

Abstract
We develop hierarchical generalized linear models and computationally efficient algorithms for genomewide analysis of quantitative trait loci (QTL) for various types of phenotypes in experimental crosses. The proposed models can fit a large number of effects, including covariates, main effects of numerous loci, and gene–gene (epistasis) and gene–environment (G × E) interactions. The key to the approach is the use of continuous prior distribution on coefficients that favors sparseness in the fitted model and facilitates computation. We develop a fast expectation-maximization (EM) algorithm to fit models by estimating posterior modes of coefficients. We incorporate our algorithm into the iteratively weighted least squares for classical generalized linear models as implemented in the package R. We propose a model search strategy to build a parsimonious model. Our method takes advantage of the special correlation structure in QTL data. Simulation studies demonstrate reasonable power to detect true effects, while controlling the rate of false positives. We illustrate with three real data sets and compare our method to existing methods for multiple-QTL mapping. Our method has been implemented in our freely available package R/qtlbim (www.qtlbim.org), providing a valuable addition to our previous Markov chain Monte Carlo (MCMC) approach.
MOST complex traits are influenced by interacting networks of multiple quantitative trait loci (QTL) and environmental factors (Carlborg and Haley 2004). Mapping QTL is to infer which genomic loci are strongly associated with the complex trait and to estimate genetic effects of these loci, i.e., main effects and gene–gene (epistasis) and gene–environment (G × E) interactions. Due to the multilocus nature of complex traits, it is desirable to simultaneously analyze multiple loci rather than one locus (or a few loci) at a time. However, QTL mapping studies usually genotype hundreds or thousands of genomic loci (markers), leading to numerous variables and a huge number of possible models, and the dependence of genotypes on a chromosome results in many correlated variables. Therefore, mapping multiple QTL requires sophisticated methods that can handle problems with high-dimensional correlated variables.
The popular approaches to mapping multiple QTL are some form of variable selection. Such techniques involve identifying a subset of all possible genetic effects (a multiple-QTL model) that best explains the phenotypic variation. Classical variable selection methods use forward or stepwise search procedures and selection criteria such as Bayesian information criteria (BIC) or modified versions to find a multiple-QTL model (Kao et al. 1999; Broman and Speed 2002; Bogdan et al. 2004; Baierl et al. 2006). Bayesian methods proceed by setting up a likelihood function for observed data and prior distributions on unobserved quantities. Two types of prior distributions have been suggested for multiple-QTL mapping. The first assumes a two-component mixture distribution for each genetic effect, typically a normal distribution with known or unknown variance and a point mass at zero. This discrete prior allows each effect to have positive probability of dropping out of the model (Yi 2004; Yi and Shriner 2008). The second formulation takes continuous prior distributions for genetic effects that favor a sparse structure with many of the effects having values close to zero and few with large values (Meuwissen et al. 2001; Xu 2003; Yi and Xu 2008). These Bayesian models are computed using Markov chain Monte Carlo (MCMC) algorithms to sample from the posterior distribution. Due to the recent development of MCMC algorithms and associated computer software, Bayesian methods have become increasingly popular in QTL mapping (Yi et al. 2005, 2007a,b; Yandell et al. 2007; Yi and Shriner 2008). The Bayesian MCMC approaches can provide comprehensive information, but they are computationally intensive in interacting-QTL analysis. Further, the existing methods rely primarily upon normal linear models.
We present a unified methodology for mapping multiple QTL based upon the generalized linear model framework. The generalized linear model is the main tool for routine statistical analysis, enjoying a body of well-developed theory, algorithms, and software and including various models as special cases. However, classical generalized linear models cannot simultaneously handle many correlated variables. We propose a Bayesian generalized linear model approach, placing continuous prior distributions on genetic effects, to deal with the high-dimensional problem. Although various priors can be used for this purpose (Griffin and Brown 2007; Park and Casella 2008), we consider the well-known Student's t-distribution and recommend the choice of hyperparameters to yield a sharp mode at zero and to induce a sparse model. Our unified framework incorporates the advantages of generalized linear models and hierarchical modeling into multiple-QTL mapping, allowing us to deal with various types of continuous and discrete phenotypes and to simultaneously analyze covariates, main effects of numerous loci, and epistatic and G × E interactions.
