

Abstract
Blood coagulation often accompanies bacterial infections and sepsis and is generally accepted as a consequence of immune responses. Though many bacterial species can directly activate individual coagulation factors, they have not been shown to directly initiate the coagulation cascade that precedes clot formation. Here we demonstrated, using microfluidics and surface patterning, that the spatial localization of bacteria substantially affects coagulation of human and mouse blood and plasma. Bacillus cereus and Bacillus anthracis, the anthrax-causing pathogen, directly initiated coagulation of blood in minutes when bacterial cells were clustered. Coagulation of human blood by B. anthracis required secreted zinc metalloprotease InhA1, which activated prothrombin and factor X directly (not via factor XII or tissue factor pathways). We refer to this mechanism as ‘quorum acting’ to distinguish it from quorum sensing—it does not require a change in gene expression, it can be rapid and it can be independent of bacterium-to-bacterium communication.
This paper describes a physical and biochemical mechanism responsible for regulating the initiation of human blood coagulation by bacteria. In vivo, coagulation often accompanies bacterial infections of the blood and is believed to be a consequence of immune and inflammatory responses1-5. Immune and inflammatory responses cause upregulation of tissue factor on the timescale of hours and lead to increased coagulation6,7. One of the few drugs available to treat septic shock, activated protein C, is also an anticoagulant8. This coagulation is believed to prevent dissemination of bacteria through the blood9,10 but also results in serious vascular damage due to blockage and injury of blood vessels8. Coagulation accompanying bacterial infections of the blood is particularly relevant for people infected with anthrax, which involves sepsis and disseminated intravascular coagulation caused by the pathogen Bacillus anthracis4. Here, we considered an alternative and complementary mechanism for the coagulation that accompanies infection: direct activation of the human coagulation cascade through activation of coagulation factors by bacteria.
Many bacteria and bacterial components can directly activate individual human coagulation factors. However, direct initiation of the coagulation cascade and the formation of a propagating clot are not typically observed11-17. These bacterial components usually activate low levels of coagulation factors, which does not result in the amplification and positive feedback necessary to form a clot that can grow and propagate. For example, Staphylococcus aureus produces coagulase, a protein that binds prothrombin stoichiometrically and leads to cleavage of fibrinogen to fibrin14. However, this conversion simply precipitates fibrin and does not result in production of thrombin, feedback or amplification of the coagulation cascade. Escherichia coli that express the protein Curli are also known to activate coagulation factors, such as factor XII (ref. 17). This process was shown to cause slower initiation of coagulation due to depletion of factor XII (ref. 17). Bacteria are also well known to directly initiate coagulation in some organisms, such as horseshoe crabs, but this mechanism of controlling infection is believed to have been lost during the evolution of vertebrates18. All of these results prompt the following simple question: are bacteria capable of directly initiating the coagulation cascade and causing coagulation of human blood?
We hypothesized that initiation of coagulation by bacteria would be regulated by the spatial localization, not the total amount, of bacteria. In other words, for bacteria that activate coagulation factors, coagulation would only occur when a cluster of bacteria forms. This hypothesis was based on previous experiments with human blood and plasma that showed that (i) stimuli must exceed a local threshold concentration to initiate coagulation19,20, and (ii) this threshold response to concentration leads to a spatial threshold response, in which coagulation initiates on a patch of stimulus above but not below a threshold size21,22. The threshold to concentration for coagulation is due to competition between production and inhibition of activated coagulation proteases. The spatial threshold response to the size of stimulus arises from the competition between the local production of activated coagulation proteases at the site of the stimulus and the diffusion of these proteases away from the stimulus. We hypothesized that individual bacteria as a stimulus of coagulation are below the threshold stimulus size needed to initiate coagulation (Fig. 1a). Therefore, a solution of uniformly dispersed bacteria would not initiate coagulation if the bacteria are spaced far apart. However, a sufficiently large cluster of bacteria would generate a high local concentration of activated coagulation factors, which would exceed the threshold concentration and initiate the coagulation cascade rapidly. In this paper we showed that coagulation can be controlled by changing the spatial distribution, or clustering, of bacteria.
Figure 1.
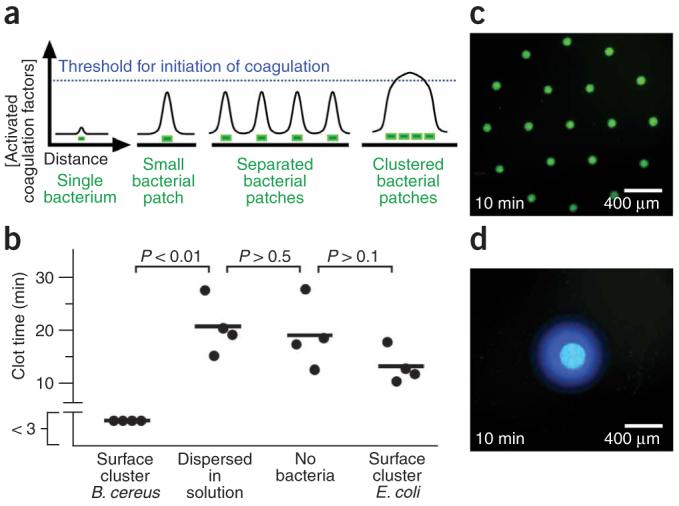
Human blood plasma coagulates on spatially localized B. cereus in the absence of flow. (a) A schematic drawing illustrates the coagulation potential of a single bacterium, a single small surface patch of bacteria, surface patches of bacteria that are separated and surface patches of bacteria that are clustered together. The concentration of activated coagulation factors only exceeds the threshold required for initiation of coagulation (blue dashed line) when a sufficient number of bacteria are clustered together as a large patch. (b) The chart compares the clotting times of human blood plasma on the same amount of bacteria either clustered in a large patch or dispersed in solution. Each data point represents an independent experiment, where clot time was measured in a microfluidic chamber using fluorescence microscopy. Imaging began 3 min after plasma was added to the surface cluster of bacteria. Clotting on the surface cluster of B. cereus occurred before the first image at 3 min, as indicated by a break in the y axis. (c,d) Fluorescent photographs of a microfluidic chamber used to test coagulation (blue fluorescence) of human blood plasma on clustered bacteria (green fluorescence) patterned as patches of different sizes. Coagulation occurs rapidly on a large patch (d; 7 independent experiments) but not on an array of smaller patches containing the same amount of bacteria (c; 7 independent experiments).
