

Abstract
Given that the integration of human papillomavirus type 16 (HPV16) into the host genome occurs preferentially with the disruption of the E2 gene, a ratio of E2 to E7 gene copies is often used as a marker for integration. It is largely undetermined, however, whether ratio estimates are affected by HPV intratypic variations. We assembled four plasmid constructs, each containing a DNA fragment from an HPV16 European, Asian-American, African-1, or African-2 variant. These constructs and nine cervical swab samples were assayed by real-time PCR with two primer-probe sets for each gene: a specific set, fully complementary to the HPV16 prototype, and a degenerate set, incorporating degenerate bases at positions where nucleotides differed among the variants. The ratio of E2 to E7 gene copies for the European variant construct was close to 1, no matter which sets of primers and probes were used. While the ratios for the African-1 and Asian-American variant constructs remained close to 1 with the degenerate sets of primers and probes, the ratios were 0.36 and 2.57, respectively, with the specific sets of primers and probes. In addition, a nucleotide alteration at the position immediately following the 3′ end of the E2 forward primer binding site was found to be responsible for an underestimation of E2 gene copies for the African-2 variant construct. Similar patterns were found in nine cervical samples. In conclusion, mismatches between the primers and probes and their targets due to HPV16 intratypic variations would introduce errors in testing for integration; this situation can be sufficiently ameliorated by incorporating degenerate bases into the primers and probes.
Infection with oncogenic human papillomavirus (HPV) is necessary but insufficient to cause invasive cervical cancer, with additional virus-host interactions needed to lead to the malignant phenotype (7, 20, 30). The integration of a high-risk HPV genome into the host chromosome is thought to be a key event in cervical carcinogenesis (23), as it may result in increased stability of HPV E6/E7 mRNA (16), augmented viral immortalization capacity (25), a high level of chromosomal instability (24), and specific alterations of cellular gene expression (1). Evidence from a study in vitro (15) indicates that cells with integrated, compared to episomal, HPV type 16 (HPV16) DNA have a selective growth advantage. Because the integration of the HPV genome occurs preferentially with the disruption of the coding sequence for the E2 protein, a ratio of E2 to E7 (or E6) gene copy numbers determined by real-time PCR has been commonly used as a marker for integration in epidemiological and clinical investigations (2, 3, 11, 13, 14, 21, 22, 26, 28, 34). Theoretically, the ratio ranges from 0 (completely integrated) to 1 (episomal only) if the integration occurs within the E2 target region; values between 0 and 1 reflect a mixed status.
Previous studies of HPV DNA integration have focused mainly on HPV16, the type that confers the highest risk of cervical cancer and also the type most commonly detected in women with normal cervical cytology (5). The reported rates of HPV16 integration (including cases of mixed integrated and episomal forms) vary from 8 to 100% in women without detectable cervical lesions and from 36 to 100% in those with high-grade cervical intraepithelial neoplasia (2, 3, 6, 9-11, 13, 14, 18, 29). Such a wide range may be explained in part by differences in study populations and/or issues in integration detection. Sequence analyses of various regions of the HPV16 genome have demonstrated a variety of naturally occurring variants. These variants are phylogenetically classified and named according to their geographic relatedness, as follows: European (E), Asian (As), Asian-American (AA), African-1 (Af1), and African-2 (Af2) (8, 33). In view of the fact that nucleotide alterations are common in the regions targeted by many previously reported primers and probes, the findings from these studies may be affected by the intragenomic diversity of the virus.
To address this issue, we assembled a set of plasmid constructs, each containing a DNA fragment derived from the HPV16 E, AA, Af1, or Af2 variant. These constructs and a set of cervical swab samples were assayed by real-time PCR to examine whether, and to what extent, testing for integration by using ratios of E2 to E7 gene copies would be affected by HPV16 intratypic variations.
MATERIALS AND METHODS
Plasmid construction.
PCR products from the HPV16 genome were generated from three cervical swab samples known to be positive for Af1, Af2, and AA variants. The amplification of the entire E2 and E7 regions was carried out using a LongRange PCR kit (Qiagen, Valentia, CA) with a pair of primers (forward primer [nucleotide positions 419 to 443], 5′-CGGAATTCTGTCAAAAGCCACTGTGTCCTGAAG, and reverse primer [nucleotide positions 3891 to 3866], 5′-CGGGATCCGCACGCCAGTAATGTTGTGGATGTA) which contained EcoRI and BamHI sites, respectively, at their 5′ termini. PCR products were digested with EcoRI and BamHI (New England Biolabs, Beverly, MA), purified with a QIAquick PCR purification kit (Qiagen, Valentia, CA), and ligated into pUC19 by using a quick-ligation kit (New England Biolabs, Beverly, MA). The Escherichia coli strain TOP10 (Invitrogen, Faraday, CA) was transfected with the ligation products.
