

Abstract
The human ribonucleoprotein telomerase is a validated anticancer drug target, and hTRP2b is a part of the human telomerase RNA (hTR) essential for its activity. Interesting ligands that bind hTR-P2b were identified by iteratively using a tandem structure-based approach: docking of potential ligands from small databases to hTR-P2b via the program MORDOR, which permits flexibility in both ligand and target, with subsequent NMR screening of high-ranking compounds. A high percentage of the compounds tested experimentally were found via NMR to bind to the U-rich region of hTR-P2b; most have MW <500 daltons, are from different compound classes, and several possess a charge of 0 or +1. Of the 48 ligands identified, 24 exhibit a decided preference to bind hTR-P2b RNA rather than A-site rRNA and 10 do not bind A-site rRNA at all. Binding affinity was measured by monitoring RNA imino proton resonances for some of the compounds that showed hTR binding preference.
Keywords: virtual screening, structure-based drug-design, MORDOR, RNA recognition, RNA-ligand interactions, small molecules
Introduction
While there have been many advances in the battle to control cancer, it remains a huge problem worldwide. For example, it is the second greatest cause of death in the U.S. Several potential new therapies and targets have arisen from increasing knowledge of processes that govern carcinogenesis. Among the viable targets is human telomerase, which maintains telomeres at the end of chromosomes, in large part by adding repeats of 5'-TTAGGG-3', thus foiling the process of normal cellular senescence and imparting a quality of immortalization to the cell. Indeed, approximately 90% of human tumors have been associated with up-regulation of telomerase. For example, in prostate cancer, telomerase activity correlates with graded tumors, being maximal in high-grade prostate carcinoma.1 Work on both validation and development of therapeutics has recently been reviewed.2
Telomerase is a eukaryotic ribonucleoprotein that synthesizes telomeric DNA.3,4 Telomerase apparently also acts by capping telomeres, so that the terminal DNA will not be susceptible to DNA damage-response. Many proteins are associated with telomerase RNA (TER, or hTRa for human telomerase RNA) depending on the stage of the cell cycle, but telomerase reverse transcriptase (TERT) provides the core enzymatic activity. The core secondary structure for vertebrate telomerase RNA exhibits some conserved regions (CR) (Figure 1).5 One conserved region corresponds to the pseudoknot domain, which has been shown to be essential for telomerase activity. Physical, functional, and structural studies have suggested that the pseudoknot domain exists in two alternative states of nearly equal stability in solution acting as a molecular switch: one is the previously proposed pseudoknot formed by pairing P3 with the loop domain of P2b, and the other is a structured P2b loop alone; interconversion between its two states appears essential for telomerase function.6,7 Mutations that stabilize either state and hence modify the equilibrium were found to reduce telomerase activity.6 In addition, phylogenetic covariation in the P2b and P3 sequences of 35 species revealed naturally occurring compensatory base pairing that served to maintain the two states at comparable energies. Molecular dynamics studies supported the dynamic nature of the noncanonical base pairs in P2b structures, being able to bind to P3 in formation of the pseudoknot.8 However, subsequent mutational analysis did not support the notion of a molecular switch.9 Regardless of whether the pseudoknot region operates as a switch or serves in some other function, it is still essential for telomerase activity.
Figure 1.

Proposed vertebrate telomerase RNA secondary structure showing conserved regions (CR). Dashed lines show the P2b stem-loop moiety and the CR4-CR5 conserved regions. The thick lines delineate the template sequence for telomere transcription, P2b, and the P3 region complementary to part of P2b.
With significant evidence that telomerase is a viable cancer target, we have consequently reasoned that hTR should be targeted, specifically, the P2b/P3 region may be a plausible target to find small nonpeptide, nonnucleotide organic molecules that would bind to either the intact pseudoknot or to P2b and thereby inhibit telomerase activity. We have not been alone in the assessment of hTR as a target: Geron Corporation has developed a promising antisense oligonucleotide, GRN163 GRN163 (1); a lipid conjugate of 1, GRN163L, is in stage 3 clinical trials, following earlier studies demonstrating inhibition of multiple forms of cancer cell growth both in vitro and in vivo.10 These studies of 1 from Geron clearly demonstrate telomerase RNA as a validated target. Our intent has been to see if a small organic compound could be found that will bind to this validated target; naturally, it would be necessary to develop any binders further and test for inhibition.
In the entire pharmacopeia there are few, if any, drugs developed with the intent to target RNA. However, some clinically useful antibiotics such as tetracyclines, macrolides, and aminoglycosides bind to conserved rRNA structural motifs as their mode of action. Certainly, RNA-binding drugs have precedence. While there are a few different ways to find small molecular weight compounds to bind to a new protein or RNA target, we have been exploring structure-based approaches. One approach to structure-based drug discovery entails computational docking (or virtual screening) of a large database of small molecules to a three-dimensional structure of a selected target. Such in silico screening has been used to discover ligands that bind three-dimensional (3D) protein structure targets.11,12 Since electrostatics and solvation are much more critical for RNA, we have been developing and applying methodologies that employ virtual screening against a 3D structure of a selected RNA target, followed by experimental testing of the more promising predicted ligands for binding and possibly inhibition of function;13,14 the structure of one promising molecule bound to its target was determined.15 Other researchers have also successfully developed their own approaches to virtual screening against RNA targets.16,17
Major challenges for the field of virtual screening largely center around two things. First, the scoring functions used to rank the ligands for binding affinity are highly imperfect. Our earlier work benefited from efforts to find a useful empirical scoring function.13,14 Secondly, most molecules adapt their structure upon binding to another molecule, i.e., both the ligand and the target are malleable such that neither the ligand nor the receptor moiety in the complex has the same structure as it did when unbound. This could be due to induced fit binding as each molecule adapts its structure in the presence of the other. Alternatively, the ligand and the receptor are dynamic, so that a particular conformation of a free receptor may be present as a fraction of the total population and that conformation is selected by the ligand for binding; of course, the same phenomenon can be presented for selecting the ligand conformation. These two concepts could be considered as the extremes of an attractive model suggested by Grünberg et al: the two partners would be bound initially by selection of shape-compatible conformers followed by a modification of the structure of one or both partners after initial binding to optimize the interaction.18 Recently, we have developed the docking program MORDOR (MOlecular Recognition with a Driven dynamics OptimizeR), which allows the ligand and receptor, either RNA or protein, to adapt to one another, modifying both of their conformations to minimize the total energy as the small molecule approaches the receptor.19 MORDOR does not, of course, strictly mimic the binding process in the physical world; it does, however, allow screening of a database of putative ligands by allowing the conformations of receptor and ligand to adjust to one another rather than requiring the receptor to be static.