RESULTS
Spatial localization of B. cereus controls coagulation
To test this hypothesis (Fig. 1a), we compared the clot time of human blood plasma exposed to bacteria dispersed in solution to the clot time of human blood plasma containing bacteria clustered on the surface of the microfluidic chamber (Fig. 1b). Bacillus cereus spatially localized to a surface cluster rapidly initiated coagulation. However, B. cereus dispersed in solution at concentrations of up to 107 colony-forming units (CFU) ml-1 did not initiate coagulation (P < 0.01, for clustered versus dispersed B. cereus). The clot times of human blood plasma exposed to the dispersed bacteria were not significantly different from the clot times of the control samples of human blood plasma that did not contain bacteria (P > 0.5). In another control experiment, clusters of E. coli did not rapidly initiate coagulation, and there was no significant difference between the clot time of clustered E. coli and the samples of plasma that did not contain bacteria (P > 0.1). These results demonstrate that not all bacterial strains initiate coagulation in this experimental setup. The E. coli control strain, which does not produce the Curli protein, was used here because it was previously shown not to activate coagulation factors17. In the solution-phase experiments (Fig. 1b), approximately 5 × 105 CFU of B. cereus in 50 μl of human blood plasma did not initiate coagulation. However, significantly fewer bacteria initiated coagulation when clustered—single clusters of approximately 4 × 103 CFU were capable of initiating coagulation in 10 ml of human blood plasma. This number was also substantially lower than the number of bacteria (approximately 108 CFU) that could not initiate coagulation when dispersed in 10 ml of human blood plasma. Control experiments confirmed that fluorescence observed in coagulation by B. cereus corresponded to true initiation of the coagulation cascade and was not due to simple cleavage of the fluorogenic substrate or to an S. aureus-type coagulase activity (Supplementary Figs. 1 and 2 online). For example, both coagulation factor X and prothrombin are required for initiation of coagulation by B. cereus, which is not expected for S. aureus-type coagulase activity.
In a second experiment, we used microfluidics23,24 and micropatterned surfaces to control the spatial distribution of bacteria (Fig. 1c,d) and to demonstrate that the size of the cluster, rather than the amount of bacteria, can control the rate of initiation of coagulation of human blood plasma. We patterned the surface of a microfluidic chamber with 90 μm patches of B. cereus expressing green fluorescent protein (GFP). We then monitored coagulation of human blood plasma on these patches in the absence of flow. On smaller patches (90 μm) spaced far apart (400 μm), coagulation was slow, initiating on the first patch in 9 min ± 1 with clotting on all the patches in the array in 22 ± 3 min (mean ± s.e.), which indicates that the individual 90 μm patches were below the size necessary to initiate coagulation rapidly (Fig. 1c). However, when the same number of bacteria were patterned closer together to form a large patch, coagulation initiated rapidly in 5 ± 1 min (mean ± s.e.) over the entire patch (Fig. 1d) (P < 0.01 in comparison with initiation on the first 90 μm patch and P < 0.005 in comparison with initiation on the entire set of patches). For this large patch, activated coagulation factors accumulated and exceeded the threshold concentration because diffusion of activated coagulation factors off of the patch was slower than the production of activated coagulation factors.
B. cereus initiates coagulation of flowing whole blood
To test whether B. cereus initiates coagulation in the presence of flow, human whole blood was flowed over localized colonies of B. cereus in microfluidic channels (Fig. 2). We wished to test this effect because flow is important in maintaining hemostasis and could affect phenomena that rely on local concentration thresholds. We demonstrated previously that thresholds to initiation and propagation of coagulation are preserved in the presence of flow25,26. In these experiments, several parameters known to contribute to the coagulation process in vivo were incorporated and carefully controlled, including flow and shear rates, the geometry and surface chemistry of channels, and the presence of platelets and cells of the blood. Other components that contribute to coagulation in vivo were not tested here, including the presence of membrane proteins and other components of the vessel wall. The components of the endothelium are known to greatly contribute to the coagulation process, and differences are likely to exist between the microfluidic system described here and the in vivo setting. Clusters of bacteria in microfluidic channels were made by encapsulating bacteria in gel microdroplets (GMDs)27. GMDs consisted of colonies of bacteria and magnetic particles 1 μm in diameter contained in agarose spheres approximately 50 μm in diameter; the magnetic particles allowed the GMDs to be trapped in the microfluidic channels by a magnet incorporated into the device near the channel (Fig. 2a)28. Clusters of B. cereus initiated coagulation of flowing human whole blood in 3-13 min (Fig. 2b,c), whereas coagulation did not occur until 48-59 min in experiments with the control strain of E. coli (Fig. 2d; P < 0.001). Previous work suggests that initiation and propagation of coagulation of human blood and plasma are sensitive to shear rate26. It predicts that coagulation induced by bacteria will be most pronounced in regions of low shear, such as in dead volumes in venous valves or in the extremities of people that are immobilized and experiencing venous stasis (for example, in intensive care units). This prediction has not yet been tested.
Figure 2.

Human whole blood coagulated on spatially localized B. cereus in the presence of flow. (a) A microphotograph shows the microfluidic device used to flow human whole blood over colonies of bacteria. The bacteria were localized in GMDs that also contained magnetic particles. Magnets were used to localize the GMDs in the device. (b) A brightfield image shows coagulation of human whole blood on GMDs containing B. cereus expressing GFP (overlaid green fluorescence). (c,d) Microphotographs of fluorescence show coagulation of human whole blood (blue) on GMDs containing B. cereus (green) (c) but not on E. coli (green) (d). White dashed lines outline the channel walls. Images were taken 11 min after blood was introduced into the device. (e) A graph shows the clot times of flowing whole blood on colonies of B. cereus and E. coli in microfluidic devices. Each data point represents an independent experiment.
Classical initiation points of the network are bypassed
There are two classical pathways to initiate coagulation (Fig. 3a). The principal pathway in vivo is initiated by tissue factor, a high-affinity receptor for coagulation factor VII. The other pathway, which is activated via factor XII, has been suggested as an initiation point for some bacteria17. We initially hypothesized that coagulation by B. cereus would occur by activation of either factor XII or factor VII. To determine which coagulation factors are essential, we tested blood plasmas that were depleted of specific coagulation factors. The clot times by B. cereus were compared to the clot times by reagents of the prothrombin time (PT) test or the activated partial thromboplastin time (APPT) test (Fig. 3b,c). The PT test initiates coagulation via the tissue factor/factor VII pathway, and the APTT test initiates coagulation via the factor XII pathway. Surprisingly, coagulation of plasma immunodepleted in either factor XII or factor VII was still rapidly initiated by B. cereus, which indicates that these factors are not essential for this process. However, coagulation did not occur within 30 min in plasma immunodepleted in either factor X or prothrombin. We performed further experiments with purified coagulation factors and found that B. cereus is capable of directly activating prothrombin (factor II) and factor X (Supplementary Figs. 3 and 4 online). The initiation point for B. cereus occurred at the major hubs of the coagulation network. It remains to be seen whether further network analysis can be used to identify inhibition points of the network that could stop coagulation initiated by bacteria while maintaining the ability of blood to initiate coagulation by tissue factor.
Figure 3.

