

Abstract
Cytochrome P450 2E1 (CYP2E1) is a microsomal enzyme that generates reactive oxygen species during its catalytic cycle. We previously found an important role for calcium in CYP2E1-potentiated injury in HepG2 cells. The possibility that CYP2E1 may oxidatively damage and inactivate the microsomal Ca2+-ATPase in intact liver cells was evaluated, in order to explain why calcium is elevated during CYP2E1 toxicity. Microsomes were isolated by differential centrifugation from two liver cells lines: E47 cells (HepG2 cells transfected with the pCI neo expression vector containing the human CYP2E1 cDNA, which overexpress active microsomal CYP2E1), and control C34 cells (HepG2 cells transfected with the pCI neo expression vector alone, which do not express significantly any cytochrome P450). The Ca2+-dependent ATPase activity was determined by measuring the accumulation of inorganic phosphate from ATP hydrolysis. CYP2E1 overexpression produced a 45% decrease in Ca2+-dependent ATPase activity (8.6 nmol Pi/min/mg protein in C34 microsomes vs 4.7 nmol Pi/min/mg prot in E47 microsomes). Saturation curves with Ca2+ or ATP showed that CYP2E1 overexpression produced a decrease in VMAX but did not affect the KM for either Ca2+ or ATP. The decrease in activity was not associated with a decrease in SERCA protein levels. The ATP-dependent microsomal calcium uptake was evaluated by fluorimetry using fluo-3 as the fluorogenic probe. Calcium uptake rate in E47 microsomes was 28% lower than in C34 microsomes. Treatment of E47 cells with 2 mM N-acetylcysteine prevented the decrease in microsomal Ca2+ ATPase found in E47 cells. These results suggest that CYP2E1 overexpression produces a decrease in microsomal Ca2+ ATPase activity in HepG2 cells mediated by reactive oxygen species. This may contribute to elevated cytosolic calcium and to CYP2E1-potentiated injury.
Keywords: CYP2E1, HepG2, calcium, SERCA, oxidative stress
1. Introduction
Calcium homeostasis is essential for cell survival, and changes in intracellular calcium are pivotal to signal transduction (Grover et al., 2003). In mammalian cells, a 10,000x concentration gradient across the plasma membrane is achieved by permanent active cytoplasmic Ca2+ extrusion, through the Na+/Ca2+ exchanger, by using the Na+ gradient generated by Na+/K+ ATPase, and is fine-tuned by the plasma membrane Ca2+-ATPase (PMCA) activity. In addition, most of the intracellular calcium is sequestered in the luminal side of the endoplasmic reticulum, by active pumping from the cytoplasm promoted by the sarcoendoplasmic reticulum Ca2+-ATPase (SERCA), creating another similar transmembrane gradient (Souza dos Santos et al., 2007).
Several reports have described that reactive oxygen species (ROS) and reactive nitrogen species (RNS) can inhibit SERCA, leading to alterations in calcium homeostasis. Oxidative damage to SERCA was reported in sarcoplasmic reticulum vesicles treated in vitro with Fe2+/H2O2/ascorbate (Castilho et al., 1996), hypoxanthine/xanthine oxidase (Barnes at al., 2000), hydrogen peroxide (Grover at al., 1997), and peroxynitrite (Gutierrez-Martin et al., 2004). Partial inactivation of SERCA caused by oxidative stress has been associated with several pathological conditions such as biological aging (Viner et al., 1999), obesity (Li et al., 2006), heart failure (Lokuta et al., 2005), and high intensity exercise (Matsunaga et al., 2003).
An active source of cellular ROS is cytochrome P450. The cytochrome P450 enzymes are a superfamily of hemeproteins that serve as terminal oxidases in the mixed function oxidase system for metabolizing various endogenous substrates such as steroids and fatty acids, and xenobiotics including drugs, toxins and carcinogens (Caro and Cederbaum, 2004). Oxygen activation by P450, necessary for the enzyme catalytic function, can also result in the production of ROS. Small amounts of the superoxide anion radical (O2•−) can be produced from decay of the oxygenated P450 complex, while hydrogen peroxide (H2O2) can form from either dismutation of O2•− or decay of the peroxy P450 complex (Puntarulo and Cederbaum, 1998). Among the different P450 isoforms, CYP2E1 has a high capacity to reduce dioxygen to reactive oxy radicals (Caro and Cederbaum, 2004).
In this work, we evaluated the possibility that CYP2E1-generated reactive oxygen species may inhibit microsomal Ca2+-ATPase activity in intact liver cells.
2. Materials and methods
2.1. Chemicals
Phosphate-buffered saline (PBS) was from Roche (Newark, NJ). G418 was from Invitrogen (Carlsbad, CA). Protein concentration was measured using the Bio-Rad DC protein assay (Hercules, CA). Most of the other chemicals used were from Sigma Chemical Co. (St Louis, MO). Antibodies for immunoblots were from Santa Cruz Biotechnology (Santa Cruz, CA).