We must experimentally test our top computational “hits” for binding. NMR is the most effective way to detect weak-binding with minimal false positives when an activity assay is not readily available: Telomerase activity is typically measured in cells or cell lysates; for first-stage efforts to discover novel compounds that might bind to the RNA target, complications from the cellular environment are not desirable. NMR also has the huge advantages that we can readily use it to define the ligand binding epitope, map the binding site on the receptor (consequently verifying binding specificity), and measure the binding affinity. We previously found conditions for saturation-transfer-difference (STD) NMR with RNA targets that are most effective for discerning binding and the binding epitope on the compound being tested.20,21,22
In the study reported here, we used the published structure of the P2b target,7 although the structure of the P2b/P3 pseudoknot23 is also a viable target for structure-based virtual screening. VS was carried out using MORDOR;19 this is the first report of ligands identified by MORDOR for a novel target. Novel small molecules were found that bind to the hTR target RNA, with some showing selectivity for that target relative to A-site rRNA.
Results and Discussion
There are no reports of small molecule ligands for telomerase RNA. Here is described a tandem virtual screening/similarity searching-NMR screening approach that was used for docking ligands to the selected hTR-P2b target (Protein Data Bank ID code 1NA2). The secondary structure of the hTR-P2b construct used as a target is shown in Figure 2, indicating the non-canonical UU and UC base pairs observed. A small number of apparent “hits”, compounds that were predicted by computation to bind to the RNA target upon screening a library of compounds, were selected and tested experimentally for binding via NMR. Those found to bind experimentally were used to search for similar compounds in the ZINC (ZINC Is Not Commercial) database24, with subsequent rounds of virtual screening and ranking of those similar compounds, and additional compounds tested via NMR.
Figure 2.

Secondary structure of the hTR-P2b construct used in these studies. NMR studies show that the U-rich region is structured with unusual hydrogen bonds forming three UU and one UC base pair.6,7
Computational screening with MORDOR and Similarity Searching
We used in this study a virtual screening program MORDOR that we developed to permit flexibility in both the putative ligand and in the receptor.19 While enabling flexibility in the target as well as the ligand is probably useful for docking of small molecules to proteins, it is particularly valuable for docking to RNA, because RNA targets (except canonical double helical RNA or certain tetraloops) typically exhibit considerable flexibility. On a test set of 57 RNA complexes (33 crystal structures and 24 NMR structures), the lowest energy conformations resulting from docking with MORDOR reproduced the experimental binding poses within an RMSD of <2.5 Å for 74% of the tested complexes; structures of 53 out the 57 complexes in the test set could be found within 2.5 Å among MORDOR's four best-scoring poses.19
For the current study, MORDOR was used to screen a small database of compounds, the FDA-drug-approved KEGG (Kyoto Encyclopedia of Genes and Genomes) library of approximately 3000 compounds (http://www.genome.jp/ligand/),25 against the hTR-P2b structure (PDB code 1NA2) as a target. This first MORDOR docking run (R1) produced many compounds that appeared to be feasible binders to different parts of the unique U-rich region of the target, including adjacent base pairs. They were reviewed and ranked according to their binding energy. Twenty compounds were obtained for testing experimentally using STD NMR (vide infra); criteria for selection included binding energy, RNA drug-like potentiality, diversity, estimated water solubility, and cost. Seven of the compounds, however, were either insoluble or caused precipitation of the RNA when added. Of the 13 for which STD NMR spectra could be acquired, a surprisingly large percentage, ten compounds, were found to bind according to STD NMR results. Mapping of the binding site via examination of spectra designed to monitor signals on the RNA (vide infra) verified that the compounds interacted with the U-loop region, including adjacent base pairs, of hTR-P2b.
Those ten “binders” were used as the basis for a similarity search among the two million compounds in the ZINC database that could be searched for similarity,24 yielding an hTR RNA ligand-like database of ~4700 compounds. These 4700 compounds were docked and ranked using MORDOR in round two (R2). Extensive review of R1 and R2 results lead to a list of putative ligands deemed worthy of screening by STD NMR. Early second round NMR screening results and further review of R1 and R2 suggested two new ZINC similarity searches (934 hits and 126 hits, respectively), the results of which were docked and ranked with MORDOR, yielding a final RNA ligand-like database of ~5750 compounds. From this ranked list, an additional 93 compounds were selected and obtained for STD NMR screening, for a total of 113 compounds tested. Although the cutoff in terms of number tested was arbitrary; the criteria for selection were as stated above.
On inspection of the 1000 highest ranking compounds in the ligand-like database, we found that these structures fit entirely within 11 classes. Table 1 provides the names and relative abundance of these 11 classes in the R2 virtual screening round. Care was taken to include examples of each structure class in the 113 test compounds.
Table 1.