B. cereus initiated coagulation by a mechanism different from that of the reagents used in either the PT test or the APPT test, bypassing factor VII and factor XII in initiating the coagulation cascade. (a) A partial network diagram for the hemostasis network. The reactions shown are those used in the numerical simulations discussed below. Inhibitory and positive feedback processes are shown in light and dark gray, respectively, and the principal forward reactions are shown in black. Blue indicates points of classical activation; red indicates points essential for activation by bacteria. (b,c) The clot times of plasmas deficient in a specific coagulation factor in the PT test (b) and APTT test (c) are plotted against the clot times after initiation by B. cereus bacteria. In these graphs, prothrombin is labeled as FII. Data points circled in blue indicate factor-deficient plasmas that show very slow clotting in either the PT or APTT test but that initiate coagulation rapidly by B. cereus, which indicates that these factors are not essential for initiation of coagulation by B. cereus. Data points circled in red indicate factors that are essential for initiation of coagulation by B. cereus. Each data point represents the average of four measurements of clot time on large (>300 μm) clusters of bacteria in microfluidic chambers.
Multiple Bacillus species initiate coagulation
In addition to B. cereus, we found that clusters of several other Bacillus species, including B. anthracis, the anthrax-causing pathogen, rapidly initiated coagulation of human blood plasma (Fig. 4a). The closely related species Bacillus thuringiensis and other species, including Bacillus subtilis and Bacillus licheniformis, also initiated coagulation. Experiments with purified coagulation factors showed a strong correlation between the ability of these strains to initiate coagulation and their ability to activate prothrombin and factor X (Supplementary Fig. 4). Furthermore, components secreted from the bacteria into solution were also capable of activating purified coagulation factors, including prothrombin and factors X and XI, but not factors VII and IX (Supplementary Fig. 5 online). It is surprising that an insect pathogen, B. thuringiensis, rapidly caused coagulation of human plasma. This result may be due to the conservation of protease cascades29,30. Further work is needed to understand these phenomena.
Figure 4.

Human blood plasma coagulates on surface clusters of many Bacillus species, including B. anthracis, but not on control species of E. coli and S. aureus. (a) Chart quantifying the clot times of human blood plasma on clusters of bacteria in a microfluidic chamber in the absence of flow. (b,c) B. anthracis strains that do not produce secreted zinc metalloproteases NprB (ΔnprB) or InhA1 (ΔinhA1) have reduced ability to activate purified human prothrombin (b) and purified human factor X (c). (d) Purified prothrombin is activated by InhA1 purified from B. anthracis. (e) A graph quantifying the clot times of human blood plasma on Ames 35, ΔnprB and ΔinhA1 strains of B. anthracis shows that the Ames 35 control strain rapidly initiated coagulation. The ΔnprB strain also initiated coagulation, whereas the ΔinhA1 strain did not accelerate coagulation relative to background clotting (P < 0.001 for Ames 35 versus ΔinhA1). (f) A graph quantifying the clot times of human blood plasma on control Ames 35 and a ΔluxS strain shows that the ΔluxS strain rapidly initiated coagulation despite its inability to secrete autoinducer-2 (1), a quorum-sensing signaling molecule. Each data point (a,e,f) represents the clot time on a single large (>300 μm) cluster of bacteria in a microfluidic chamber that was measured by fluorescence microscopy. Clotting on some surface clusters occurred before the first images obtained at 3 min (a) or 2 min (e and f), as indicated by breaks in the y axis.
Coagulation by B. anthracis requires metalloprotease InhA1
To identify the molecular components responsible for activating coagulation factors and initiating coagulation, we screened a small library of B. anthracis Ames 35 mutants; most of these mutants are deficient in secretion of a specific protease. We chose to investigate mutants of B. anthracis for two reasons. First, B. anthracis secretes a much smaller number of proteases than B. cereus, resulting in a smaller screening library. Second, there is currently a demand for identifying the essential molecular components responsible for the pathophysiology of anthrax31,32. It has been shown previously that the B. anthracis protein lethal factor does not initiate coagulation33. Inthe mutants used here, the genes corresponding to the proteases were removed by Cre recombinase (Supplementary Fig. 6 online)34. We hypothesized that secreted proteases were involved because solutions containing secreted components from B. anthracis cells were found to activate purified coagulation factors (Supplementary Fig. 5), but with activities below the threshold needed for coagulation.
We found that bacteria that did not produce either metalloprotease NprB or InhA1 displayed reduced ability to activate purified human prothrombin or factor X and reduced ability to initiate coagulation of human blood plasma compared with B. anthracis Ames 35 (Fig. 4b-e). NprB is highly homologous to bacillolysin proteases of other Bacillus species. InhA1 is a homolog of the B. thuringiensis immune inhibitor A (ref. 16), and its expression has been observed during growth in minimal aerobic medium35. InhA1 was essential for initiation of coagulation of human blood plasma by B. anthracis (P < 0.001 for Ames 35 versus ΔinhA1). To ensure that InhA1 is capable of activating human coagulation factors, we purified this enzyme from the B. anthracis Ames 35 ΔnprB strain. As reported, InhA1 was purified as a 46 kDa and 18 kDa complex (Supplementary Fig. 7 online)16. Purified InhA1 indeed cleaved prothrombin and generated active thrombin (Fig. 4d). In addition, control experiments showed that heat-inactivated InhA1 was not active, and the measured activity of purified InhA1 was due to activation of prothrombin, not direct cleavage of the fluorescent substrate by InhA1. Recent experiments have found that von Willebrand factor, a regulator of platelet aggregation, is also a substrate for InhA1 (ref. 15). Although B. anthracis could still rapidly initiate coagulation in human blood plasma deficient in von Willebrand factor and platelets (see Supplementary Methods online), interactions with this factor further supported the notion that B. anthracis may target the coagulation process during infection.
The dependence of coagulation on the spatial arrangement of bacteria could suggest a quorum-sensing mechanism, and B. anthracis has been previously shown to exhibit quorum sensing36. However, two results contradict such a hypothesis. First, for the experiments described in Figure 1b, the bacteria in each sample were subjected to the same conditions until the human blood plasma was introduced. Then, within one minute, the bacteria were either dispersed into the plasma or clustered in the plasma, leading to the significant difference in initiation of coagulation (Fig. 1b). It is not likely that quorum sensing could induce changes in phenotype within one minute. The production of InhA1 by B. anthracis was not strongly influenced by clustering on the timescale that coagulation occurs (less than 2 h), as determined by immunoblot analysis using mouse anti-InhA1 serum (Supplementary Fig. 8 online). In a second experiment, mutants of B. anthracis that had reduced quorum-sensing ability still triggered rapid coagulation of human blood plasma. We used a previously characterized quorum-sensing mutant strain of B. anthracis (34F2) that lacks functional luxS activity and production of autoinducer-2 (1)36.Similar to the B. anthracis Ames 35 control strain, this mutant strain rapidly initiated coagulation of human blood plasma in less than 3 min (Fig. 4f). The alternative mechanism that we are proposing here is that individual bacteria are below the critical size necessary to initiate coagulation (Fig. 1a), but clusters of bacteria exceed the threshold size necessary for initiation21,22,37. We used numerical simulations to test the feasibility of this mechanism.
