386a. [DOI] [PubMed] [Google Scholar]
- Zhang C, Hazarika P, Ni X, Weidner D, Duvic M. Induction of apoptosis by bexarotene in cutaneous T-cell lymphoma cells: Relevance to mechanism of therapeutic action. Clin Cancer Res. 2002;8:1234–1240. [PubMed] [Google Scholar]