Structure classes comprising the 1000 top-ranked compounds from the R2 virtual screen.
| Class Number | Structure Class Name | Percent Abundance |
|---|---|---|
| 1 | Monoaromatics | 2 |
| 2 | Chained Polyaromatics | 5 |
| 3 | Tetrahydroisoquinolines | 6 |
| 4 | 4-Aminoquinolines | 4 |
| 5 | 9-Aminoacridines | 2 |
| 6 | Phenothiazines | 5 |
| 7 | Thiaxanthenones | 7 |
| 8 | Anthracyclines | 59 |
| 9 | Bis-tetrahydroisoquinoline macrocycles | 3 |
| 10 | 6-7-6 Ring systems | 5 |
| 11 | Misc fused ring systems | 1 |
NMR screening
From the MORDOR-ranked list of compounds, 113 compounds were experimentally tested for binding using saturation transfer difference NMR. STD NMR, especially in D2O, has been proven to be efficient in identifying and characterizing weakly binding compounds to small RNA constructs.20,21 STD signals reflect the transfer of saturation from the irradiated RNA to the ligand while in the bound state, but the effect is manifest for the exchange-averaged signal from the ligand that is interconverting between free or bound states. In addition to simple unequivocal detection of ligand binding, this method allows the determination of the binding epitope of the ligand: ligand protons that are close to the RNA surface when bound exhibit stronger STD NMR signal intensities and belong to ligand moieties involved in or very near the binding interaction.
The library tested in the present study contains some small compounds, <300 daltons, so it is expected that they will bind weakly and consequently manifest weaker signals in the STD NMR spectra. Small, weak binders are still of interest, however, as they may be improved by chemical elaboration or by linking weak binding moieties.26
With the intent to acquire STD NMR spectra, attempts to prepare aqueous solutions of the 113 compounds, generally with 20 mM sodium phosphate (pH=6.7) and 50 mM NaCl, were made; most formed solutions, but some did not and other solution conditions did not result in sufficient improvement. RNA was added to form a final concentration of 25 μM of the hTR-P2b construct with 20 times excess putative ligand for STD experiments. However, many of the compounds resulted in formation of a precipitate, which was found to contain both RNA and the added compound. Of those mixtures remaining in solution, NMR spectra were acquired and analyzed. Some of the spectra manifest resonance linewidths much greater than expected for a complex, indicative of aggregation. STD NMR results for each of the 70 compounds that did not cause precipitation or aggregation in the presence of hTR-P2b were obtained. Subsequently, we found that 15 of these were insufficiently pure for publication. Results for the remaining 55 compounds are shown in Figure 3; the apparent affinity of each of the binders for hTR-P2b was divided into three categories (weak, medium or strong) according to STD NMR spectral intensities, as a very rough indicator of the strength of the interaction with the RNA. Results are summarized in Table 2. Slight line broadening was also evident for ligand protons in close proximity to RNA in the binding site, as one would expect for binding to the modestly large RNA construct. Experimentally it was found that 48 of the compounds (87%) could bind to hTR-P2b out of the 55 compounds in Figure 3. Figure 4 shows reference and STD spectra of two compounds selected from the library as examples. Compound 20 in Figure 3 exhibited line broadening effects and substantial STD enhancements in the presence of hTR-P2b (Figure 4A). Compounds with these characteristics were categorized as strong binders. On the other hand, compound 13 in Figure 3 manifests little line broadening and only a weak STD effect at the same ligand and RNA concentrations as for compound 20 (Figure 4B). Compounds showing these characteristics were categorized as weak binders. Non-binders exhibited no line broadening or STD signals for the ligand resonances in the presence of hTR-P2b.
Figure 3.



STD NMR results for compounds tested for binding to hTR-P2b RNA. The first number simply refers to its number in this table, which is used in the text for identification, and the second number is the compound identification number in the ZINC database; “+” signifies that the compound shows a distinct preference for binding to hTR-P2b RNA relative to A-site rRNA as assessed by STD NMR; “++” signifies that the compound shows no indication of binding to A-site rRNA but binds hTRP2b RNA; “=” indicates binding is comparable to the two RNA species, and “-“ indicates a preference for binding to the A-site rRNA transcript. Colors indicate STD NMR spectral intensities, which roughly correlate with binding affinity: weak (yellow), medium (green), strong (blue) or no STD effect observed (white).
Table 2.
STD NMR screening of compounds (500 μM) from MORDOR-ranked list of compounds for binding to hTR-P2b (25 μM).
| STD NMR | Count | Percent |
|---|---|---|
| Insoluble, causes precipitation or aggregation, insufficiently pure | 58 | |
| No binding | 7 | 13% |
| Weak binding | 17 | 31% |
| Medium binding | 14 | 25% |
| Strong binding | 17 | 31% |
| TOTAL | 113 | 100% |
Figure 4.

Representative 500 MHz NMR spectra of two compounds from Figure 3. (A) Compound 20 from Figure 3. (B) Compound 13 from Figure 3. The two top spectra are reference spectra of the compound alone. The two bottom spectra are STD NMR spectra of the compound in the presence of hTR-P2b RNA. For each spectrum, the concentration of the compound was 500 μM and the concentration of RNA was 25 μM.
Structure Classes
The 55 compounds reported in Figure 3 include members of each class listed in Table 1 except the 9-aminoacridines (class 5). Although the 9-aminoacridines show STD effects, its members have a strong tendency for precipitation and/or aggregation and were thus eliminated from further study. In fact, all classes showed STD effects except class 1. We remain interested in class 1 structures, however, since their low molecular weights make them attractive for fragment-based drug design.