Clones containing target inserts were identified by digestion with appropriate restriction enzymes, followed by electrophoresis. Plasmid DNA was purified with a QIAWell-8 plasmid kit (Qiagen, Valentia, CA). The purified DNA templates were sequenced using a BigDye Terminator cycle sequencing kit (Applied Biosystems, Foster City, CA). The pBR322 plasmid with the full length of the HPV16 prototype sequence (i.e., the E variant) was kindly provided by Denise Galloway (Fred Hutchison Cancer Research Center, Seattle, WA).
Clinical specimens.
Nine HPV16-positive cervical swab samples from a screening population (17) were selected for the determination of ratios of E2 to E7 gene copies. Of these samples, two were from women with normal cytology, five were from women with atypical squamous cells of undetermined significance (ASC-US), and two were from women with low-grade squamous intraepithelial lesions (LSIL). HPV16 variants in these samples, including six samples with E variants, one with an Af1 variant, one with an Af2 variant, and one with an AA variant, were previously characterized by sequencing part of the long control region and the entire E6 region (unpublished data).
Quantitative measurement of HPV16 E2 and E7 gene copy numbers by real-time PCR.
Listed in Table 1 are primers and probes for real-time PCR which were designed using Primer Express 3.0 (Applied Biosystems, Foster City, CA). Because the purpose of the present study was to examine the impact of HPV16 intratypic sequence variations on the detection of viral DNA integration rather than to assess the impact of the number and locations of the mismatches, the primers and probes were designed according to sequence variations found in the natural variants. The type-specific primers and probes were fully complementary to HPV16 prototype sequences, whereas the degenerate primers and probes incorporated degenerate bases at positions where the nucleotides differ among the variants. We additionally designed three modified degenerate E2 forward primers for the Af2 variant construct.
TABLE 1.
Primers and probes
| Primer or probea | Sequenceb (5′-3′) | Nucleotide position |
|---|---|---|
| E7 forward primer | CGGACAGAGCCCATTACAATATT | 701-723 |
| E7 reverse primer | CGCACAACCGAAGCGTAGA | 766-748 |
| E7-specific probe | FAM-TAACCTTTTGTTGCAAGTGT-MGBNFQ | 725-744 |
| E7 degenerate probe | FAM-TAACCTTYTGTTGCAAGTGT-MGBNFQ | 725-744 |
| E2-specific forward primer | GCCGCGACCCATACCAAA | 3413-3430 |
| E2 degenerate forward primer | GCCGCGACCCATWCCAAA | 3413-3430 |
| Modified E2 degenerate forward primer 1 | CGCCGCGACCCATWCCAA | 3412-3429 |
| Modified E2 degenerate forward primer 2 | CCGCCGCGACCCATWCCA | 3411-3428 |
| Modified E2 degenerate forward primer 3 | YCCGCCGCGACCCATWCC | 3410-3427 |
| E2 reverse primer | TCGCTGGATAGTCGTCTGTGTT | 3475-3454 |
| E2-specific probe | TET-CGTCGCCTTGGGCACCGAA-TAMRA | 3433-3451 |
| E2 degenerate probe | TET-CGTCGCCTTGGGCACCRAA-TAMRA | 3433-3451 |
Specific sets of primers and probes were as follows: for E2, E2-specific forward primer, E2 reverse primer, and E2-specific probe, and for E7, E7 forward and reverse primers and E7-specific probe. Degenerate sets of primers and probes were as follows: for E2, E2 degenerate forward primer, E2 reverse primer, and E2 degenerate probe, and for E7, E7 forward and reverse primers and E7 degenerate probe. In modified degenerate sets, the E2 degenerate forward primer was replaced with a modified E2 degenerate forward primer.
W, A or T; R, A or G; and Y, T or C. FAM, 6-carboxyfluorescein; MGBNFQ, minor groove binding nonfluorescent quencher; TET, tetrachloro-6-carboxy-fluorescein; TAMRA, 6-carboxytetramethylrhodamine.
Real-time PCR was performed on the ABI Prism 7900HT sequence detection system (Applied Biosystems, Foster City, CA). Briefly, the assay was set up in a 15-μl reaction volume containing 1× TaqMan universal PCR master mix (Applied Biosystems, Foster City, CA), 0.1 μM (each) primers and probe, and 1 μl of the DNA template. PCR amplification was carried out with a cycling program of holding at 50°C for 2 min and then at 95°C for 10 min, followed by a two-step cycle of 95°C for 15 s and 60°C for 1 min for a total of 40 cycles.