Numerical simulations reproduce the coagulation dynamics
To examine the physical mechanism responsible for the initiation of coagulation on localized clusters of bacteria, we used a two-dimensional numerical simulation that considered 40 reactions of the human coagulation cascade, the activation of prothrombin and factor X by B. cereus or B. anthracis, the spatial localization of bacteria, and diffusion. The components and rate constants for the coagulation reactions were chosen based on an established numerical model for the human coagulation network. The rates of activation of prothrombin and factor X per bacterium were determined from kinetic assays that used B. cereus, purified prothrombin or factor X, and fluorogenic substrates for thrombin or factor Xa with known rates of cleavage38. Simulations mimicked 2 × 106 bacteria in 50 μl of human blood plasma, either dispersed throughout the volume or clustered. When dispersed, the coagulation cascade was not activated, and only 0.13 pmol of thrombin, 0.2 fmol of factor Xa, 8.1 fmol of factor Va and 0.28 fmol of factor VIIIa were produced after 1,500 s (Fig. 5a). The majority (74%) of thrombin generated was due to direct proteolytic cleavage of prothrombin by bacterial proteases, thereby confirming that the coagulation cascade was not turned on. When bacteria were clustered into a 500-μm-diameter patch in the same volume of plasma, an intense local burst of thrombin and fibrin was generated in the simulation, which indicates the initiation of coagulation (Fig. 5a). Much larger amounts of activated clotting factors were produced: 18 pmol of thrombin, 9.9 fmol of factor Xa, 310 fmol of factor Va and 11 fmol of factor VIIIa were produced after 1,500 s. The majority (>99.9%) of thrombin was produced by the coagulation cascade itself (via the cleavage of prothrombin by the Xa-Va complex), thereby confirming that the coagulation cascade was initiated. In addition, the entire coagulation cascade, including factor X, was activated by bacteria even when competition between factor X and prothrombin for the bacterial protease was included in the simulation, which further supports experiments indicating that factor X and prothrombin are essential for initiation of coagulation by bacteria. Note we compared total amounts of activated factors produced, rather than their local concentrations. When local concentrations were compared, the difference between dispersed and clustered bacteria became even more pronounced (Fig. 5b,c). The maximum concentration of active thrombin generated after 140 s was 4.1 pM when the bacteria were dispersed and 0.44 μM when the bacteria were clustered. We also observed obvious propagation of the clot initiated on the cluster of bacteria (Fig. 5c). To simplify the interpretation, we did not allow diffusion of bacterial proteases in the simulation; therefore, the propagation of coagulation could be attributed exclusively to the activation of the coagulation cascade. We performed a control simulation where proteases were allowed to diffuse and observed similar results.
Figure 5.
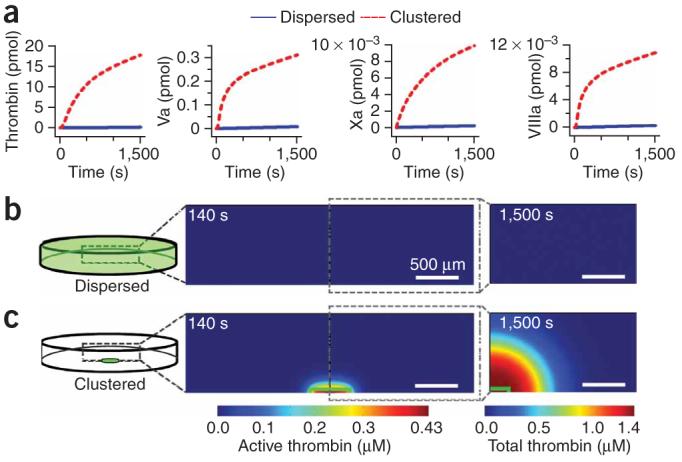
Two-dimensional simulations of the human blood coagulation cascade comparing the generation of activated coagulation factors by bacteria dispersed in solution versus bacteria clustered in a surface patch. The overall number of bacteria was the same in all simulations. (a) Graphs of the total amount (not concentrations) of thrombin and factors Xa, Va and VIIIa generated in the simulation by dispersed (blue line) and clustered (red dashed line) bacteria. The total amounts included both active (free) forms of the enzyme and active forms that have been inhibited after activation. (b,c) Two-dimensional plots show the thrombin concentration in a simulated microfluidic chamber (represented in schematic drawings at the left). Plots at 140 s show the concentration of active thrombin, and plots at 1,500 s show the total thrombin concentration (both active and inhibited). When bacteria (green) were dispersed in solution, little thrombin was produced and coagulation did not initiate within 1,500 s (b). However, when the same number of bacteria were localized to a surface cluster (inside green outline), thrombin was generated at a high concentration, coagulation was initiated and the clot propagated away from the cluster (c).
Clusters of B. anthracis initiate coagulation in mice
We used a mouse model to test whether the correlation between clustering of B. anthracis and local coagulation was also observed in vivo. Mice were injected with B. anthracis vegetative bacteria, and then lung, heart, spleen and liver tissues were harvested quickly (30 or 90 min) after the injection. Rapid harvesting was used to minimize initiation of coagulation by the immune response1,7, so we could observe more clearly coagulation caused by direct initiation by bacteria. After harvesting, histological sections of the mouse tissues were scored for the percentage of vessels showing fibrin clots. In the control experiment, two mice were injected with 104 bacteria per mouse; we hypothesized that clusters of bacteria would be less likely to form at such a low dose. In these control mice (Fig. 6a), no clusters of bacteria and no fibrin clots were observed in any tissue. Then, two mice were injected with a higher dose of 108 bacteria per mouse; we hypothesized that the formation of bacterial clusters would be more likely at this high dose. In the mice receiving 108 bacteria, clusters of B. anthracis were observed in the microvasculature of the lungs (Fig. 6b), but not in any other organs 30 min after the injection. By analyzing two whole-organ sections per mouse, ten fields per section, with a minimum of 50 vessels per field, we determined that 45 and 80% of pulmonary vessels were clotted in these mice (Supplementary Fig. 9a online). Bacteria were never observed in the vessels without fibrin clots, and clotting was associated with the presence of bacterial clusters 100% of the time in every vessel analyzed at high magnification (100x objective).
Figure 6.