Class 8, the anthracyclines, deserves special comment. In addition to their long-standing clinical relevance as anti-cancer agents, MORDOR picks more structures of this class as potential hTR-P2b ligands than all other classes combined. Unfortunately, poor solubility and a tendency for aggregation prevented testing of most of the anthracyclines in the 113 compounds we selected. Nevertheless, the two we could test were strong binders. Based on the foregoing, one could speculate that some members of this class may have additional modes of action, such as binding hTR, in addition to intercalating GC-rich DNA. This possibility needs further study.
NMR Characterization of Binding
For the more promising binders, imino proton chemical shifts and line broadening were monitored to identify and characterize the location of the small molecule binding to the 27-nt construct of hTR-P2b (50 μM). As an example, Figure 5 shows 800 MHz imino spectra of hTR-P2b as compound 33 of Figure 3 is added incrementally. As in Figure 5, all binders were found to perturb resonances from, and therefore to bind to, the U-rich region (including proximal base pairs) in the hTR-P2b construct. Addition of up to twenty times excess ligand failed to cause chemical shift changes beyond the U-rich region, precluding significant non-specific binding, unless it is mentioned in the discussion below that non-specific binding was observed.
Figure 5.

Imino proton signals (800 MHz) of hTR-P2b (see Figure 2 for secondary structure) as a function of increasing concentration of compound 33 from Figure 3, as an example. The concentration of RNA is 50 μM in a buffer of 20 mM sodium phosphate, pH 6.7, 50 mM NaCl and a 90/10% H2O/D2O mixture. The concentration of ligand is shown with the spectra.
Specificity of Binding
The binders listed in Figure 3 for hTR-P2b were experimentally tested for specificity via STD NMR using the 16S aminoacyl-tRNA site (A-site) of ribosomal RNA as another potential target. The model A-site rRNA is a 27-nt construct with an asymmetric A-rich internal loop (Figure 6).
Figure 6.
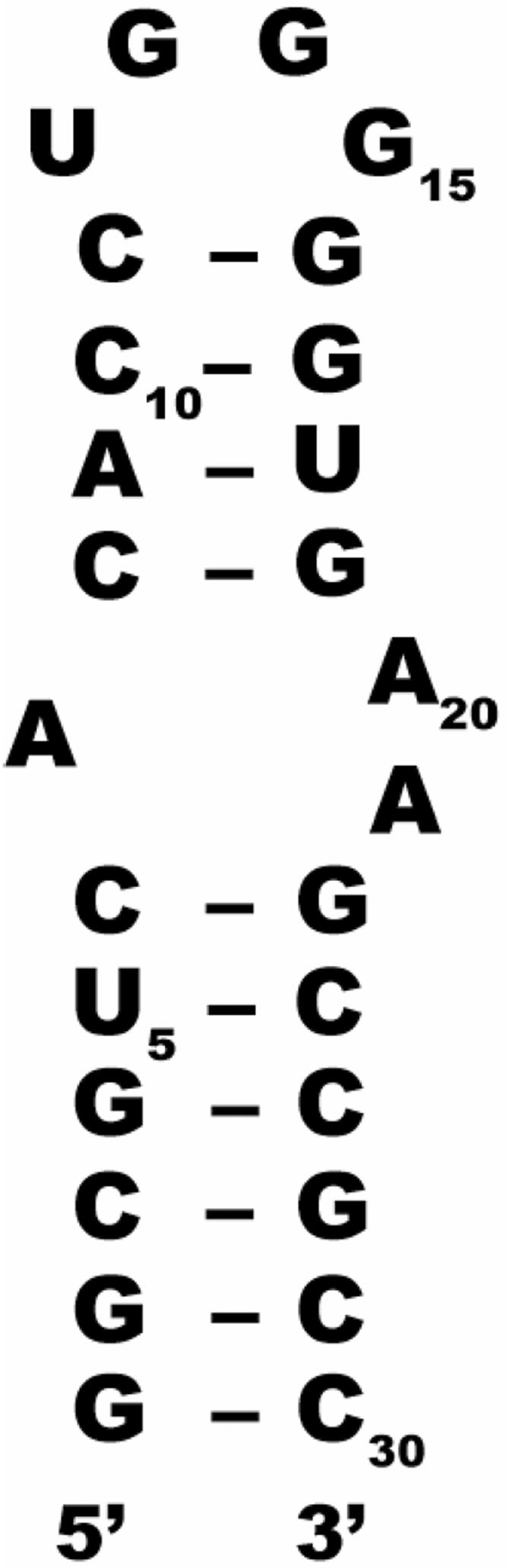
Secondary structure of the A-site rRNA construct used to test compounds found to bind to hTR for specificity of binding.
Among the 48 compounds found to bind to hTR-P2b, 24 compounds showed a distinct preference for binding to hTR-P2b RNA relative to A-site rRNA with 10 of those exhibiting no binding at all to A-site rRNA. Those 24 compounds could be separated into four classes as shown in Figure 7. For each of the 24 compounds, chemical shifts demonstrated binding in the vicinity of the U-rich region in the hTR RNA construct. Some titrations were carried out monitoring imino resonance chemical shift changes, which were used to estimate the dissociation constant of the individual compounds to hTR-P2b RNA. The calculated KD values for some of the compounds showing a preference for binding to hTR-P2b relative to the A-site rRNA construct are shown in Table 3.
Figure 7.

The 24 compounds that showed a distinct preference for binding to hTR-P2b RNA, relative to A-site rRNA, belong primarily to one of four classes of structure. Representatives of these four classes are shown.
Table 3.