2.2. Cell culture
Two human hepatoma HepG2 cell sublines described in Chen and Cederbaum (1998) were used as models in this study: 1) E47 cells, which constitutively express human CYP2E1, and 2) C34 cells, which are HepG2 cells transfected with the empty pCI vector. Both cell sublines were grown in minimal essential medium (MEM) containing 10% fetal bovine serum and 0.5 mg/ml G418 supplemented with 100 units/ml penicillin and 100 μg/ml streptomycin in a humidified atmosphere in 5% CO2 at 37°C. Cells were subcultured at a 1:5 ratio once a week. For the experiments, cells were plated at a density of 100,000 cells/ml and incubated for 7 days in MEM supplemented with 5% fetal bovine serum, 100 units/ml penicillin, and 100 μg/ml streptomycin (MEMexps). The 7 days in culture allowed formation of sufficient cells to isolate microsomes for the assays described below.
2.3. Microsomal preparation
Cells (2x107) on a 75 cm2 flask were rinsed twice with PBS, and then detached by scraping into 20 mL of microsomal buffer (0.32M sucrose, 20 mM HEPES-KOH, pH 7.0, containing 0.4 mM phenylmethylsulfonyl fluoride, leupeptin (0.8 μg/mL), and aprotinin (100 units/mL). The cells were homogenized at 4 °C using 20 up and down strokes of a rotating Potter-Elvehjem homogenizer. The homogenate was centrifuged at 11,000g for 20 min to spin down nuclei, mitochondria, and cellular debris. The supernatant from this step was centrifuged in an ultracentrifuge at 105,000g for 90 min at 4°C. The resulting pellet was resuspended in 100 mM Hepes buffer pH 7.4 using a Potter-Elvehjem homogenizer. Sonication was avoided as it depleted SERCA as evaluated by western blot, and decreased microsomal Ca2+-ATPase activity (data not shown).
2.4. Ca2+-ATPase assays
The microsomal Ca2+-ATPase activity was determined by measuring the inorganic phosphate (Pi) released from ATP, as a colored complex of malachite green-phosphomolybdenum (Chan et al., 1986). The microsomal Ca2+-ATPase activity was assayed in a medium containing 20-100 μg/ml of protein with 200 μM EGTA, varying concentrations of CaCl2 (to give up to 50 μM free Ca2+), 1 mM ATP, 5 mM sodium azide, 2.5 mM ruthenium red, 100 mM KCl, and 50 mM imidazole-HCl, pH 6.8 at 37 °C. The concentration of free Ca2+ was estimated using the program WEBMAXCLITE V1.15 (Chris Patton, Stanford University, http://www.stanford.edu/~cpatton/webmaxc/webmaxclite115.htm, October 2006). The reaction was started with the addition of ATP and samples were incubated for 15 min or as indicated in the legends. For the colorimetric determination of released Pi, 500 μL of the assay volume was mixed with 2 mL of the mixed reagent. The composition of the mixed reagent was: 2 parts of malachite green (0.812g malachite green/1L water) + 1 part of polyvinyl alcohol (11.6g polyvinyl alcohol/500 mL water) + 1 part ammonium molybdate (28.6g ammonium molybdate/500 mL 6N HCl) + 2 parts of water. The absorbance of the color complex was measured at 630 nm against a mixed reagent blank. A calibration curve in our conditions, using potassium phosphate as standard, produced an absortivity of 2600 M-1cm-1.
2.5. Calcium uptake experiments
Calcium uptake was measured as described by Mezna and Michelangeli (1996). Typically, 300 μg of microsomes were suspended in 2 mL of 40 mM Tris/phosphate buffer pH 7.2, at 37 °C in the presence of 10 mM phosphocreatine, 10 μg/mL creatine kinase, and 1.25 mM fluo-3. The mixture was incubated at 37 °C, and Ca2+ uptake was initiated by the addition of 1.5 mM MgATP. Ca2+ transport across the microsomal membrane was followed by monitoring the fluorescence change of fluo-3 in a BioRad fluorescence microplate reader at 505 nm excitiation/525 nm emission, under constant shaking.
2.6. Western blots
After washing the cells with PBS, cells were incubated on ice for 15 min in extraction buffer [250 mM Tris-HCl, pH 8.0, 150 mM NaCl, and 1% IGEPAL CA-630 (Sigma Chemical Co.)] containing 1 mM sodium orthovanadate, 50 μg/ml aprotinin, and 0.1 mg/ml phenylmethylsulfonyl fluoride. Lysates were centrifuged at 15,000 rpm at 4°C to remove insoluble material, and the protein concentration in whole-cell lysates was measured using the Bio-Rad DC protein assay kit. Protein samples (30 μg) were separated by SDS-polyacrylamide gel electrophoresis, transferred to nitrocellulose membranes, and incubated with primary and secondary antibodies. Antibodies were used at the following dilutions: anti-β-actin (1:10,000), anti-SERCA 1, 2, 3 (1:1000), and anti-CYP2E1 (1:1000).