The log-phase five-point standard curves for a known number of HPV16 prototype copies (from 102 to 106 in a 10-fold dilution) were implemented in each set of the assays. The standard curve was generated by each set of primers and probe. The PCR run efficiency, which measures amplification rates, was calculated using the following formula (12):
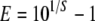 |
where E is the run efficiency and S is the slope of the standard curve. To minimize assay-to-assay variation, all reagents except for the PCR master mix were freshly made from the same stock solution. The numbers of E2 and E7 gene copies were measured in separate reactions but on the same plate to avoid potential competition and minimize variation in sample loading. Each sample was assayed in triplicate, and a mean value of three measurements was used for analysis.
Statistical analyses.
The reliability of the mean of the triplicate measures was evaluated by one-way analysis of variance with random effects. The ratios of HPV16 E2 to E7 gene copy numbers in four plasmid samples were independently measured five times. The coefficient of variation (CV) was used to describe interassay variations.
RESULTS
Nucleotide alterations in HPV16 variant sequences within the plasmid constructs.
Table 2 shows sequence variations in part of the E2 gene (from nucleotide position 3224 to 3852) and the entire E7 gene (from nucleotide position 562 to 858) within the plasmid constructs. Compared to the HPV16 prototype sequence, the Af1, Af2, and AA variants had a total of 13 consensus changes in the regions examined, including G to A, A to G, C to G, C to T, G to A, C to A, T to G, C to A, T to A, G to T, C to A, T to C, and T to G at positions 3249, 3362, 3377, 3410, 3449, 3516, 3566, 3684, 3694, 3778, 3787, 789, and 795, respectively. Each of these variants had several additional unique changes. Within the E2 probe binding site, there was a G-to-A change at position 3449 in all three non-E variants. In addition, the Af1 variant had an A-to-T change at position 3425 in the E2 forward primer binding site, and the AA variant had a T-to-C change at position 732 in the E7 probe binding site.
TABLE 2.
Nucleotide alterations from positions 3224 to 3852 in the E2 region and 562 to 858 in the E7 region of HPV16 variants in four plasmid constructsa
| Plasmid construct | Nucleotide alteration at position in:
|
||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E2 region
|
E7 region
|
||||||||||||||||||||||||
| 3224 | 3235 | 3249 | 3362 | 3377 | 3387 | 3410 | 3425 | 3431 | 3449 | 3516 | 3517 | 3538 | 3566 | 3664 | 3684 | 3694 | 3706 | 3778 | 3784 | 3787 | 3805 | 732 | 789 | 795 | |
| E (prototype) | T | T | G | A | C | T | C | A | G | G | C | T | A | T | T | C | T | T | G | T | C | T | T | T | T |
| Af1 | − | C | A | G | G | − | T | T | − | A | A | − | − | G | − | A | A | − | T | G | A | − | − | C | G |
| Af2 | − | − | A | G | G | − | T | − | A | A | A | C | C | G | − | A | A | C | T | − | A | G | − | C | G |
| AA | A | − | A | G | G | C | T | − | − | A | A | C | C | G | C | A | A | C | T | − | A | G | C | C | G |
The nucleotide positions are numbered according to those documented in Human Papillomavirus 1997 (19), corresponding to the numbering of the original HPV16 sequence (27). Positions in boldface (3410, 3425, 3449, and 732) were within the binding sites of the modified E2 degenerate forward primers, the E2 forward primer, the E2 probe, and the E7 probe, respectively. −, identical to the prototype sequence.
Ratios of HPV16 E2 to E7 gene copy numbers in plasmid samples.
The numbers of E2 and E7 gene copies in four plasmid samples were measured in triplicate with the same equipment at five different times, with the known copy number of the HPV16 prototype as the standard. The overall reliability of the mean of the triplicate measures was 0.988, with an intraclass correlation of 0.965 (95% confidence interval, 0.951 to 0.978). The R2 values that describe the correlation between the threshold cycle and the log of the starting copy number of the standard (1.00 indicates perfect correlation) were no less than 0.99 for both E2 and E7 genes with either specific or degenerate sets of primers and probes. The efficiencies of five independent runs for the amplification of prototype HPV16 E2 and E7 genes varied from 0.84 to 0.88 and 0.83 to 0.86, respectively, with the specific sets of primers and probes and from 0.86 to 0.88 and 0.83 to 0.88, respectively, with the degenerate sets of primers and probes.