Dissociation constants measured for some compounds of Figure 3 measured by monitoring chemical shifts of hTR-P2b imino protons. Compounds selected show a preference for binding to hTR-P2b relative to A-site rRNA.
| Classes | Compound | Dissociation Constant |
|---|---|---|
|
Class 2 Chained Polyaromatics |
6 | 570 +/- 40 μM |
| 7 | 1.1 +/- 0.2 mM | |
| 12 | 1.5 +/- 0.3 mM | |
| 13 | 180 +/- 30 μM | |
|
Class 4 4-Aminoquinolines |
26 | 84 +/- 31 μM |
|
Class 6 Phenothiazines |
33 | 310 +/- 120 μM |
| 34 | 1.1 +/- 0.2 mM | |
| 38 | 450 +/- 80 μM | |
|
Class 10 6-7-6 Ring Systems |
46 | 1.3 +/- 0.2 mM |
| 48 | 1.1 +/- 0.1 mM | |
| 49 | 1.5 +/- 0.4 mM |
Class 2
The 13 compounds in class 2 exhibit mostly weak-to-medium STD effects. The STD spectra imply that the aromatic rings of these structures are in close contact with the RNA protons; no STD effects are observed for protons of the sidechains, so no conclusions about their direct interaction with the RNA can be made. Analysis of the imino proton chemical shifts shows that in all cases the only resonances experiencing changes are those of the U region. Kd values calculated for those compounds that preferentially bind to hTR-P2b are approximately 200 μM for strong binders, 0. 5 mM for medium binders and >1 mM for weak binders. In general, this class tends to be specific for hTR.
Class 4
Of the eleven compounds in this class, three show a preference for binding to hTR-P2b. Compound 26 manifests a strong STD effect, and the Kd value was calculated to be ~100 μM. Imino proton resonance chemical shift changes upon addition of the ligands arise from residues in the U region as well as G110 and G98. This means that the binding mode is not solely to the U region. In general, this class tends to bind strongly but nonspecifically.
Class 6
The STD spectra for the compounds of class 6 with hTR-P2b RNA do not give completely accurate information, because they have a signal near the secondary irradiation frequency (5.5 ppm). Consequently, that ligand proton experiences some perturbation in magnetization directly, which can have an effect on the other ligand protons via spin diffusion. Therefore, we cannot discern whether the STD effect is strong, medium or weak. Of course, we could change our STD NMR screening protocol and select a different irradiation frequency that would work for this class of compounds, but we set up the protocol to be fairly generally applicable to RNA targets. We can still surmise from the STD spectra that the aromatic rings of the structures have close contact with the RNA protons, but weaker STD effects are observed for protons of the sidechains. The STD spectra might be deemed unreliable, but analysis of the imino proton chemical shifts from the RNA unequivocally demonstrates binding. This analysis shows that in all cases the resonances experiencing chemical shift changes are those of the U region. Dissociation constants were calculated for those compounds exhibiting preferential binding to hTR RNA. The measured Kd values vary from 0.3-1.1 mM. In the present context, most can be classified as strong binders. Only compound 34 manifests a weak STD effect, and the Kd value was calculated to be 1.1 mM. This class shows mixed specificity.
We have previously found that phenothiazines can bind to some loop regions of different RNAs.14,15,21,22 Interestingly, different phenothiazine derivatives manifest distinctly different binding preferences; some will not bind at all to one or more loops but will bind to another loop.
Class 10
Six of the eight compounds in this class manifest a weak STD effect. The STD spectra imply that the aromatic rings of these structures are in close contact with the RNA protons; no STD effects are observed for protons of the sidechains. Imino proton chemical shift changes are observed only for resonances emanating from the U region. Kd values calculated for those compounds that preferentially bind to hTR RNA vary from 1.1 to 1.5 mM for these weak binders. Although this class tends to bind weakly, it also tends to be specific for hTR.
Binding Affinity
As seen in Table 3, the ligands we have identified do not bind tightly to hTR. However, they are not so poor as it might appear, since (a) ligand binding to RNA is generally not as tight as to proteins, (b) known RNA-binding ligands typically have multiple positive charges while most of the binders discovered here have a charge of 0 or +1, and (c) most of compounds identified here are smaller than the best-known RNA ligands. With the exception of a few aptamers, most known ligands, including clinically useful drugs, bind to RNA with micromolar affinity and rarely with a Kd value smaller than 10-7 M. Ryu and Rando27 give several examples. Most RNA binders with micromolar affinity have molecular weights greater than 500 daltons. Very roughly, the efficiency of known RNA-binding ligands to the free energy of binding is about 12-16 cal/dalton. The ligands identified in the present study to bind with the best affinity exhibit about the same efficiency. Of course, significant improvements in affinity are likely to result if additional charged groups are incorporated into new derivatives.
Summary
We have been successful in identifying interesting new ligands using the approach we present here, which starts with flexible-ligand, flexible-receptor docking of potential ligands from a small database to the selected hTR-P2b RNA target via MORDOR with subsequent STD NMR screening of high-ranking compounds. To enrich and diversify our database of compounds that could bind to the target, experimental binders from the first round were used as the basis to search for similar compounds in a very large database, yielding a database of potential ligands that was subjected to docking and ranking via MORDOR with additional high-ranking compounds selected for NMR screening. A remarkably high percentage of the compounds tested experimentally were found via NMR mapping to bind to the U-rich region of hTR-P2b; since we did not do experimental screening of a set of randomly selected compounds, there remains a possibility that the U-rich region has a high propensity for binding to small molecules with little or no charge, unlike any other RNA motif studied to date. Among the newly discovered ligands, most of which have molecular weight <500 daltons and are in many different compound classes, are several interesting compounds with a charge of either 0 or +1. The largest STD effects are observed for some of the ring systems, implying that the aromatic groups of the compounds maintain contact with the RNA, probably due to stacking with bases in RNA. Many of the ligands exhibit a distinct preference to bind hTR-P2b RNA rather than A-site rRNA. The binding affinity to the hTR-P2b RNA target depends on ligand characteristics, and the affinity can evidently be modulated by addition or deletion of certain chemical groups. This gives hope that eventually these scaffolds can be fine-tuned to bind to a variety of unique tertiary RNA structures with the required affinity and specificity. In the relatively new field of targeting RNA with small molecules, it remains to be seen if one will be able to generate a ligand of high affinity and specificity from a low affinity lead compound as has been accomplished for protein targets.