2.7. Intracellular ROS Measurement
Intracellular ROS were monitored with 2’,7’-dichlorofluorescein diacetate (DCFH-DA) as the probe. 10,000 E47 cells were plated in 10-mm Petri dishes, and after 7 days of incubation in the presence or absence of 2 mM NAC, the medium was replaced with fetal bovine serum-free MEMexps supplemented with 5 μM DCFH-DA. The cells were incubated for 30 min at 37 °C, and after this incubation, they were washed with PBS, trypsinized, and resuspended in 1 ml of PBS. Fluorescence was immediately read in a fluorescence spectrometer at 490 nm for excitation and 525 nm for emission. Background readings from cells incubated without DCFDA were substracted. Results are expressed as arbitrary units of fluorescence.
2.8. Statistics
Data are expressed as mean ± S.E.M. from three to four independent experiments run in duplicate. One-way analysis of variance with subsequent post hoc comparisons by Scheffe was performed. A p < 0.05 was considered as statistically significant.
3. Results
3.1. Microsomal Ca2+-ATPase activity in CYP2E1 overexpressing cells
Previous work has shown that E47 cells overexpress active microsomal CYP2E1 and generate higher levels of reactive oxygen species with respect to control C34 cells, which do not express significantly any cytochrome P450 (Caro and Cederbaum, 2004). In this work, we measured the microsomal Ca2+ ATPase activity in E47 versus C34 cells. The selected assay for microsomal Ca2+-ATPase activity measures inorganic phosphate (Pi) released by the ATPase activity in one step, without removing proteins from the solution (one-step protocol). The removal of proteins using trichloroacetic acid before the determination of Pi (two-step protocol) caused erratic, low absorbance readings (data not shown). It has been suggested that removal of proteins in this assay can cause other reagents to precipitate and that the subsequent removal of the precipitate by centrifugation can reduce color intensity thereby causing erroneous absorbance readings (Chan et al., 1986). In the one-step protocol described in Materials and Methods, the polyvinyl alcohol in the mixed reagent prevents the precipitation of proteins, and therefore was used in this work. The Ca2+-ATPase activity was linear with incubation time over the time range studied, with squares of correlation coefficients, r2 of 0.9894 for C34- and 0.9668 for E47-derived microsomes (Fig 1A). The Ca2+-ATPase activity was linear with protein concentration over the concentration range studied, with squares of correlation coefficients, r2 of 0.9951 for C34- and 0.9956 for E47-derived microsomes (Fig 1B). The microsomal Ca2+-ATPase activity (i.e. the slope of the straight lines in Fig 1A and B) was lower (-45%) in E47-derived microsomes with respect to C34-derived microsomes (4.7 nmol Pi/min/mg protein in E47 microsomes versus 8.6 nmol Pi/min/mg protein in C34 microsomes). Microsomal Ca2+-ATPase activity in C34 cells was inhibited by thapsigargin, a specific SERCA inhibitor, with an IC50 of 25 μM (data not shown). To evaluate if the observed decrease in microsomal Ca2+-ATPase activity in E47 vs C34 cells was due to a decrease in levels of SERCA protein, we assesed SERCA protein levels. SERCA protein levels in C34 and E47 cells microsomes were determined by western blot after loading 30 μg protein. Using an antibody against SERCA 1, 2 and 3, a single band was detected at 107 kDa, compatible with the liver-expressed endoplasmic reticulum Ca2+-ATPase (Fig 2A) (Jong et al., 1990; Bennett and Williams, 1993). The intensity of the band (normalized against Gβ, a microsomal loading control) was the same in C34 and E47 derived microsomes (Fig 2A). Membranes were stripped and reprobed with an anti-CYP2E1 antibody; immunoreactivity was only detected in E47 microsomes, confirming that E47 cells overexpress microsomal CYP2E1 (Fig 2B).
Figure 1.
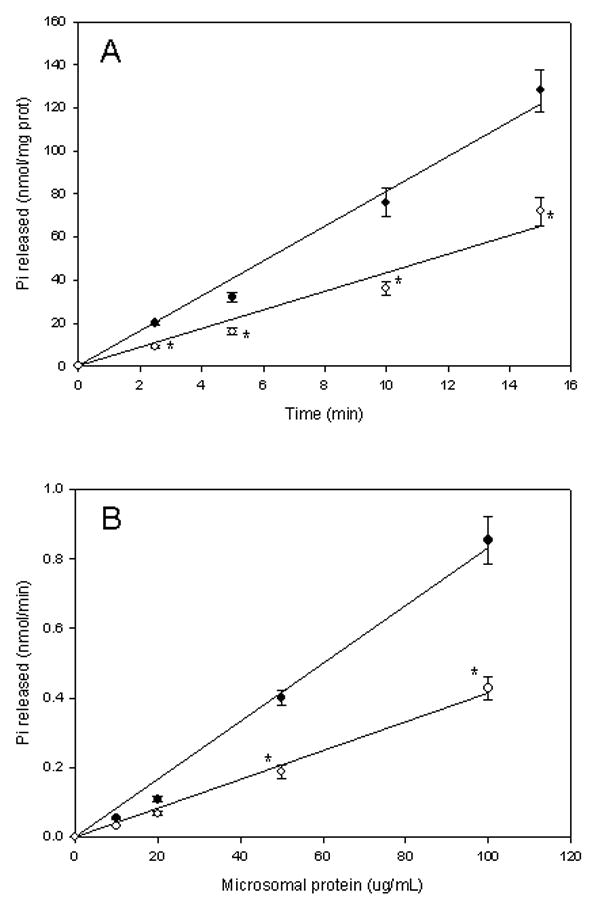
Microsomal Ca2+-ATPase activity in HepG2 cells. (A) Time curve. Microsomes (0.1 mg protein/mL derived from C34 (black circles) or E47 cells (white circles) were incubated in the presence of 1 mM ATP, 20 μM free Ca2+ for variable periods (up to 15 min). The release of inorganic phosphate was measured following Materials and Methods; (B) Microsomal protein concentration curve. Microsomes (variable protein concentration up to 0.1 mg/mL) derived from C34 (black circles) or E47 cells (white circles) were incubated in the presence of 1 mM ATP, 20 uM free Ca2+ for 15 min. The release of inorganic phosphate was measured as described in Materials and Methods.