In principle, we can fit our hierarchical generalized linear model by adapting the MCMC algorithms of Yi and Xu (2008) for normal regressions. However, it is desirable to have a quick calculation that estimates the posterior mode of genetic effects (a point estimate that maximizes the posterior distribution). Although the posterior mode estimate provides less information than a fully Bayesian MCMC analysis, such analyses are similar to classical QTL practice, are easy to understand, and can be valuable for identifying significant variables. Various methods have been proposed for the purpose, but they are built upon special models and optimization algorithms (Tibshirani 1996; Figueiredo 2003; Kiiveri 2003; Efron et al. 2004; Genkin et al. 2007) and cannot be easily set up for multiple-QTL mapping. Recently, some mode-finding methods for mapping multiple QTL have been suggested (Zhang and Xu 2005; Xu 2007). However, these methods have dealt with only continuous traits and have not been effectively implemented in computer software.
We present a fast expectation-maximization (EM) algorithm to fit our hierarchical generalized linear models by estimating the posterior modes of coefficients. We develop our algorithm by making use of the two-level formulation of Student's t-density and expressing the prior information on coefficients as additional “data points.” This strategy allows us to incorporate our algorithm into the usual iteratively weighted least squares for generalized linear models as implemented in R (R Development Core Team 2006). This implementation takes advantage of the well-developed existing algorithm and software, extending available tools for fitting generalized linear models to multiple-QTL mapping.
Although in principle our hierarchical model and algorithm can fit all possible effects in a single model, we do not recommend this and rather propose a novel model search method to build a parsimonious model by seeking significant genetic effects. This procedure provides a flexible and convenient way to deal with large-scale QTL data and also has the advantage of accommodating the correlation structure in QTL data. Simulation studies demonstrate that our method provides reasonable power to detect true effects and can control the rate of extraneous effects. Real data analyses show that the proposed approach compares favorably to existing sophisticated methods.
METHODS
Generalized linear models of multiple QTL:
We consider experimental crosses derived from two inbred lines (for example, F2, backcross, and recombinant inbred lines). Observed data in QTL studies consist of phenotypic values of a complex trait, genetic markers across the genome, and/or some relevant environmental factors (covariates). The marker data include the genotypes and the genomic positions of markers. Mapping QTL is to identify genomic loci that associate with the phenotype and to estimate their genetic effects. For simplicity, we describe our methods by considering observed markers as potential QTL. The methods can be extended to consider loci between the observed markers.
We use the generalized linear model framework to analyze various types of complex traits. A generalized linear model consists of three components: the linear predictor, the link function, and the distribution of the outcome variable (McCullagh and Nelder 1989; Gelman et al. 2003). We simultaneously fit environmental effects, main effects of markers, epistatic effects, and gene–environment (G × E) interactions. Therefore, the linear predictor can be expressed as
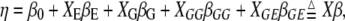 |
(1) |
where 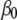 is the intercept,
is the intercept, 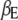 and
and  represent the vectors of environmental effects and all possible main effects, respectively,
represent the vectors of environmental effects and all possible main effects, respectively,  and
and 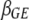 represent the vectors of all possible epistatic and G × E interactions, respectively, and
represent the vectors of all possible epistatic and G × E interactions, respectively, and  ,
, 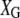 ,
, 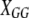 , and
, and  are the corresponding design matrices of effect predictors. We describe the construction of these design matrices in the next section.
are the corresponding design matrices of effect predictors. We describe the construction of these design matrices in the next section.