Experimental Procedures
Virtual Screening with MORDOR and Similarity Searching
The MORDOR program has been optimized for docking quality rather than for docking speed; the time spent by MORDOR to dock one ligand currently takes 0.5-3 hrs, depending on ligand and receptor characteristics, on a single AMD Athlon 3200 MHz, so we can realistically dock only modest-sized libraries of compounds at present. The strategy used in our docking is a succession of docking/experimental testing cycles.
Initially, MORDOR was used to screen the FDA-drug-approved KEGG library of 2,669 compounds (http://www.genome.jp/ligand/)25 for binding to hTR-P2b RNA (Protein Data Bank ID code 1NA2)7 in round R1. Twenty of the best-ranking compounds resulting from this search were obtained; criteria for selection included binding energy (VDW, electrostatics and solvation energy) and VDW interaction energy, RNA drug-like potentiality, diversity, estimated water solubility, and cost. The thirteen that were soluble and did not cause precipitation/aggregation when added to the RNA were screened via STD NMR; binding solely to a single site was verified by examining the imino proton changes upon binding for ten compounds. To enrich the pool of potential ligands, the 2-D similarity searching feature in ZINC (http://blaster.docking.org/zinc8/)24 was used to search the full ZINC library of more than two million “drug-like” molecules, amenable to searching. In effect, each of the binders provides a scaffold for the similarity search, but the search turns up compounds with a diversity of atom heterogeneity in the scaffold as well as substituents decorating the scaffold. This resulted in ~4700 compounds that could feasibly bind to hTR-P2b. These 4700 compounds were docked and ranked using MORDOR in round two (R2). Examination of R2 results lead to selection of additional compounds for screening by STD NMR. Early second round NMR screening results and further review of R1 and R2 lead to two more ZINC similarity searches, yielding another 934 hits and 126 hits, respectively, leading to a final RNA ligand-like database of ~5750 compounds. The compounds in this database were docked and ranked with MORDOR. In total, we docked ~14000 compounds, including the original KEGG library subset of 2669 compounds From this ranked list, an additional 93 compounds were selected and obtained for NMR screening and mapping.
MORDOR docking procedure
The details of using MORDOR have been described in detail,19 so a brief description should suffice here. At first, the target is prepared for docking. For this purpose, the high-resolution structure of hTR-P2b RNA (PDB Code 1NA2) was energy-minimized within an RMSD of 0.5 Å from the experimental structure using the CHARMM-27 force field28 and an all-atom implicit solvent model, the Generalized Born/Molecular Volume (GBMV) model,29 for calculating the solvent energy. Ten spheres defining the binding pocket in the U-rich region of the receptor were calculated and subsequently used at a starting point to initially place an atom of a putative ligand at the surface of the receptor prior to the docking search.19 An atomic RMSD penalty is added to the potential energy, which acts as a driving force to constantly change the ligand from its initial position in small increments of 0.1 Å. The vicinity of this binding pocket is extensively sampled, with total energy (receptor + ligand) as well as interaction energy being recorded at each step. This procedure is repeated for each heavy atom of the ligand for a given sphere. The whole procedure is repeated for each of the ten spheres. The ligand is thus placed in various orientations at ten different hot spots on the receptor surface, with numerous explorations of adaptive fitting of ligand and receptor, enabling a broad sampling of ligand binding to the receptor.
One difference from the published docking procedure19 is that in the current study, we used the interaction energy to discriminate and rank the “best” docking poses. Subsequent to the calculations in the present study, we found that total energy (receptor + ligand) is a better metric than the interaction energy for selecting and ranking binding poses.19
Preparation of RNA Samples
The RNA constructs were synthesized by in vitro transcription using T7 RNA polymerase and a synthetic DNA template.30 The DNA templates consisted of a double-stranded 18-base pair T7 promoter sequence plus a single-stranded coding sequence. By using this method we obtained not only the desired RNA but also undesired products, such as the addition of one or more nontemplated nucleotides at the 3'terminus of the nascent RNA (the N+1 product). Modification in the last two nucleotides at the 5'terminus of the DNA template with methoxy moieties at the ribose C2' position significantly reduce N+1 product formation by T7 RNA polymerase and also increase the abundance of the desired RNA,31 so this technique was employed. Samples were purified by polyacrylamide gel electrophoresis and electroelution, and dialyzed. NMR samples were suspended in NMR buffer that contained 20 mM sodium phosphate (pH=6.7) and 50 mM sodium chloride with either 99.9% D2O or a 90/10% H2O/D2O mixture. RNA solutions were heated to 90 °C for 5 minutes and snap-cooled in an ice bath. Final RNA concentrations were 25 μM for STD NMR experiments and 50 μM for imino proton NMR titration studies. Reagents were purchased from various companies.
NMR Spectroscopy
All NMR spectra were acquired in either a Bruker DRX 500 MHz or Bruker 800 MHz NMR spectrometer; both are equipped with a Cryoprobe. In all STD experiments, on-resonance irradiation was set to 5.5 ppm and off-resonance irradiation was set to 30 ppm, where no RNA resonances are present. The temperature was set to 15 °C in all experiments. STD experiments were processed with the XWIN-NMR software. Typically, a spectrum of the compound alone at 500 μM concentration was acquired, and then RNA was added to produce a 25 μM final RNA concentration. For STD experiments, 256 scans were acquired. Presaturation of RNA resonances was achieved by an appropriate number of band-selective G4 Gaussian cascade pulses to give a saturation time of 2 seconds. In 1D NMR spectra of imino signals in H2O, water suppression was achieved by including a WATERGATE module in the pulse sequence before acquisition, with the excitation profile optimized for maximum intensity at 13 ppm.