* significantly different (p<0.05, ANOVA) with respect to control C34 cells.
Figure 2.
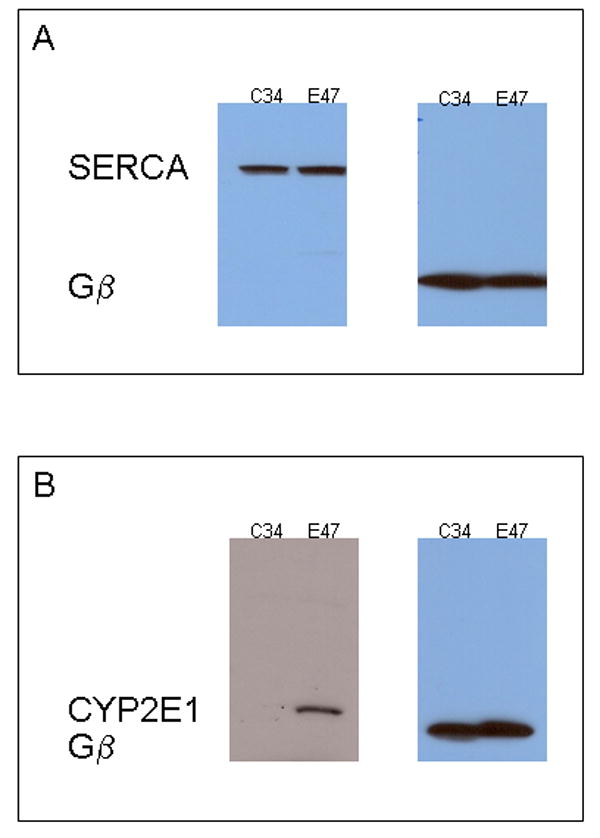
SERCA and CYP2E1 expression in C34 and E47 cells. Microsomes derived from C34 or E47 cells were loaded (30 μg prot) in a SDS-polyacrylamide gel, and western-blotted using anti-SERCA 1,2,3 (A) or anti-CYP2E1 antibodies (B). Protein Gβ was used as microsomal loading control.
3.2. Ca2+ and ATP saturation curves
In the absence of added calcium, a low ATPase activity was detected in both C34 and E47 microsomes; no significant difference in this activity was detected between C34 and E47 microsomes (Fig 3A). Increasing the concentration of free Ca2+ produced an increase in ATPase activity in both C34 and E47 cells (Fig 3A). Differences in the Km for Ca2+ in C34 and E47 microsomes were non-statistically significant (3.7 μM and 3.9 μM for C34 and E47 cells, respectively). However, Vmax was significantly lower in E47 with respect to C34 microsomes (4.9 nmol/min/mg prot vs 8.8 nmol/min/mg prot, respectively) (Fig 3C). Saturation curves for ATP showed a similar trend: C34 and E47 microsomes showed non-statistical differences in the Km for ATP (1.3 mM and 1.4 mM for C34 and E47 cells, respectively), but lower Vmax in E47 cells with respect to C34 cells (4.6 nmol/min/mg prot vs 10.8 nmol/min/mg prot, respectively), in the Ca2+-dependent ATPase assay (Figs 3B and D).
Figure 3.