The invertible link function g relates the linear predictor 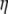 to the mean of the outcome variable y,
to the mean of the outcome variable y,
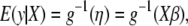 |
(2) |
where the vector  represents the phenotypic values. The distribution of y depends on the linear predictor
represents the phenotypic values. The distribution of y depends on the linear predictor  and also on a dispersion (or variance) parameter
and also on a dispersion (or variance) parameter 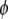 (but some models do not require
(but some models do not require 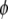 ), and can be expressed as
), and can be expressed as
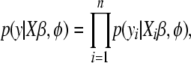 |
(3) |
where  is the linear predictor for the ith individual. Generalized linear modeling provides a unified framework for statistical analysis; by choosing appropriate link functions and data distributions, some commonly used models, e.g., normal linear (Gaussian), logistic, probit, and Poisson regressions, become special cases.
is the linear predictor for the ith individual. Generalized linear modeling provides a unified framework for statistical analysis; by choosing appropriate link functions and data distributions, some commonly used models, e.g., normal linear (Gaussian), logistic, probit, and Poisson regressions, become special cases.
Construction of the design matrices:
There is some discussion in the QTL mapping literature on the best way to construct contrasts for genetic predictors (Zeng et al. 2005). Among those, due to its orthogonal property, the Cockerham genetic model is widely used in QTL mapping and is applied in this study. For a backcross design, the Cockerham model defines the values of the main-effect contrast as −0.5 or 0.5 for two genotypes at any locus. For an intercross (F2) design, there are two types of main effects, called additive and dominance effects, and the original Cockerham model defines additive contrast as −1, 0, and 1 for three genotypes and dominance contrast as −0.5 and 0.5 for homozygotes and heterozygotes, respectively. To make additive and dominance contrasts have a common scale, however, we rescale additive contrast as −2−0.5, 0, and 2−0.5. For loci with missing genotypic values, we replace the values of the above contrasts by their conditional expectations given the observed marker data (Haley and Knott 1992). The conditional expectations can be calculated using the multipoint method (Jiang and Zeng 1997).
For each covariate, we transform the raw values to have a mean of 0 and a standard deviation of 0.5, by subtracting the mean and dividing by 2 × SD (the standard deviation of the raw values) (Gelman et al. 2008). This transformation standardizes all the covariates to a common scale, the scale of all the genetic main effects described above. Finally, the epistatic contrasts  or the G × E contrasts
or the G × E contrasts  are constructed by multiplying two corresponding main-effect contrasts.
are constructed by multiplying two corresponding main-effect contrasts.
Prior and posterior distributions:
Mapping QTL is equivalent to estimating coefficients  in the above model, including environmental effects, main effects of markers, and epistatic and G × E interactions. The number of coefficients in the model can be large and the predictors are highly correlated, precluding the use of classical maximum-likelihood methods. This problem could be solved by setting up a prior distribution on
in the above model, including environmental effects, main effects of markers, and epistatic and G × E interactions. The number of coefficients in the model can be large and the predictors are highly correlated, precluding the use of classical maximum-likelihood methods. This problem could be solved by setting up a prior distribution on  to capture the notion that most of the components of
to capture the notion that most of the components of  are likely to be zero or at least negligible; such prior distributions are often referred to as shrinkage priors. For the intercept
are likely to be zero or at least negligible; such prior distributions are often referred to as shrinkage priors. For the intercept 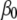 and the dispersion parameter
and the dispersion parameter 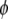 if present, we use the uniform priors,
if present, we use the uniform priors, 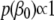 and
and 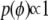 .
.
We assume independent Student's t-priors 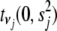 on coefficients
on coefficients  , with
, with  and
and 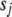 chosen to give each coefficient a high probability of being near zero while still allowing for occasional large effects. We are motivated to use the t-distribution because it allows for robust inference, shrinkage estimation, and easy computation (Gelman et al. 2003). There is no easy way to estimate the coefficients directly using the t densities, but it is straightforward to deal with the two-level formulation of t distribution (Gelman et al. 2003, 2008). The t-distribution
chosen to give each coefficient a high probability of being near zero while still allowing for occasional large effects. We are motivated to use the t-distribution because it allows for robust inference, shrinkage estimation, and easy computation (Gelman et al. 2003). There is no easy way to estimate the coefficients directly using the t densities, but it is straightforward to deal with the two-level formulation of t distribution (Gelman et al. 2003, 2008). The t-distribution  can be expressed as a mixture of normal distributions with mean 0 and variance distributed as scaled inverse-
can be expressed as a mixture of normal distributions with mean 0 and variance distributed as scaled inverse- ,
,
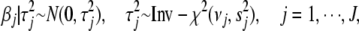 |
(4) |
where J is the number of the coefficients, and the hyperparameters 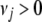 and
and 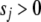 represent the degrees of freedom and the scale of the distribution, respectively.