The CVs of the ratios of E2 to E7 gene copies for five independent runs were less than 10% for all except for the AA variant construct (CV = 18%) with the specific sets of primers and probes (Table 3). The mean ratio for the E variant construct was close to 1, no matter which sets of primers and probes were used. However, while the ratios for the Af1 and AA variant constructs remained close to 1 with the degenerate sets of primers and probes, they differed substantially from 1 with the specific sets of primers and probes, with mean ratios of 0.36 for the Af1 construct and 2.57 for the AA construct. The numbers of E2 and E7 gene copies estimated by the degenerate sets of primers and probes were substantially larger than those estimated by the specific sets of primers and probes. The mean ratio for the Af2 variant construct estimated by the degenerate sets of primers and probes, although larger than that estimated by the specific sets of primers and probes (0.48 versus 0.17), reflected an approximately twofold difference between the numbers of E2 and E7 gene copies. The result remained similar when this construct was assayed another five times (data not shown).
TABLE 3.
Ratios of HPV16 E2 to E7 gene copy numbers in four plasmid samplesa
| Primer-probe set | HPV16 variant | Result from run:
|
Mean ratio (CV [%]) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1
|
2
|
3
|
4
|
5
|
||||||||
| E2/E7 gene copy no. | Ratio | E2/E7 gene copy no. | Ratio | E2/E7 gene copy no. | Ratio | E2/E7 gene copy no. | Ratio | E2/E7 gene copy no. | Ratio | |||
| Specific | E | 9,984/10,293 | 0.97 | 10,421/11,116 | 0.94 | 9,961/10,174 | 0.98 | 9,473/10,114 | 0.94 | 9,414/10,034 | 0.94 | 0.95 (2.2) |
| Af1 | 5,986/15,397 | 0.39 | 5,882/15,352 | 0.38 | 5,098/16,778 | 0.30 | 5,850/16,358 | 0.36 | 6,148/16,915 | 0.36 | 0.36 (9.4) | |
| AA | 5,999/2,487 | 2.41 | 5,562/2,740 | 2.03 | 7,735/2,338 | 3.31 | 5,657/2,201 | 2.57 | 6,197/2,360 | 2.63 | 2.59 (17.9) | |
| Af2 | 1,853/10,883 | 0.17 | 1,951/11,718 | 0.16 | 2,090/12,369 | 0.17 | 1,974/12,049 | 0.16 | 2,020/12,136 | 0.17 | 0.17 (1.7) | |
| Degenerate | E | 9,321/9,415 | 0.99 | 9,023/8,714 | 1.04 | 11,084/9,916 | 1.12 | 9,521/9,770 | 0.97 | 10,101/10,847 | 0.93 | 1.01 (7.0) |
| Af1 | 14,445/14,545 | 0.99 | 15,396/14,041 | 1.10 | 16,312/15,371 | 1.06 | 15,494/15,294 | 1.01 | 15,997/15,370 | 1.04 | 1.04 (3.8) | |
| AA | 11,989/12,822 | 0.94 | 11,038/12,139 | 0.91 | 12,554/12,789 | 0.98 | 11,815/14,020 | 0.84 | 11,166/12,375 | 0.90 | 0.91 (5.5) | |
| Af2 | 5,018/11,015 | 0.46 | 5,073/10,250 | 0.49 | 6,120/11,848 | 0.52 | 5,868/11,391 | 0.52 | 5,552/12,746 | 0.44 | 0.48 (7.5) | |
Gene copies were detected in five separate runs with the specific and degenerate sets of primers and probes. Copy numbers shown are averages of triplicate measures.
The sequence variations of the Af2 variant in the plasmid construct were reconfirmed by DNA sequencing. We noticed that the Af2 variant carried a unique nucleotide alteration at the position immediately following the 3′ end of the initially designed E2 forward primer (a G-to-A change at position 3431). To examine whether this change would introduce errors into the determination of the E2 gene copy number, we tentatively redesigned three E2 forward primers by moving the original primer sequence 1 to 3 bases away from that alteration. With the replacement of the E2 degenerate forward primer by these newly designed forward primers, the ratios of E2 to E7 gene copies gradually approached 1, with the value of 0.97 (mean copy numbers of 9,899 for the E2 gene and 10,212 for the E7 gene) reached by using modified E2 degenerate forward primer 3, the 3 ′ end of which was 3 bases away from the alteration. The modification of the E2 degenerate forward primer did not appreciably affect ratio estimates for the other three variant constructs (data not shown).
Ratios of HPV16 E2 to E7 gene copy numbers in cervical swab samples.