Clusters of B. anthracis rapidly initiate coagulation in mice. (a,b) H&E-stained histological sections of mouse lung. (a) Pulmonary vessels from control mice injected with a low dose of bacteria show no clusters and no coagulation. (b) Pulmonary vessels from mice injected with a higher dose of bacteria show clustering of bacteria. Coagulation in vessels (large magenta regions) occurred on clusters of bacteria (chains of rod-like bacteria are seen, blue) within 30 min. Digitally magnified portions of a vessel are shown in images on the left and right. Two mice were sampled at each dose. See text for details.
The correlation between clustering of bacteria and coagulation in vivo was strong. These results support the overall mechanism observed in vitro with human plasma (Fig. 1a), but the most conclusive experiment in this mouse model—comparing the same amount of dispersed bacteria to clustered bacteria—was not performed owing to the difficulty in controlling clustering in vivo. In vitro, coagulation of mouse plasma was accelerated by clustering of bacteria (Supplementary Fig. 9b), and this was also true in human plasma.
We emphasize that the characterization of the interaction of Bacillus cells and proteases (Fig. 4 and Supplementary Figs. 4-6) with the coagulation cascade was performed by using human (not mouse) whole blood and plasma. Therefore, the details of the in vivo experiment performed with the mouse model (Fig. 6) should be interpreted with caution. Mouse models have been useful for studying the pathophysiology of B. anthracis infection, but they have been shown to have major differences in response to B. anthracis infection compared with humans39. In addition, there are known differences between proteolytic activation of mouse and human clotting cascades40,41. Rapid clotting in the mouse model strongly suggests that direct activation of coagulation occurs; however, we did not identify the key proteases responsible for initiation of coagulation by B. anthracis in mouse, rather than human, blood. The ΔnprB and ΔinhA1 strains of B. anthracis were still able to rapidly initiate coagulation in mice, which indicates that coagulation directly induced by B. anthracis is a robust process in mice. In addition, this result implies either that clotting of the mouse blood is initiated by an unknown rapid mechanism that is sensitive to spatial distribution and clustering of bacteria, or that proteases other than InhA1 and NprB are involved in the activation of the mouse coagulation cascade. In support of the latter (and simpler) argument, we found differences in the rates and specificity of the B. anthracis proteases for human and mouse coagulation proteins. For example, we tested activation of human and mouse factor X under the same conditions by B. anthracis cells in vitro. Deletion of either InhA1 or NprB substantially decreased the rate of human factor X activation by a factor of 20 to low levels, but these deletions decreased the rate of activation of mouse factor X by only a factor of 5 and 2.5, with substantial activation remaining, which suggests that other B. anthracis proteases can also activate mouse factor X. These proteases may be active in the B. anthracis Ames 35 strain or upregulated in response to the loss of a protease in the mutant strains. Though the overall dynamics of rapid activation of clotting factors by the parent Ames 35 strain were reproducible, the exact amount of decrease of activation in mutant strains varied with conditions. These results were consistent with the differences in the sequence of peptides of human and mouse factor X, which is substantially different in the region that is proteolysed to convert factor X to factor Xa (ref. 42). These differences make the mouse model less appropriate for the detailed biochemical studies that are necessary to understand induction of coagulation in the human blood in vivo. In addition, these results suggest that, in addition to InhA1, other proteases could potentially affect induction of coagulation of human blood (for example, by cross-activation). An important next step for understanding the role of coagulation induced in humans by bacterial proteases would be identifying an animal model of blood coagulation that responds to bacterial proteases in a manner similar to human coagulation. Rabbit and primate models may be suitable, but these experiments are beyond the scope of this paper.
DISCUSSION
The first area of interest related to this work is the connection between bacterial infections and blood coagulation1,4,43 and the hypothesis that clusters of bacteria may directly initiate coagulation of human blood during infection, bypassing inflammation. This hypothesis should be considered for immunosuppressed and immunocompromised people for whom infection and septicemia by B. cereus is a threat44. It should also be considered in the context of inflammatory responses that are triggered by activated coagulation factors2,5,43. Our results reported a physical and biochemical mechanism in support of this hypothesis and provide the motivation to test this mechanism further by using in vivo models of infection. This mechanism predicts that inhibiting clustering of bacteria, inhibiting local accumulation of coagulation factors on surfaces of bacteria17, and inhibiting expression, transport or processing of bacterial proteases may help reduce coagulation during infection. To choose the appropriate in vivo model, one would have to characterize carefully the interactions between bacteria and the coagulation cascade of the model organism to ensure relevance to the human coagulation system. In vivo bioluminescent imaging of B. anthracis45 should be a useful tool for understanding further the role of this mechanism in anthrax by characterizing the effects of clusters, biofilms and local infection sites on coagulation.
The second area of interest related to this work is understanding the dynamics of groups or clusters of bacteria46,47. Quorum sensing is perhaps the best known example of such dynamics46. In quorum sensing, bacteria send out a diffusible signal (Fig. 7a). At a low concentration of bacteria, the signal diffuses away and does not accumulate, and bacteria do not detect the ‘quorum’. As bacteria are brought together to a higher (quorum) concentration, the diffusible signal accumulates above a threshold concentration and is sensed by the bacteria. A linear system cannot exhibit a spatially dependent phenomenon such as a quorum sensing25, and a nonlinearity, such as a threshold response, must be present either in the system itself or in its environment. In quorum sensing, the nonlinearity required for the threshold response is provided by the bacterial regulatory network, and no nonlinearity in the environment is required. Once quorum sensing takes place, it drives changes in gene expression to produce effector molecules required for bacteria to take appropriate actions (Fig. 7a).
Figure 7.
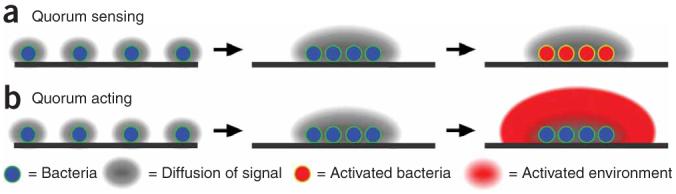
Even without quorum sensing, clustering of bacteria may elicit large-scale action. (a) During quorum sensing, the localization of bacteria (blue) at a high concentration elicits phenotypic changes in the bacteria themselves (red). (b) When bacteria interact with an environment capable of nonlinear responses, upon clustering, bacteria may act without having to undergo phenotypic changes. Action is illustrated here as activation of the environment (large red area).