represent the degrees of freedom and the scale of the distribution, respectively.
The priors (4) introduce coefficient-specific variances, resulting in distinct shrinkage for different coefficients. The variances 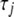 are not the parameters of interest, but they are useful intermediate quantities to allow easy and efficient computation. The hyperparameters
are not the parameters of interest, but they are useful intermediate quantities to allow easy and efficient computation. The hyperparameters  and
and  affect the amount of shrinkage in the coefficient estimates and should be carefully chosen. We discuss our choice of
affect the amount of shrinkage in the coefficient estimates and should be carefully chosen. We discuss our choice of  and
and  after describing our computational algorithm. Our algorithm highlights how these hyperparameters affect the estimates of the parameters.
after describing our computational algorithm. Our algorithm highlights how these hyperparameters affect the estimates of the parameters.
With the above prior distributions, we can express the log-posterior distribution of the parameters (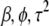 ) as
) as
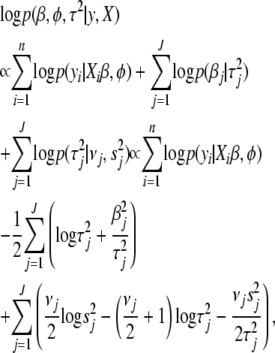 |
(5) |
where 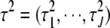 .
.
EM algorithm for computing the posterior mode:
Our hierarchical generalized linear model can be fit using MCMC algorithms, applied to the joint posterior distribution 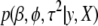 (Gelman et al. 2003). However, it is desirable to have a faster computation that estimates the posterior mode of
(Gelman et al. 2003). However, it is desirable to have a faster computation that estimates the posterior mode of 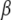 and
and  by maximizing the marginal posterior
by maximizing the marginal posterior 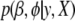 rather than the full posterior distribution. The two-level hierarchical model described above allows us to obtain the posterior mode by using the EM algorithm (Gelman et al. 2003, 2008). Following Gelman et al. (2008), we develop our approach built upon the standard algorithm for classical generalized linear models.
rather than the full posterior distribution. The two-level hierarchical model described above allows us to obtain the posterior mode by using the EM algorithm (Gelman et al. 2003, 2008). Following Gelman et al. (2008), we develop our approach built upon the standard algorithm for classical generalized linear models.
The analysis of classical generalized linear models uses iterative weighted linear regression to obtain approximate maximum-likelihood estimates for the parameters (as implemented in the R routine glm, for example). The basic method is to approximate the generalized linear model by a normal linear model and then apply the algorithm for normal linear models to estimate the parameters  (Gelman et al. 2003, 2008). At each iteration, the algorithm proceeds by calculating pseudodata
(Gelman et al. 2003, 2008). At each iteration, the algorithm proceeds by calculating pseudodata  and pseudovariances
and pseudovariances  for each observation i on the basis of the current estimates of the parameters
for each observation i on the basis of the current estimates of the parameters 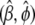 , approximating the generalized likelihood
, approximating the generalized likelihood  by the weighted normal likelihood
by the weighted normal likelihood 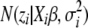 , and then updating the parameters
, and then updating the parameters  by weighted linear regression. The iteration proceeds until convergence. The pseudodata
by weighted linear regression. The iteration proceeds until convergence. The pseudodata 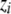 and pseudovariances
and pseudovariances 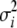 are calculated by
are calculated by
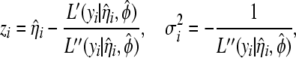 |
(6) |
where 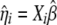 ,
,  ,
, 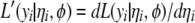 ,
, 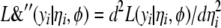 , and
, and 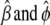 are the current estimates of
are the current estimates of 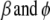 , respectively.