Nine cervical swab samples were assayed in triplicate by real-time PCR with the specific and modified degenerate sets of primers and probes. The overall reliability of the mean of the triplicate measures was 0.997, with an intraclass correlation of 0.992 (95% confidence interval, 0.987 to 0.996). The ratios of E2 to E7 gene copies in six cervical samples that were positive for the E variant were close or equal to 1 with either set of primers and probes (Table 4). However, the ratios for the three non-E variants differed substantially: in contrast to the ratio of ≥0.94 for the Af1 or AA variant with the modified degenerate sets of primers and probes, the ratio for the Af1 variant was 0.41 and that for the AA variant was 2.42 with the specific sets of primers and probes. The ratio of E2 to E7 gene copies for the Af2 variant was 0.56 with the modified degenerate sets of primers and probes and 0.07 with the specific sets of primers and probes.
TABLE 4.
Ratios of HPV16 E2 to E7 gene copy numbers in nine cervical swab samplesa
| Cervical sample | Cervical cytology | HPV16 variant | Results with specific sets for:
|
Results with modified degenerate sets for:
|
||
|---|---|---|---|---|---|---|
| E2/E7 gene copy no. | Ratio | E2/E7 gene copy no. | Ratio | |||
| 1 | Normal | E | 13,196/13,351 | 0.99 | 12,722/13,162 | 0.97 |
| 2 | LSIL | Af1 | 11,812/28,932 | 0.41 | 26,250/27,265 | 0.96 |
| 3 | ASC-US | Af2 | 48/729 | 0.07 | 412/732 | 0.56 |
| 4 | LSIL | E | 17,137/18,730 | 0.91 | 16,699/16,894 | 0.99 |
| 5 | Normal | AA | 201/83 | 2.42 | 419/445 | 0.94 |
| 6 | ASC-US | E | 1,827/1,941 | 0.94 | 1,991/1,967 | 1.01 |
| 7 | ASC-US | E | 43,915/44,300 | 0.99 | 43,054/42,961 | 1.00 |
| 8 | ASC-US | E | 2,231/2,301 | 0.97 | 2,127/2,213 | 0.96 |
| 9 | ASC-US | E | 13,921/14,031 | 0.99 | 14,456/14,121 | 1.02 |
Gene copies were detected with specific and modified degenerate sets of primers and probes. Copy numbers are averages of triplicate measures.
DISCUSSION
In this study, we quantitatively evaluated the impacts of mismatches between the primers and probes and their binding sites on the detection of HPV16 DNA integration by real-time PCR. Because the numbers of E2 and E7 gene copies within the plasmid construct are known to be equal, comparisons of ratios of E2 to E7 gene copies estimated by the specific versus the degenerate sets of primers and probes reflect to what extent the physical status of the virus would be misinterpreted simply because of base mismatches.
We have assumed that the estimates derived by using primers and probes that were fully complementary to the targets were the actual copy numbers. As expected, the specific sets of primers and probes, which had no mismatches with the E variant construct, gave a ratio of E2 to E7 gene copies of close to 1 for this construct. The ratio of 1 for the E variant construct, derived by using the degenerate sets of primers and probes, suggested that the incorporation of degenerate bases, relative to those fully complementary, did not appreciably affect the determination of copy number. Impressively, results for the three non-E variant constructs differed substantially between tests that used the specific versus the degenerate sets of primers and probes. While the ratio estimates remained close to 1 for all except the Af2 variant construct with the degenerate sets of primers and probes, the estimates were significantly different from 1 with the specific sets of primers and probes. These differences cannot be explained by variations in specimen loading or any other assay procedures, because the aliquots for E2 and E7 that were tested on the same plate were from the same premix and the results from five independent runs were comparable.
The departure from the ratio of 1 for the Af1 and AA variant constructs with the specific sets of primers and probes is likely to be a result of the mismatch-related reduction of amplification efficiency (32). For example, the ratio of <1 for the Af1 plasmid construct could be explained by the presence of mismatches in the E2 forward primer and probe binding sites but not in the E7 primer and probe binding sites. The AA variant had a G-to-A change in the E2 probe target and a T-to-C change in the E7 probe target. Both mismatches reduced the amplification efficiency, but the impact of the E7 mismatch was more substantial than that of the E2 mismatch, thereby leading to the reduced copy numbers of both E2 and E7 genes and the ratio of >1. The ratio of <1 for the Af2 variant construct that was derived by using the degenerate sets of primers and probes was unexpected because this variant had only a single mismatch in the E2 probe binding site, the one that was also present in the Af1 and AA variant constructs. The ratio of close to 1 obtained by testing with the modified degenerate sets of primers and probes suggested that the underestimation of E2 gene copies by the degenerate set may be attributed to a nucleotide alteration at the position immediately following the 3′ end of the E2 forward primer: from guanine at position 3431 in the prototype that was used to generate the standard curve to adenine in the Af2 variant construct. Knowing about this pattern, although the underlying mechanism is unclear, is useful for a better design of primers and probes for real-time PCR.