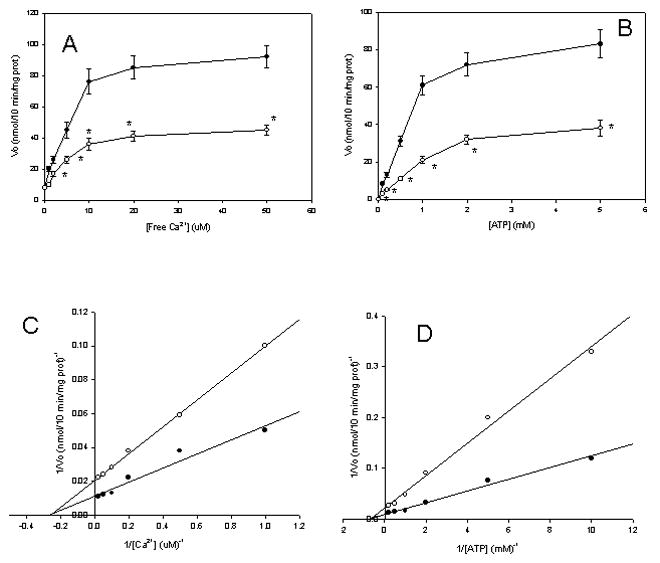
Microsomal Ca2+-ATPase saturation curves. (A and C): Microsomes (0.1 mg protein/mL derived from C34 (black circles) or E47 cells (white circles) were incubated in the presence of 1 mM ATP, variable free Ca2+ concentration (0-50 μM), for 10 min. The release of inorganic phosphate was measured as described in Materials and Methods; (B and D): Microsomes (0.1 mg protein/mL derived from C34 (black circles) or E47 cells (white circles) were incubated in the presence of 50 μM free Ca2+, variable ATP concentration (0-5 mM), for 10 min. The release of inorganic phosphate was measured as described in Materials and Methods.
* Significantly different (p<0.05, ANOVA) with respect to control C34 cells.
3.3. Microsomal calcium incorporation
We evaluated if the inhibition of the Ca2+-dependent ATPase activity in E47 cells is associated with a decrease in microsomal calcium uptake. Calcium uptake was undetectable in the absence of ATP (Fig 4). Addition of ATP induced a decrease in extraluminal calcium, which was linear with incubation time (up to 14 min). The decline in extracellular calcium was significantly lower (-28%) in E47 cells with respect to C34 cells.
Figure 4.
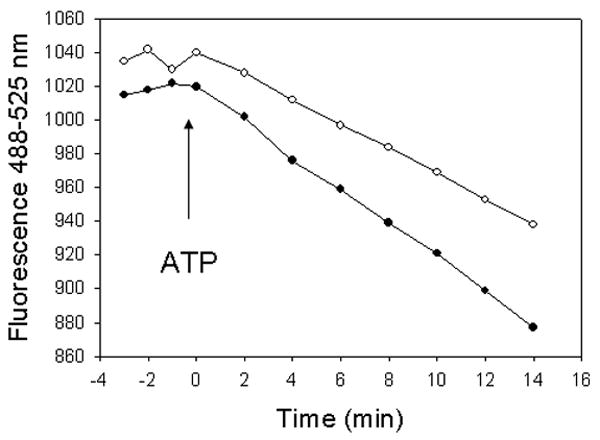
Microsomal calcium uptake experiments. Calcium uptake was initiated with MgATP (arrow), in a system containing cell microsomes (C34-derived, black circles or E47-derived, white circles), an ATP regenerating system (phosphocreatine/creatine kinase) and fluo3 (membrane impermeable). The reaction was followed in a microplate reader with appropriate fluorescence filters under constant shaking.
3.4. Effect of antioxidants on microsomal Ca2+ ATPase
If the decrease in microsomal Ca2+ ATPase activity in E47 cells is associated with increased oxidative stress in these cells, then this decrease should be prevented by antioxidants. We incubated the cells for the 7 day culture period after subculturing the cells with 2 mM N-acetylcysteine (an antioxidant per se, and a precursor of glutathione, the most abundant water soluble antioxidant in HepG2 cells), which was used at a concentration which decreased ROS levels in HepG2 cells (Gong and Cederbaum, 2006). This concentration did not produce any toxicity over the 7 day culture period (data not shown). NAC partially prevented the decrease in microsomal Ca2+ ATPase observed in E47 cells, while having no effect in C34 cells (Fig 5A). The level of reactive oxygen species was assessed by measuring the oxidation of DCFH-DA as described in Materials and Methods. NAC-treated E47 cells showed a lower dichlorofluorescein cellular fluorescence than non-treated E47 cells (140 relative units versus 251 relative units, respectively), suggesting decreased ROS levels in NAC-treated E47 cells in our experimental conditions. We incubated the cells for the 7 day culture period after subculturing the cells with 0.2 mM vitamin E (equivalent to 0.095 IU/mL), a concentration which we previously showed to inhibit arachidonic acid-induced lipid peroxidation (Caro and Cederbaum, 2002). Vitamin E did not show any significant effect on microsomal Ca2+ ATPase in C34 or E47 cells (Fig 5B).
Figure 5.