Initiation of coagulation by clusters of bacteria (Fig. 1) can occur by a mechanism distinct from that of quorum sensing. In the proposed mechanism (Fig. 7b), a diffusible molecule is also generated by an individual bacterium. For initiation of coagulation discussed in this paper, two classes of molecules satisfy the requirement of this diffusible molecule: (i) a diffusible molecule secreted directly by the bacterium—for example, protease InhA1—and (ii) a diffusible molecule from the environment that can be activated near or on the surface of the bacterium—for example, thrombin. The molecule does not accumulate at low concentrations of bacteria. When bacteria are brought closer together into a cluster, the molecule reaches a threshold concentration. At this point, there are three critical differences between this mechanism and quorum sensing. First, the nonlinearity that established the threshold can come from the environment around the bacteria, not necessarily from the bacterial regulatory network. In the example discussed here, the threshold comes from the nonlinearities of the coagulation network. Second, sensing by bacteria is not required for this mechanism to operate, because ‘sensing’ is essentially performed by the environment via its threshold. This also differs from the sensing mechanism used by bacteria, such as E. feacalis, to probe their environment for host mammalian cells48. Third, this mechanism does not require either changes in bacterial gene expression or any other bacterial mechanism for production of the effector molecules. The secreted molecule is the effector molecule required for action, and it triggers the response of the environment once bacteria reach a sufficiently high local density. To emphasize these three differences, we refer to this mechanism as ‘quorum acting’. This distinction is supported by the rapid initiation of coagulation by the B. anthracis luxS mutant deficient in quorum sensing (Fig. 4f). We predict that other bacterial species that activate coagulation factors may demonstrate this quorum-acting mechanism, although this prediction remains to be tested. Porphyromonas gingivalis, a causative agent of gum disease, is one likely candidate. Purified proteases of P. gingivalis are particularly potent and known to activate many coagulation factors and reduce coagulation times in standard assays11. P. gingivalis infections have also been linked to cardiovascular disease, although the nature of this connection is still under investigation49.
Further work is required to differentiate the connections between quorum sensing and quorum acting, as the two mechanisms are likely to be coupled and are likely to feedback to one another. Though the quorum-acting mechanism does not require a change in bacterial phenotype to function, it is not likely that it is constitutively turned on, independently of the phenotype. What regulates the ‘coagulation phenotype’ and secretion of proteases responsible for the initiation of coagulation? What is the role of the environment relative to the role of bacterial communication, for example by oligopeptides46, in this regulation? Other bacteria such as streptococci and Yersinia pestis, the plague agent, are known to break apart clots3,41,43. Is avoiding entrapment by coagulation10 a better strategy? Or does initiation of coagulation benefit B. anthracis by shielding it from the host’s immune system and, coincidentally, from administered antibiotics?
Although we do not know whether quorum acting is as widespread as quorum sensing, more examples are likely to be found in environments capable of nonlinear responses. Such environments could range from interactions of communities of microorganisms in soils and in biofilms, to secretion of toxins and virulence factors, to interactions of microorganisms with the gut, the immune system and the coagulation cascade of a mammalian host. One may expect confined environments to enhance quorum acting, in analogy to quorum sensing47. Quorum acting may be especially beneficial when a rapid response to aggregation of microorganisms is needed, either as a defensive response, or an opportunistic response. Quorum acting could also serve as a driving force for the evolution of cooperation within and among the bacterial groups, by facilitating kin and group selection as well as reciprocity—the common themes for collaborative selections50.
In conclusion, this work demonstrates that bacteria can directly initiate coagulation of human blood and plasma, a process that was previously thought to be lost during vertebrate evolution. This process relies on a quorum-acting mechanism that is distinct from quorum-sensing processes. These results emphasize the importance of spatial distribution, rather than average concentration, in the function of nonlinear biochemical networks21,25. We expect spatial distribution to also be critical in initiation of coagulation by other mechanisms that are distinct from proteolytic activation by bacterial proteases. These results may also have implications for improving our understanding of coagulation during bacterial infections and of the role of spatial organization of bacteria in their interactions with nonlinear environments.
METHODS
General methods
See Supplementary Methods for additional experimental protocols, description of the bacterial strains and details of the numerical simulation. A summary of the methods is given below.
Patterning bacteria using microfluidic techniques
For experiments measuring the initiation of coagulation of human blood and plasma on clusters of bacteria that were not spatially patterned (that is, not Fig. 1c,d), bacteria were concentrated to a pellet, and then droplets of the concentrated bacteria (∼50 nl) were deposited onto a plastic coverslip in the bottom of a microfluidic chamber. The bacteria were either dispersed in human blood plasma by mixing for ∼2 s or allowed to remain localized in a patch. Bacteria remained localized owing to weak adhesive forces between themselves and the plastic coverslip.
To prepare spatially patterned bacteria (Fig. 1c,d), micropatterning techniques were used24. Bacteria were patterned on substrates consisting of alumina membranes (200 nm pore size) coated with patterned photoresist. A gentle vacuum was applied from under the substrate to pull the bacteria to the open pores. Control experiments confirmed that both patterned and nonpatterned substrates were relatively inert; bacteria were able to grow on them, and, in the absence of bacteria, these surfaces did not initiate coagulation of human blood for >30 min.
Measuring clot times of human whole blood and plasma
Citrated human platelet-poor plasma was obtained from George King Biomedical, Inc. Citrated immunodepleted plasmas and measurements of their PT and APTT times were obtained from Haematologic Technologies, Inc. Human whole blood was obtained from individual healthy donors in accordance with the guidelines set by the Institutional Review Board (protocol # 12502A) at The University of Chicago. Written informed consent was obtained from donors. All human blood and plasma samples were incubated with corn trypsin inhibitor to inhibit the factor XII pathway of initiation of coagulation, with the exception of the experiment testing clustered bacteria versus dispersed bacteria with human plasma (Fig. 1b) and the experiment with immunodepleted plasmas (Fig. 3b,c). Human whole blood and plasma were recalcified by adding a solution containing CaCl2 and a thrombin-sensitive fluorescent substrate. Experiments were performed at 37 °C. In all experiments, clot times were determined by monitoring the formation of thrombin and fibrin by fluorescence and brightfield microscopy, respectively.
Microfluidic device fabrication
All devices were fabricated by using rapid prototyping in polydimethylsiloxane (Dow Corning Corporation). Before adding the GMDs or human blood, the microfluidic channels were coated with inert phospholipids by flowing vesicles of l-α-phosphatidylcholine (Avanti) through the device. GMDs containing bacteria were flowed into the device, localized near magnets and grown for 14-24 h. GMDs27 (Fig. 2) consisted of ∼50-μm-sized droplets of solid agarose containing bacteria and magnetic particles. They were prepared separately using a droplet-based microfluidic approach. After bacterial colonies were present, human whole blood was flowed through the device and the formation of thrombin and fibrin was monitored.