The present study further showed evidence of the variant-associated misinterpretation of HPV16 integration by testing a set of cervical swab samples. Because proportions of HPV16 non-E variants are related to geography and vary from one population to another, the findings of this study may in part explain the wide range of integration rates reported so far. Given that HPV16 variants differ biologically and etiologically (31), the variant-related base mismatches may somewhat bias estimates of the integration-related risk of cervical neoplasia. We noticed a ratio of <1 estimated by the modified degenerate sets of primers and probes for a clinical sample that was positive for the HPV16 Af2 variant. One interpretation for this result is that the variant might have existed in both episomal and integrated forms. Alternatively, this variant may carry additional nucleotide alterations that were not covered by our modified degenerate sets of primers and probes.
To our knowledge, this is one of the first studies, if not the first, to quantitatively address the impact of HPV16 variants on the detection of viral integration. Although the influence of HPV16 E2 polymorphisms on the detection of HPV16 integration was reported previously (4), the findings of the study were limited by the fact that the evaluation was based only on clinical samples in which a true integration status was unknown. The use of plasmid constructs for illustration avoided any integration-related misinterpretation that might arise with the use of clinical samples. We are, however, aware that our degenerate sets of primers and probes, although better than the specific sets, were designed based on limited sequence information. In natural infections, some HPV16 variants may carry more nucleotide alterations than those currently identified. Because of the intratypic diversity of many HPV types, the careful design of primers and probes is a prerequisite for a valid assay to test the integration of the HPV genome. One way to achieve this design is to choose a conserved region as the target (4). However, it is sometimes difficult to find such a region, particularly when one needs to take into account the appropriate amplicon size (usually less than 100 bp for real-time PCR) and comparable efficiencies of amplification. Alternatively, as shown in the present study, we may incorporate degenerate bases into the primers and probes at sites where nucleotides differ among the variants.
The implications of the results of this study go beyond studying viral DNA integration. Currently, quantitative analysis of the HPV DNA load by real-time PCR has been widely used in many clinical and epidemiological investigations. The primers and probes for the assay are usually designed based on the prototype sequence. It is likely that errors in viral load estimation may arise if the sample is positive for nonprototype HPV variants.
In summary, our data indicate that mismatches between the primers and probe and their targets can introduce significant errors into the determination of copy number. Incorporating degenerate bases into the primers and probe can sufficiently compensate for the mismatch-reduced efficiency of amplification.
Acknowledgments
This work was supported by Public Health Service grants CA34493 and CA84396 from the National Cancer Institute. L.A.K. has received research funds from Merck Research Laboratories. Other authors have no commercial or other associations that might pose a conflict of interest.
We thank the participants who provided biologic specimens and clinical data to this study.
Footnotes
Published ahead of print on 30 December 2008.
REFERENCES
- 1.Alazawi, W., M. Pett, B. Arch, L. Scott, T. Freeman, M. A. Stanley, and N. Coleman. 2002. Changes in cervical keratinocyte gene expression associated with integration of human papillomavirus 16. Cancer Res. 626959-6965. [PubMed] [Google Scholar]
- 2.Andersson, S., H. Safari, M. Mints, I. Lewensohn-Fuchs, U. Gyllensten, and B. Johansson. 2005. Type distribution, viral load and integration status of high-risk human papillomaviruses in pre-stages of cervical cancer (CIN). Br. J. Cancer 922195-2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arias-Pulido, H., C. L. Peyton, N. E. Joste, H. Vargas, and C. M. Wheeler. 2006. Human papillomavirus type 16 integration in cervical carcinoma in situ and in invasive cervical cancer. J. Clin. Microbiol. 441755-1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azizi, N., J. Brazete, C. Hankins, D. Money, J. Fontaine, A. Koushik, A. Rachlis, K. Pourreaux, A. Ferenczy, E. Franco, and F. Coutlee. 2008. Influence of human papillomavirus type 16 (HPV-16) E2 polymorphism on quantification of HPV-16 episomal and integrated DNA in cervicovaginal lavages from women with cervical intraepithelial neoplasia. J. Gen. Virol. 891716-1728. [DOI] [PubMed] [Google Scholar]