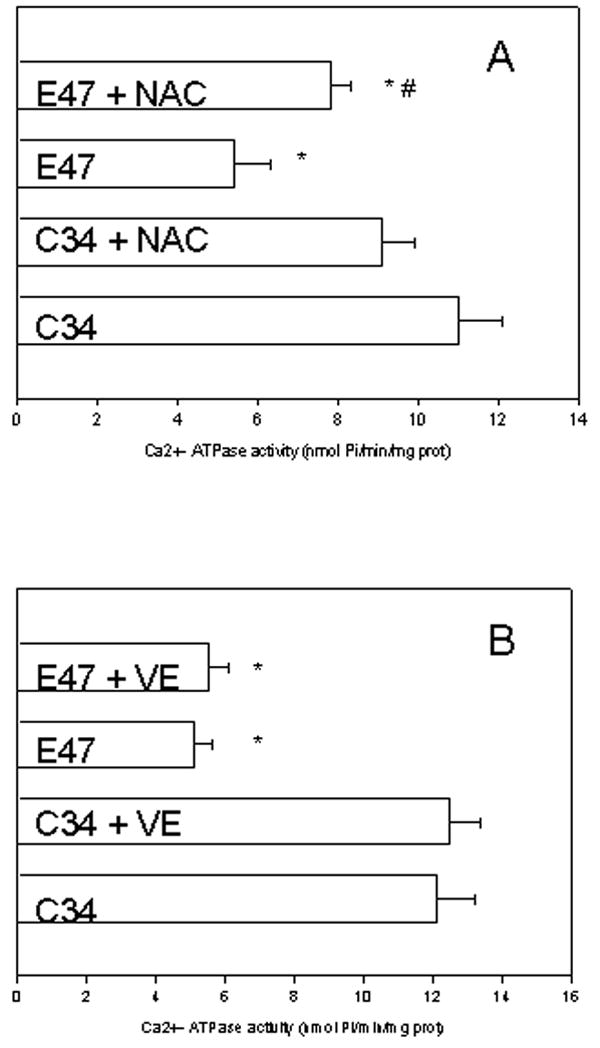
Effect of antioxidants on microsomal Ca2+-ATPase activity. C34 and E47 cells were incubated for 7 days with 2 mM NAC (A) or 0.2 mM vitamin E (VE) (B), or solvent controls (PBS or ethanol, respectively). Microsomes were isolated from treated cells, and the Ca2+-dependent ATPase activity was determined as described in Materials and Methods.
*significantly different (p<0.05) with respect to untreated C34 cells.
#significantly different (p<0.05) with respect to untreated E47 cells.
4. Discussion
Microsomal ATPase activity in HepG2 cells showed two components: 1) a Ca2+-independent activity (around 10% of the total), which did not show significant differences between C34 and E47 cells; and 2) a Ca2+-dependent activity (around 90% of the total), which showed a significant 55% decrease in E47 with respect to C34 cells. This suggests that CYP2E1 overexpression inhibits microsomal Ca2+-dependent ATPase activity in intact HepG2 cells. The ATPase activity of rat liver microsomes also showed Ca2+ dependent and independent components, the Ca2+-dependent component being involved in Ca2+ pumping across the microsomal membrane (Dawson and Fulton, 1983). In our model, ATP-dependent microsomal Ca2+ uptake was significantly lower in E47 cells with respect to control C34 cells (Fig 4), suggesting that the decreased Ca2+-dependent ATPase activity in CYP2E1 overexpressing cells is associated with decreased calcium pumping activity across the microsomal membrane.
Several possibilities may explain the loss of Ca2+-ATPase activity with oxidation: (i) sensitive cysteine sulfhydryl groups, and/or other critical amino acids (such as met, lys, tyr and others) of the Ca2+-ATPase pump may be oxidized; (ii) membrane lipids may be oxidized and may indirectly oxidize the Ca2+-ATPase pump, or lipid oxidation products may induce a structural change in the lipid bilayer which indirectly inhibits the Ca2+-ATPase pump activity; (iii) oxidation-related protein fragmentation or protein cross-linking may inhibit the activity of the pump. A review of the literature shows that the mechanism for oxidative inhibition of Ca2+-ATPase activity depends on the model system under study (enzyme isoform, enzymatic source, type of oxidants, oxidant exposure). For example, AAPH-derived radicals (peroxyl, alkoxyl) decreased SERCA activity in rabbit muscle SR vesicles, associated with the formation of intermolecular bityrosine cross-links, but not with lipid peroxidation, protein fragmentation, or oxidation of sulfhydryl groups (Viner et al., 1997). Fe2+/ascorbate/H2O2 induced SERCA fragmentation and inhibition; inhibition of enzymatic activity was not associated with lipid peroxidation or oxidation of sulfhydryl groups (Castilho et al., 1996). Peroxynitrite inactivated SR membranes by oxidation of thiol groups and tyrosine nitration (Gutierrez-Martin et al., 2004). Hydrogen peroxide and peroxydisulfate inhibited Ca2+-ATPase in SR microsomes by oxidation of sulfhydryl groups, independent of lipid oxidation or protein crosslinking (Scherer and Deamer, 1986). High intensity exercise produced SERCA inactivation associated to oxidation of amino acids other than cysteine (Matsunaga et al., 2003). In our model of cellular CYP2E1 overexpression, inhibition of microsomal Ca2+ ATPase activity was not associated with a decrease in SERCA levels, or SERCA protein fragmentation (Fig 4). The inhibition of microsomal Ca2+ ATPase in CYP2E1 overexpressing cells was significantly prevented by N-acetylcysteine which also decreased intracellular ROS levels, but not by vitamin E. This result suggests that the CYP2E1-dependent effects on Ca2+-ATPase activities are not mediated by lipid peroxidation byproducts, but probably by other reactive oxygen species. Oxidative damage of the microsomal Ca2+ ATPase activity was associated with changes in Vmax but not in Km, suggesting that the apparent affinity of the enzyme for calcium and ATP remains intact after CYP2E1 overexpression. The exact nature of the CYP2E1-dependent oxidative damage on microsomal Ca2+-ATPase will be evaluated in future experiments.