Measuring coagulation by B. anthracis in mice
All animal experiments and protocols were approved by and conducted according to the guidelines of the US National Institute of Allergy and Infectious Diseases (NIAID) Animal Care and Use Committee. Solutions containing B. anthracis (vegetative cells) were injected intravenously into the tail vein of DBA/2J mice (Jackson Laboratories). After 30 or 90 min, organs were harvested and immediately fixed in a neutral-buffered 10% formalin solution for hematoxylin and eosin (H&E) staining.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the US National Institutes of Health (NIH) Director’s Pioneer Award (grant number DP1OD003584), the US National Science Foundation CAREER Award (grant number CHE-0349034), the US Office of Naval Research (grant number N000140610630), the Camille Dreyfus Teacher-Scholar Awards Program and the Cottrell Scholar of Research Corporation Awards Program to R.F.I., by NIH grants (GM 62548 and GM 81539) to W.-J.T., and by the Intramural Research Program of the NIAID. We thank J. Alverdy, B. Bishop, S. Crosson, C. Esmon, M. Mock, M. Runyon, J. Shapiro, U. Spitz, T. Van Ha, D. Wiebel and O. Zaborina for helpful discussions; O. Zaborina (The University of Chicago) for the gift of the EGPF plasmid for E. coli and for assisting in the transformation procedure; M. Mock (Institut Pasteur) for the gift of the mouse anti-InhA1 serum; L. Cheng and D. Crown for assisting in the animal studies; H. Herwald (Lund University) for the gift of the E. coli Ymel-1 strain; J. Handelsman (University of Wisconsin) for the gift of the B. cereus GFP strain; M. Blaser (New York University) for the gift of the B. anthracis ΔluxS strain; and J. Price for contributions in writing and editing this manuscript. We thank C. Tallant (Institut de Biologia Molecular de Barcelona) for assistance in construction of the protease gene knockout strains.
References
- 1.Opal SM, Esmon CT. Bench-to-bedside review: functional relationships between coagulation and the innate immune response and their respective roles in the pathogenesis of sepsis. Crit. Care. 2003;7:23–38. doi: 10.1186/cc1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Niessen F, et al. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature. 2008;452:654–658. doi: 10.1038/nature06663. [DOI] [PubMed] [Google Scholar]
- 3.Sun H. The interaction between pathogens and the host coagulation system. Physiology (Bethesda) 2006;21:281–288. doi: 10.1152/physiol.00059.2005. [DOI] [PubMed] [Google Scholar]
- 4.Stearns-Kurosawa DJ, Lupu F, Taylor FB, Kinasewitz G, Kurosawa S. Sepsis and pathophysiology of anthrax in a nonhuman primate model. Am. J. Pathol. 2006;169:433–444. doi: 10.2353/ajpath.2006.051330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Esmon CT. The interactions between inflammation and coagulation. Br. J. Haematol. 2005;131:417–430. doi: 10.1111/j.1365-2141.2005.05753.x. [DOI] [PubMed] [Google Scholar]
- 6.Tang H, et al. Sepsis-induced coagulation in the baboon lung is associated with decreased tissue factor pathway inhibitor. Am. J. Pathol. 2007;171:1066–1077. doi: 10.2353/ajpath.2007.070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pawlinski R, et al. Regulation of tissue factor and inflammatory mediators by Egr-1 in a mouse endotoxemia model. Blood. 2003;101:3940–3947. doi: 10.1182/blood-2002-07-2303. [DOI] [PubMed] [Google Scholar]
- 8.Levi M, de Jonge E, van der Poll T. New treatment strategies for disseminated intravascular coagulation based on current understanding of the pathophysiology. Ann. Med. 2004;36:41–49. doi: 10.1080/07853890310017251. [DOI] [PubMed] [Google Scholar]
- 9.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 10.Mullarky IK, et al. Infection-stimulated fibrin deposition controls hemorrhage and limits hepatic bacterial growth during listeriosis. Infect. Immun. 2005;73:3888–3895. doi: 10.1128/IAI.73.7.3888-3895.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Imamura T, Potempa J, Tanase S, Travis J. Activation of blood coagulation factor X by arginine-specific cysteine proteinases (gingipain-Rs) from Porphyromonas gingivalis. J. Biol. Chem. 1997;272:16062–16067. doi: 10.1074/jbc.272.25.16062. [DOI] [PubMed] [Google Scholar]
- 12.Narasaki R, et al. Bacillolysin MA, a novel bacterial metalloproteinase that produces angiostatin-like fragments from plasminogen and activates protease zymogens in the coagulation and fibrinolysis systems. J. Biol. Chem. 2005;280:14278–14287. doi: 10.1074/jbc.M500241200. [DOI] [PubMed] [Google Scholar]
- 13.Smith SA, et al. Polyphosphate modulates blood coagulation and fibrinolysis. Proc. Natl. Acad. Sci. USA. 2006;103:903–908. doi: 10.1073/pnas.0507195103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedrich R, et al. Structural basis for reduced staphylocoagulase-mediated bovine prothrombin activation. J. Biol. Chem. 2006;281:1188–1195. doi: 10.1074/jbc.M507957200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung MC, et al. Degradation of circulating von Willebrand factor and its regulator ADAMTS13 implicates secreted Bacillus anthracis metalloproteases in anthrax consumptive coagulopathy. J. Biol. Chem. 2008;283:9531–9542. doi: 10.1074/jbc.M705871200. [DOI] [PubMed] [Google Scholar]
- 16.Chung MC, et al. Secreted neutral metalloproteases of Bacillus anthracis as candidate pathogenic factors. J. Biol. Chem. 2006;281:31408–31418. doi: 10.1074/jbc.M605526200. [DOI] [PubMed] [Google Scholar]
- 17.Herwald H, et al. Activation of the contact-phase system on bacterial surfaces - a clue to serious complications in infectious diseases. Nat. Med. 1998;4:298–302. doi: 10.1038/nm0398-298. [DOI] [PubMed] [Google Scholar]
- 18.Muta T, Iwanaga S. The role of hemolymph coagulation in innate immunity. Curr. Opin. Immunol. 1996;8:41–47. doi: 10.1016/s0952-7915(96)80103-8. [DOI] [PubMed] [Google Scholar]
- 19.Jesty J, Rodriguez J, Beltrami E. Demonstration of a threshold response in a proteolytic feedback system: control of the autoactivation of factor XII. Pathophysiol. Haemost. Thromb. 2005;34:71–79. doi: 10.1159/000089928. [DOI] [PubMed] [Google Scholar]
- 20.van’t Veer C, Mann KG. Regulation of tissue factor initiated thrombin generation by the stoichiometric inhibitors tissue factor pathway inhibitor, antithrombin-III, and heparin cofactor-II. J. Biol. Chem. 1997;272:4367–4377. doi: 10.1074/jbc.272.7.4367. [DOI] [PubMed] [Google Scholar]