Calculation of Kd
Ligand binding was monitored by changes in chemical shifts of RNA imino protons. In the limit of rapid exchange of free and bound ligand, at any given total concentration of the ligand, x, the observed imino chemical shift is a linear function of the bound concentration of RNA:
| (1) |
where δ(x) is the observed chemical shift of RNA at a total concentration of the ligand x, δf is the chemical shift of free RNA, δb is the chemical shift of bound RNA, r is the total concentration of RNA (50 μM), and c is the concentration of the RNA-ligand complex. Since the dissociation constant Kd is
| (2) |
we obtain
| (3) |
Kd values were estimated by nonlinear least squares fitting of equations (1) and (3) via the Python function leastsq (module scipy), which uses the Levenburg-Marquardt algorithm, for observed values {x; δ(x)}.
To estimate errors in the Kd estimates, we used the Monte Carlo approach. Observed chemical shifts were randomly perturbed 10000 times using Gaussian distributions generated for each chemical shift value. The standard deviations for the Gaussian distributions were estimated as experimental uncertainties in chemical shift measurements, which increase at higher ligand concentrations as imino proton linewidths become broader. The following standard deviation values were used: 0.001 ppm for x ≤ 250 mM, 0.0025 ppm for 250 mM < x ≤ 750 mM, and 0.005 ppm for x > 750 mM. The Monte Carlo results of the binding curve fitting were combined for at least 2 (typically 3) imino proton peaks monitored for each ligand, so the standard deviation was obtained from 20,000-40,000 simulations.
Supplementary Material
Acknowledgment
This work was partially supported by grant AI46967 from the National Institutes of Health and by a grant from the UCSF Prostate Cancer Center Development Research Program. We are grateful to Abram Calderon with aid on preparation of T7 RNA polymerase, Dr. Alejandra Gallardo-Godoy for running some of the LC-MS experiments for compound characterization, and to Dr. John Irwin for discussions and help with ZINC. We thank Dr. Conrad Huang for implementing MORDOR visualization in the VIEWDOCK module of CHIMERA. We thank OpenEye for generously providing the software suite that was used to regularize the ligand and the Drug Synthesis and Chemistry Branch, Developmental Therapeutics Program, Division of Cancer Treatment and Diagnosis, National Cancer Institute for some of the compounds tested (http://dtp.nci.nih.gov/docs/3d_database/dis3d.html).
Footnotes
Supporting Information Available: 1H NMR and LC/MS data for the compounds in Figure 3. These materials are available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: hTR, human telomerase RNA; TERT, telomerase reverse transcriptase; CRconserved region; hTR-P2b, the P2b moiety of hTR as shown in Figure 1; P3, the P3 moiety of hTR as shown in Figure 1; VS, virtual screening; ACD, Available Chemicals Directory; MORDOR, MOlecular Recognition with a Driven dynamics OptimizeR; STD NMR, saturation-transfer-difference NMR; KEGG, Kyoto Encyclopedia of Genes and Genomes; ZINC; ZINC Is Not Commercial; A-site rRNA, aminoacyl-tRNA site on ribosomal RNA; nt, nucleotide; VDW, van der Waals; RMSD, root-mean-square-deviation
References
- (1).Iczkowski KA, Pantazis CG, McGregor DH, Wu Y, Tawfik OW. Telomerase reverse For Review. Confidential - ACS transcriptase subunit immunoreactivity: a marker for high-grade prostate carcinoma. Cancer. 2002;95:2487–2493. doi: 10.1002/cncr.10988. [DOI] [PubMed] [Google Scholar]
- (2).Shay JW, Wright WE. Telomerase therapeutics for cancer: challenges and new directions. Nat. Rev. Drug Discov. 2006;5:577–584. doi: 10.1038/nrd2081. [DOI] [PubMed] [Google Scholar]
- (3).Blackburn EH. The end of the (DNA) line. Nature Struct Biol. 2000;7:847–850. doi: 10.1038/79594. [DOI] [PubMed] [Google Scholar]
- (4).Blackburn EH. Telomeres and telomerase: their mechanisms of action and the effects of altering their functions. FEBS Lett. 2005;579:859–862. doi: 10.1016/j.febslet.2004.11.036. [DOI] [PubMed] [Google Scholar]
- (5).Chen JL, Blasco MA, Greider CW. Secondary structure of vertebrate telomerase RNA. Cell. 2000;100:503–514. doi: 10.1016/s0092-8674(00)80687-x. [DOI] [PubMed] [Google Scholar]
- (6).Comolli LR, Smirnov I, Xu L, Blackburn EH, James TL. A molecular switch underlies a human telomerase disease. Proc Natl Acad Sci U S A. 2002;99:16998–17003. doi: 10.1073/pnas.262663599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Theimer CA, Finger LD, Trantirek L, Feigon J. Mutations linked to dyskeratosis congenita cause changes in the structural equilibrium in telomerase RNA. Proc Natl Acad Sci USA. 2003;100:449–454. doi: 10.1073/pnas.242720799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Yingling YG, Shapiro BA. Dynamic behavior of the telomerase RNA hairpin structure and its relationship to dyskeratosis congenita. J Mol Biol. 2005;348:27–42. doi: 10.1016/j.jmb.2005.02.015. [DOI] [PubMed] [Google Scholar]