Several authors have previously shown that SERCA 2b (the isoform present in liver) is particularly susceptible to oxidative stress (Grover et al., 2003) with respect to other ATPases; our data showing the differential sensitivity of the Ca2+-dependent versus Ca2+ independent ATPase towards CYP2E1 overexpression suggests that the Ca2+ dependent ATPase activity in HepG2 cells is more sensitive to oxidative stress than the Ca2+-independent ATPase.
The Ca2+-ATPase of sarcoplasmic or endoplasmic reticulum is the major active calcium transport protein responsible for the maintenance of normal intracellular calcium levels in a variety of cell types. Hence the potential oxidative damage of these Ca2+-ATPases by reactive oxygen species may contribute to the increased levels of intracellular calcium that lead to cellular damage. In previous work, CYP2E1 overexpression has been associated with changes in calcium metabolism, mainly increase in cytosolic calcium (Caro and Cederbaum 2002; Caro and Cederbaum 2007). Arachidonic acid, or arachidonic acid + iron treatment of CYP2E1 overexpressing cells induced an increase in cytosolic calcium, associated with calpain and/or phospholipase A2 activation, and partially linked to increased calcium influx from intracellular stores (Caro and Cederbaum 2002; Caro and Cedebaum 2003; Caro and Cederbaum 2007). Preventing the increase in cytosolic calcium or the activities of calcium-stimulated hydrolases such as calpain and phospholipase A2 prevented the CYP2E1-dependent toxicity to the cells. SERCA may be a target for CYP2E1-generated ROS as shown in this report. A decreased Ca2+-ATPase activity in CYP2E1 expressing cells may contribute to changes in calcium homeostasis in conditions where CYP2E1 is induced.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barnes KA, Samson SE, Grover AK. Sarco/endoplasmic reticulum Ca2+-pump isoform SERCA3a is more resistant to superoxide damage than SERCA2b. Mol Cell Biochem. 2000;203:17–21. doi: 10.1023/a:1007053802481. [DOI] [PubMed] [Google Scholar]
- Bennett AM, Williams GM. Alteration of rat liver endoplasmic reticulum Ca2+-ATPase thiol integrity by ciprofibrate, a peroxisome proliferator. Biochem Pharmacol. 1993;45:2093–2098. doi: 10.1016/0006-2952(93)90021-n. [DOI] [PubMed] [Google Scholar]
- Caro AA, Cederbaum AI. Role of calcium and calcium-activated proteases in CYP2E1-dependent toxicity in HEPG2 cells. J Biol Chem. 2002;277:104–113. doi: 10.1074/jbc.M107864200. [DOI] [PubMed] [Google Scholar]
- Caro AA, Cederbaum AI. Role of phospholipase A2 activation and calcium in CYP2E1-dependent toxicity in HepG2 cells. J Biol Chem. 2003;278:33866–33877. doi: 10.1074/jbc.M300408200. [DOI] [PubMed] [Google Scholar]
- Caro AA, Cederbaum AI. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- Caro AA, Cederbaum AI. Role of intracellular calcium and phospholipase A2 in arachidonic acid-induced toxicity in liver cells overexpressing CYP2E1. Arch Biochem Biophys. 2007;457:252–263. doi: 10.1016/j.abb.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho RF, Carvalho-Alves PC, Vercesi AE, Ferreira ST. Oxidative damage to sarcoplasmic reticulum Ca2+-pump induced by Fe2+/H2O2/ascorbate is not mediated by lipid peroxidation or thiol oxidation and leads to protein fragmentation. Mol Cell Biochem. 1996;159:105–114. doi: 10.1007/BF00420912. [DOI] [PubMed] [Google Scholar]
- Chan KM, Delfert D, Junger KD. A direct colorimetric assay for Ca2+- stimulated ATPase activity. Anal Biochem. 1986;157:375–380. doi: 10.1016/0003-2697(86)90640-8. [DOI] [PubMed] [Google Scholar]
- Chen Q, Cederbaum AI. Cytotoxicity and apoptosis produced by cytochrome P450 2E1 in Hep G2 cells. Mol Pharmacol. 1998;53:638–648. doi: 10.1124/mol.53.4.638. [DOI] [PubMed] [Google Scholar]