- 5.Bosch, F. X., M. M. Manos, N. Munoz, M. Sherman, A. M. Jansen, J. Peto, M. H. Schiffman, V. Moreno, R. Kurman, K. V. Shah, et al. 1995. Prevalence of human papillomavirus in cervical cancer: a worldwide perspective. J. Natl. Cancer Inst. 87796-802. [DOI] [PubMed] [Google Scholar]
- 6.Briolat, J., V. Dalstein, M. Saunier, K. Joseph, S. Caudroy, J. L. Pretet, P. Birembaut, and C. Clavel. 2007. HPV prevalence, viral load and physical state of HPV-16 in cervical smears of patients with different grades of CIN. Int. J. Cancer 1212198-2204. [DOI] [PubMed] [Google Scholar]
- 7.Castellsague, X., F. X. Bosch, and N. Munoz. 2002. Environmental co-factors in HPV carcinogenesis. Virus Res. 89191-199. [DOI] [PubMed] [Google Scholar]
- 8.Chan, S. Y., L. Ho, C. K. Ong, V. Chow, B. Drescher, M. Durst, J. ter Meulen, L. Villa, J. Luande, H. N. Mgaya, et al. 1992. Molecular variants of human papillomavirus type 16 from four continents suggest ancient pandemic spread of the virus and its coevolution with humankind. J. Virol. 662057-2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung, J. L., K. W. Lo, T. H. Cheung, J. W. Tang, and P. K. Chan. 2006. Viral load, E2 gene disruption status, and lineage of human papillomavirus type 16 infection in cervical neoplasia. J. Infect. Dis. 1941706-1712. [DOI] [PubMed] [Google Scholar]
- 10.Cricca, M., A. M. Morselli-Labate, S. Venturoli, S. Ambretti, G. A. Gentilomi, G. Gallinella, S. Costa, M. Musiani, and M. Zerbini. 2007. Viral DNA load, physical status and E2/E6 ratio as markers to grade HPV16 positive women for high-grade cervical lesions. Gynecol. Oncol. 106549-557. [DOI] [PubMed] [Google Scholar]
- 11.De Marco, L., A. Gillio-Tos, L. Bonello, V. Ghisetti, G. Ronco, and F. Merletti. 2007. Detection of human papillomavirus type 16 integration in pre-neoplastic cervical lesions and confirmation by DIPS-PCR and sequencing. J. Clin. Virol. 387-13. [DOI] [PubMed] [Google Scholar]
- 12.Fink, L., W. Seeger, L. Ermert, J. Hanze, U. Stahl, F. Grimminger, W. Kummer, and R. M. Bohle. 1998. Real-time quantitative RT-PCR after laser-assisted cell picking. Nat. Med. 41329-1333. [DOI] [PubMed] [Google Scholar]
- 13.Fujii, T., N. Masumoto, M. Saito, N. Hirao, S. Niimi, M. Mukai, A. Ono, S. Hayashi, K. Kubushiro, E. Sakai, K. Tsukazaki, and S. Nozawa. 2005. Comparison between in situ hybridization and real-time PCR technique as a means of detecting the integrated form of human papillomavirus 16 in cervical neoplasia. Diagn. Mol. Pathol. 14103-108. [DOI] [PubMed] [Google Scholar]
- 14.Guo, M., N. Sneige, E. G. Silva, Y. J. Jan, D. E. Cogdell, E. Lin, R. Luthra, and W. Zhang. 2007. Distribution and viral load of eight oncogenic types of human papillomavirus (HPV) and HPV 16 integration status in cervical intraepithelial neoplasia and carcinoma. Mod. Pathol. 20256-266. [DOI] [PubMed] [Google Scholar]
- 15.Jeon, S., B. L. Allen-Hoffmann, and P. F. Lambert. 1995. Integration of human papillomavirus type 16 into the human genome correlates with a selective growth advantage of cells. J. Virol. 692989-2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jeon, S., and P. F. Lambert. 1995. Integration of human papillomavirus type 16 DNA into the human genome leads to increased stability of E6 and E7 mRNAs: implications for cervical carcinogenesis. Proc. Natl. Acad. Sci. USA 921654-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kulasingam, S. L., J. P. Hughes, N. B. Kiviat, C. Mao, N. S. Weiss, J. M. Kuypers, and L. A. Koutsky. 2002. Evaluation of human papillomavirus testing in primary screening for cervical abnormalities: comparison of sensitivity, specificity, and frequency of referral. JAMA 2881749-1757. [DOI] [PubMed] [Google Scholar]
- 18.Kulmala, S. M., S. M. Syrjanen, U. B. Gyllensten, I. P. Shabalova, N. Petrovichev, P. Tosi, K. J. Syrjanen, and B. C. Johansson. 2006. Early integration of high copy HPV16 detectable in women with normal and low grade cervical cytology and histology. J. Clin. Pathol. 59513-517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meissner, J. 1997. Sequencing errors in reference HPV clones, p. 110-123. In G. Myers, C. Baker, K. Munger, et al. (ed.), Human papillomavirus 1997: a compilation and analysis of nucleic acid and amino acid sequences, 3rd ed. Theoretical Biology and Biophysics, Los Alamos National Laboratory, Los Alamos, NM.