- (9).Chen JL, Greider CW. Functional analysis of the pseudoknot structure in human telomerase RNA. Proc Natl Acad Sci U S A. 2005;102:8080–8085. doi: 10.1073/pnas.0502259102. discussion 8077-8089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Herbert BS, Gellert GC, Hochreiter A, Pongracz K, Wright WE, Zielinska D, Chin AC, Harley CB, Shay JW, Gryaznov SM. Lipid modification of GRN163, an N3'-->P5' thiophosphoramidate oligonucleotide, enhances the potency of telomerase inhibition. Oncogene. 2005;24:5262–5268. doi: 10.1038/sj.onc.1208760. [DOI] [PubMed] [Google Scholar]
- (11).Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- (12).Krumrine J, Raubacher F, Brooijmans N, Kuntz ID. Principles and methods of docking and ligand design. Methods Biochem Anal. 2003;44:443–476. [PubMed] [Google Scholar]
- (13).Filikov AV, Mohan V, Vickers TA, Griffey RH, Cook PD, Abagyan RA, James TL. Identification of ligands for RNA targets via structure-based virtual screening: HIV-1 TAR. J. Comp.-Aided Molec. Design. 2000;14:593–610. doi: 10.1023/a:1008121029716. [DOI] [PubMed] [Google Scholar]
- (14).Lind KE, Du Z, Fujinaga K, Peterlin BM, James TL. Structure-Based Computational Database Screening, In vitro Assay, and NMR Assessment of Compounds that Target TAR RNA. Chem. & Biol. 2002;9:185–193. doi: 10.1016/s1074-5521(02)00106-0. [DOI] [PubMed] [Google Scholar]
- (15).Du Z, Lind KE, James TL. Structure of TAR RNA Complexed with a Tat-TAR Interaction Nanomolar Inhibitor that Was Identified by Computational Screening. Chem. & Biol. 2002;9:707–712. doi: 10.1016/s1074-5521(02)00151-5. [DOI] [PubMed] [Google Scholar]
- (16).Morley SD, Afshar M. Validation of an empirical RNA-ligand scoring function for fast flexible docking using Ribodock. J. Comput.-Aided Mol. Design. 2004;18:189–208. doi: 10.1023/b:jcam.0000035199.48747.1e. [DOI] [PubMed] [Google Scholar]
- (17).Detering C, Varani G. Validation of automated docking programs for docking and database screening against RNA drug targets. J. Med. Chem. 2004;47:4188–4201. doi: 10.1021/jm030650o. [DOI] [PubMed] [Google Scholar]
- (18).Grünberg R, Leckner J, Nilges M. Complementarity of structure ensembles in protein-protein binding. Structure. 2004;12:2125–2136. doi: 10.1016/j.str.2004.09.014. [DOI] [PubMed] [Google Scholar]
- (19).Guilbert C, James TL. Docking to RNA via root-mean-square-deviation-driven energy minimization with flexible ligands and flexible targets. J. Chem. Inf. Model. 2008;48:1257–1268. doi: 10.1021/ci8000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Mayer M, James TL. Detecting ligand binding to a small RNA target via saturation transfer difference NMR experiments in D2O and H2O. J. Am. Chem. Soc. 2002;124:13376–13377. doi: 10.1021/ja027526z. [DOI] [PubMed] [Google Scholar]
- (21).Mayer M, James TL. NMR-Based Characterization of Phenothiazines as a RNA Binding Scaffold. J. Am. Chem. Soc. 2004;126:4453–4460. doi: 10.1021/ja0398870. [DOI] [PubMed] [Google Scholar]
- (22).Mayer M, Lang PT, Gerber S, Madrid PB, Pinto IG, Guy RK, James TL. Synthesis and testing of a focused phenothiazine library for binding to HIV-1 TAR RNA. Chem. & Biol. 2006;13:993–1000. doi: 10.1016/j.chembiol.2006.07.009. [DOI] [PubMed] [Google Scholar]
- (23).Theimer CA, Blois CA, Feigon J. Structure of the human telomerase RNA pseudoknot reveals conserved tertiary interactions essential for function. Mol Cell. 2005;17:671–682. doi: 10.1016/j.molcel.2005.01.017. [DOI] [PubMed] [Google Scholar]
- (24).Irwin JJ, Shoichet BK. ZINC-a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005;45:177–182. doi: 10.1021/ci049714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Goto S, Okuno Y, Hattori M, Nishioka T, Kanehisa M. LIGAND: database of chemical compounds and reactions in biological pathways. Nucleic Acids Res. 2002;30:402–404. doi: 10.1093/nar/30.1.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jahnke W, Erlanson DA. Fragment-based Approaches in Drug Discovery. Wiley-VCH; New York: 2006. [Google Scholar]
- (27).Ryu DH, Rando RR. Aminoglycoside binding to human and bacterial A-Site rRNA decoding region constructs. Bioorg Med Chem. 2001;9:2601–2608. doi: 10.1016/s0968-0896(01)00034-7. [DOI] [PubMed] [Google Scholar]
- (28).Brooks BR, Bruccoleri RE, Olafsen BD, States DJ, Swaminathan S, Karplus M. J. Comp. Chem. 1983;4:187–217. [Google Scholar]
- (29).Feig M, Im W, Brooks CL., 3rd Implicit solvation based on generalized Born theory in different dielectric environments. J Chem Phys. 2004;120:903–911. doi: 10.1063/1.1631258. [DOI] [PubMed] [Google Scholar]
- (30).Milligan JF, Uhlenbeck OC. Synthesis of small RNAs using T7 RNA polymerase. In: Dahlberg JE, Abelson JN, editors. Meth. Enzym. Vol. 180. Academic Press; New York: 1989. pp. 51–62. [DOI] [PubMed] [Google Scholar]
- (31).Kao C, Zheng M, Rudisser S. A simple and efficient method to reduce nontemplated nucleotide addition at the 3 terminus of RNAs transcribed by T7 RNA polymerase. RNA. 1999;5:1268–1272. doi: 10.1017/s1355838299991033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.