- 21.Kastrup CJ, Runyon MK, Shen F, Ismagilov RF. Modular chemical mechanism predicts spatiotemporal dynamics of initiation in the complex network of hemostasis. Proc. Natl. Acad. Sci. USA. 2006;103:15747–15752. doi: 10.1073/pnas.0605560103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kastrup CJ, Shen F, Runyon MK, Ismagilov RF. Characterization of the threshold response of initiation of blood clotting to stimulus patch size. Biophys. J. 2007;93:2969–2977. doi: 10.1529/biophysj.107.109009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weibel DB, DiLuzio WR, Whitesides GM. Microfabrication meets microbiology. Nat. Rev. Microbiol. 2007;5:209–218. doi: 10.1038/nrmicro1616. [DOI] [PubMed] [Google Scholar]
- 24.Whitesides GM, Ostuni E, Takayama S, Jiang XY, Ingber DE. Soft lithography in biology and biochemistry. Annu. Rev. Biomed. Eng. 2001;3:335–373. doi: 10.1146/annurev.bioeng.3.1.335. [DOI] [PubMed] [Google Scholar]
- 25.Pompano RR, Li HW, Ismagilov RF. Rate of mixing controls rate and outcome of autocatalytic processes—theory and microfluidic experiments with chemical reactions and blood coagulation. Biophys. J. 2008;95:1531–1543. doi: 10.1529/biophysj.108.129486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Runyon MK, Kastrup CJ, Johnson-Kerner BL, Ha TG, Ismagilov RF. The effects of shear rate on propagation of blood clotting determined using microfluidics and numerical simulations. J. Am. Chem. Soc. 2008;130:3458–3464. doi: 10.1021/ja076301r. [DOI] [PubMed] [Google Scholar]
- 27.Weaver JC, Williams GB, Klibanov A, Demain AL. Gel microdroplets - rapid detection and enumeration of individual microorganisms by their metabolic-activity. Bio/Technology. 1988;6:1084–1089. [Google Scholar]
- 28.Siegel AC, et al. Cofabrication of electromagnets and microfluldic systems in poly(dimethylsiloxane) Angew. Chem. Int. Ed. 2006;45:6877–6882. doi: 10.1002/anie.200602273. [DOI] [PubMed] [Google Scholar]
- 29.Jiang Y, Doolittle RF. The evolution of vertebrate blood coagulation as viewed from a comparison of puffer fish and sea squirt genomes. Proc. Natl. Acad. Sci. USA. 2003;100:7527–7532. doi: 10.1073/pnas.0932632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Krem MM, Di Cera E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem. Sci. 2002;27:67–74. doi: 10.1016/s0968-0004(01)02007-2. [DOI] [PubMed] [Google Scholar]
- 31.Min DH, Tang WJ, Mrksich M. Chemical screening by mass spectrometry to identify inhibitors of anthrax lethal factor. Nat. Biotechnol. 2004;22:717–723. doi: 10.1038/nbt973. [DOI] [PubMed] [Google Scholar]
- 32.Bugge TH, Leppla SH. Anthrax target in macrophages unveiled. Nat. Genet. 2006;38:137–138. doi: 10.1038/ng0206-137. [DOI] [PubMed] [Google Scholar]
- 33.Moayeri M, Haines D, Young HA, Leppla SH. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 2003;112:670–682. doi: 10.1172/JCI17991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pomerantsev AP, Sitaraman R, Galloway CR, Kivovich V, Leppla SH. Genome engineering in Bacillus anthracis using Cre recombinase. Infect. Immun. 2006;74:682–693. doi: 10.1128/IAI.74.1.682-693.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gat O, et al. Search for Bacillus anthracis potential vaccine candidates by a functional genomic-serologic screen. Infect. Immun. 2006;74:3987–4001. doi: 10.1128/IAI.00174-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jones MB, Blaser MJ. Detection of a luxS-signaling molecule in Bacillus anthracis. Infect. Immun. 2003;71:3914–3919. doi: 10.1128/IAI.71.7.3914-3919.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beltrami E, Jesty J. The role of membrane patch size and flow in regulating a proteolytic feedback threshold on a membrane: possible application in blood coagulation. Math. Biosci. 2001;172:1–13. doi: 10.1016/s0025-5564(01)00064-5. [DOI] [PubMed] [Google Scholar]
- 38.Kawabata SI, et al. Highly sensitive peptide-4-methylcoumaryl-7-amide substrates for blood-clotting proteases and trypsin. Eur. J. Biochem. 1988;172:17–25. doi: 10.1111/j.1432-1033.1988.tb13849.x. [DOI] [PubMed] [Google Scholar]
- 39.Loving CL, Kennett M, Lee GM, Grippe VK, Merkel TJ. Murine aerosol challenge model of anthrax. Infect. Immun. 2007;75:2689–2698. doi: 10.1128/IAI.01875-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407:258–264. doi: 10.1038/35025229. [DOI] [PubMed] [Google Scholar]
- 41.Sun HM, et al. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science. 2004;305:1283–1286. doi: 10.1126/science.1101245. [DOI] [PubMed] [Google Scholar]
- 42.Heidtmann HH, Kontermann RE. Cloning and recombinant expression of mouse coagulation factor X. Thromb. Res. 1998;92:33–41. doi: 10.1016/s0049-3848(98)00110-8. [DOI] [PubMed] [Google Scholar]
- 43.Degen JL, Bugge TH, Goguen JD. Fibrin and fibrinolysis in infection and host defense. J. Thromb. Haemost. 2007;5:24–31. doi: 10.1111/j.1538-7836.2007.02519.x. [DOI] [PubMed] [Google Scholar]
- 44.Akiyama N, et al. Fulminant septicemic syndrome of Bacillus cereus in a leukemic patient. Intern. Med. 1997;36:221–226. doi: 10.2169/internalmedicine.36.221. [DOI] [PubMed] [Google Scholar]
- 45.Glomski IJ, Piris-Gimenez A, Huerre M, Mock M, Goossens PL. Primary involvement of pharynx and Peyer’s patch in inhalational and intestinal anthrax. PLoS Pathog. 2007;3:e76. doi: 10.1371/journal.ppat.0030076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bassler BL, Losick R. Bacterially speaking. Cell. 2006;125:237–246. doi: 10.1016/j.cell.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 47.Redfield RJ. Is quorum sensing a side effect of diffusion sensing? Trends Microbiol. 2002;10:365–370. doi: 10.1016/s0966-842x(02)02400-9. [DOI] [PubMed] [Google Scholar]
- 48.Coburn PS, Pillar CM, Jett BD, Haas W, Gilmore MS. Enterococcus faecalis senses target cells and in response expresses cytolysin. Science. 2004;306:2270–2272. doi: 10.1126/science.1103996. [DOI] [PubMed] [Google Scholar]
- 49.Demmer RT, Desvarieux M. Periodontal infections and cardiovascular disease - the heart of the matter. J. Am. Dent. Assoc. 2006;137:14S–20S. doi: 10.14219/jada.archive.2006.0402. [DOI] [PubMed] [Google Scholar]
- 50.Nowak MA. Five rules for the evolution of cooperation. Science. 2006;314:1560–1563. doi: 10.1126/science.1133755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.