- Dawson AP, Fulton DV. Some properties of the Ca2+-stimulated ATPase of a rat liver microsomal fraction. Biochem J. 1983;210:405–410. doi: 10.1042/bj2100405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong P, Cederbaum AI. Nrf2 is increased by CYP2E1 in rodent liver and HepG2 cells and protects against oxidative stress caused by CYP2E1. Hepatology. 2006;43:144–153. doi: 10.1002/hep.21004. [DOI] [PubMed] [Google Scholar]
- Grover AK, Kwan CY, Samson SE. Effect of peroxynitrite on sarco/endoplasmic reticulum Ca2+ pump isoforms SERCA2b and SERCA3a. Am J Physiol Cell Physiol. 2003;285:1537–1543. doi: 10.1152/ajpcell.00299.2003. [DOI] [PubMed] [Google Scholar]
- Grover AK, Samsom SE, Misquitta CM. Sarco(endo)plasmic reticulum Ca2+ pump isoform SERCA3 is more resistant than SERCA2b to peroxide. Am J Physiol. 1997;273:C420–C425. doi: 10.1152/ajpcell.1997.273.2.C420. [DOI] [PubMed] [Google Scholar]
- Gutierrez-Martin Y, Martin-Romero FJ, Inesta-Vaquera FA, Gutierrez-Merino C, Henao F. Modulation of sarcoplasmic reticulum Ca2+-ATPase by chronic and acute exposure to peroxynitrite. Eur J Biochem. 2004;271:2647–2657. doi: 10.1111/j.1432-1033.2004.04193.x. [DOI] [PubMed] [Google Scholar]
- Jong YJ, Sheldon A, Zhang GH, Kraus-Friedmann N. Purification of the microsomal Ca2+-ATPase from rat liver. J Membr Biol. 1990;118:49–53. doi: 10.1007/BF01872203. [DOI] [PubMed] [Google Scholar]
- Li SY, Yang X, Ceylan-Isik AF, Du M, Sreejayan N, Ren J. Cardiac contractile dysfunction in Lep/Lep obesity is accompanied by NADPH oxidase activation, oxidative modification of sarco(endo)plasmic reticulum Ca2+-ATPase and myosin heavy chain isozyme switch. Diabetologia. 2006;49:1434–1446. doi: 10.1007/s00125-006-0229-0. [DOI] [PubMed] [Google Scholar]
- Lokuta AJ, Maertz NA, Meethal SV, Potter KT, Kamp TJ, Valdivia HH, Haworth RA. Increased nitration of sarcoplasmic reticulum Ca2+-ATPase in human heart failure. Circulation. 2005;111:988–995. doi: 10.1161/01.CIR.0000156461.81529.D7. [DOI] [PubMed] [Google Scholar]
- Matsunaga S, Inashima S, Yamada T, Watanabe H, Hazama T, Wada M. Oxidation of sarcoplasmic reticulum Ca2+-ATPase induced by high-intensity exercise. Pflugers Archiv- Eur J Physiol. 2003;446:394–399. doi: 10.1007/s00424-003-1040-0. [DOI] [PubMed] [Google Scholar]
- Mezna M, Michelangeli F. The effects of inositol 1,4,5-trisphosphate (InsP3) analogues on the transient kinetics of Ca2+ release from cerebellar microsomes. J Biol Chem. 1996;271:31818–31823. doi: 10.1074/jbc.271.50.31818. [DOI] [PubMed] [Google Scholar]
- Puntarulo S, Cederbaum AI. Production of reactive oxygen species by microsomes enriched in specific human cytochrome P450 enzymes. Free Radic Biol Med. 1998;24:1324–1330. doi: 10.1016/s0891-5849(97)00463-2. [DOI] [PubMed] [Google Scholar]
- Scherer NM, Deamer DW. Oxidative stress impairs the function of sarcoplasmic reticulum by oxidation of sulfhydryl groups in the Ca2+-ATPase. Arch Biochem Biophys. 1986;246:589–601. doi: 10.1016/0003-9861(86)90314-0. [DOI] [PubMed] [Google Scholar]
- Souza dos Santos P, Saraiva DF, Da Costa DCF, Scofano HM, De Carvalho-Alves PC. Trifluoperazine protects brain plasma membrane Ca2+-ATPase from oxidative damaging. Exp Brain Res. 2007;177:347–357. doi: 10.1007/s00221-006-0678-1. [DOI] [PubMed] [Google Scholar]
- Viner RI, Ferrington DA, Williams TD, Bigelow DJ, Schoneich C. Protein modification during biological aging: selective tyrosine nitration of the SERCA2a isoform of the sarcoplasmic reticulum Ca2+-ATPase in skeletal muscle. Biochem J. 1999;340:657–659. [PMC free article] [PubMed] [Google Scholar]
- Viner RI, Krainev AG, Williams TD, Schoneich C, Bigelow DJ. Identification of oxidation-sensitive peptides within the cytoplasmic domain of the sarcoplasmic reticulum Ca2+-ATPase. Biochemistry. 1997;36:7706–7716. doi: 10.1021/bi970058z. [DOI] [PubMed] [Google Scholar]