- 20.Munoz, N., F. X. Bosch, S. de Sanjose, R. Herrero, X. Castellsague, K. V. Shah, P. J. Snijders, and C. J. Meijer. 2003. Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 348518-527. [DOI] [PubMed] [Google Scholar]
- 21.Nagao, S., M. Yoshinouchi, Y. Miyagi, A. Hongo, J. Kodama, S. Itoh, and T. Kudo. 2002. Rapid and sensitive detection of physical status of human papillomavirus type 16 DNA by quantitative real-time PCR. J. Clin. Microbiol. 40863-867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peitsaro, P., B. Johansson, and S. Syrjanen. 2002. Integrated human papillomavirus type 16 is frequently found in cervical cancer precursors as demonstrated by a novel quantitative real-time PCR technique. J. Clin. Microbiol. 40886-891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pett, M., and N. Coleman. 2007. Integration of high-risk human papillomavirus: a key event in cervical carcinogenesis? J. Pathol. 212356-367. [DOI] [PubMed] [Google Scholar]
- 24.Pett, M. R., W. O. Alazawi, I. Roberts, S. Dowen, D. I. Smith, M. A. Stanley, and N. Coleman. 2004. Acquisition of high-level chromosomal instability is associated with integration of human papillomavirus type 16 in cervical keratinocytes. Cancer Res. 641359-1368. [DOI] [PubMed] [Google Scholar]
- 25.Romanczuk, H., and P. M. Howley. 1992. Disruption of either the E1 or the E2 regulatory gene of human papillomavirus type 16 increases viral immortalization capacity. Proc. Natl. Acad. Sci. USA 893159-3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rousseau, M. N., V. Costes, I. Konate, N. Nagot, V. Foulongne, A. Ouedraogo, P. Van de Perre, P. Mayaud, and M. Segondy. 2007. Viral load and genomic integration of HPV 16 in cervical samples from HIV-1-infected and uninfected women in Burkina Faso. J. Med. Virol. 79766-770. [DOI] [PubMed] [Google Scholar]
- 27.Seedorf, K., G. Krammer, M. Durst, S. Suhai, and W. G. Rowekamp. 1985. Human papillomavirus type 16 DNA sequence. Virology 145181-185. [DOI] [PubMed] [Google Scholar]
- 28.Si, H. X., S. W. Tsao, C. S. Poon, Y. C. Wong, and A. L. Cheung. 2005. Physical status of HPV-16 in esophageal squamous cell carcinoma. J. Clin. Virol. 3219-23. [DOI] [PubMed] [Google Scholar]
- 29.Tonon, S. A., M. A. Picconi, P. D. Bos, J. B. Zinovich, J. Galuppo, L. V. Alonio, and A. R. Teyssie. 2001. Physical status of the E2 human papilloma virus 16 viral gene in cervical preneoplastic and neoplastic lesions. J. Clin. Virol. 21129-134. [DOI] [PubMed] [Google Scholar]
- 30.Walboomers, J. M., M. V. Jacobs, M. M. Manos, F. X. Bosch, J. A. Kummer, K. V. Shah, P. J. Snijders, J. Peto, C. J. Meijer, and N. Munoz. 1999. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J. Pathol. 18912-19. [DOI] [PubMed] [Google Scholar]
- 31.Wang, S. S., and A. Hildesheim. 2003. Viral and host factors in human papillomavirus persistence and progression, p. 35-40. In Journal of the National Cancer Institute monographs, no. 31. National Cancer Institute, Bethesda, MD. [DOI] [PubMed]
- 32.Whiley, D. M., and T. P. Sloots. 2005. Sequence variation in primer targets affects the accuracy of viral quantitative PCR. J. Clin. Virol. 34104-107. [DOI] [PubMed] [Google Scholar]
- 33.Yamada, T., M. M. Manos, J. Peto, C. E. Greer, N. Munoz, F. X. Bosch, and C. M. Wheeler. 1997. Human papillomavirus type 16 sequence variation in cervical cancers: a worldwide perspective. J. Virol. 712463-2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yoshida, T., T. Sano, T. Kanuma, N. Owada, S. Sakurai, T. Fukuda, and T. Nakajima. 2008. Quantitative real-time polymerase chain reaction analysis of the type distribution, viral load, and physical status of human papillomavirus in liquid-based cytology samples from cervical lesions. Int. J. Gynecol. Cancer 18121-127. [DOI] [PubMed] [Google Scholar]