, respectively.
Given the variances 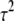 , the prior information on
, the prior information on 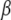 [i.e.,
[i.e., 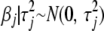 ] can be included in the classical generalized linear model as J additional “observations” of value 0, with “predictor matrix”
] can be included in the classical generalized linear model as J additional “observations” of value 0, with “predictor matrix” 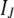 and “residual variance matrix”
and “residual variance matrix” 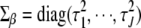 , where
, where  is the J × J identity matrix (Gelman et al. 2003, 2008). Thus, the generalized linear model with the normal prior on
is the J × J identity matrix (Gelman et al. 2003, 2008). Thus, the generalized linear model with the normal prior on 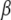 can be approximated by
can be approximated by
 |
(7) |
where
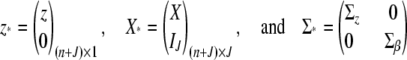 |
with 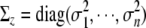 . Thus, we can use the standard iteratively weighted least-squares computation to estimate
. Thus, we can use the standard iteratively weighted least-squares computation to estimate 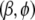 by performing a weighted regression of the augmented data vector
by performing a weighted regression of the augmented data vector  on the augmented predictor matrix
on the augmented predictor matrix  with augmented weight vector
with augmented weight vector 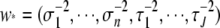 .
.
To implement the EM algorithm, we treat the unknown variances 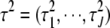 as missing data and average over them by replacing the terms involving both
as missing data and average over them by replacing the terms involving both 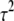 and
and  in the joint posterior (5) by their expected values conditional on the current estimate of
in the joint posterior (5) by their expected values conditional on the current estimate of  . Thus, we must evaluate the conditional expectations of
. Thus, we must evaluate the conditional expectations of  for all j. It can be easily shown that the conditional posterior distribution of
for all j. It can be easily shown that the conditional posterior distribution of  =
= 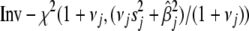 , and thus the conditional expectation of
, and thus the conditional expectation of  =
= 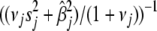 . Therefore, the E-step of our EM algorithm is equivalent to replacing the variances by
. Therefore, the E-step of our EM algorithm is equivalent to replacing the variances by
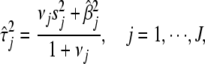 |
(8) |
where 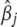 is the current estimate of
is the current estimate of  .
.
We have incorporated our algorithm into the standard package glm in R for fitting generalized linear models. We altered the glm function by inserting the steps for calculating the augmented data (7) and updating the variances (8) into the iterative procedure in the glm function. Our algorithm is initialized by setting each 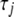 to a small value (say
to a small value (say 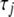 = 0.1) and
= 0.1) and 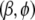 to the starting value provided by the glm function. At each step of our algorithm, we average over the variances
to the starting value provided by the glm function. At each step of our algorithm, we average over the variances  and then update
and then update 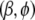 by maximizing the posterior density (5). In summary, our algorithm proceeds as follows:
by maximizing the posterior density (5). In summary, our algorithm proceeds as follows:
On the basis of the current value of
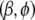 , calculate pseudodata
, calculate pseudodata 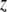 and pseudovariances
and pseudovariances 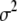 using (6).
using (6).E-step: replace each variance
 by its conditional expectation using (8).
by its conditional expectation using (8).M-step: perform the weighted least-squares regression based on the augmented data to obtain the estimate
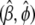 using (7).
using (7).Repeat steps 1–3 until convergence.
We apply the criterion in the glm function to assess convergence. In practice our algorithm converges rapidly. At convergence of the algorithm, we obtain all the outputs produced by the glm function, including the latest estimate 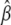 , their standard deviations and P-values (for testing
, their standard deviations and P-values (for testing  ), and some additional values (e.g., for the variances). The outputs are automatically stored as a standard glm object.
), and some additional values (e.g., for the variances). The outputs are automatically stored as a standard glm object.
Choosing the hyperparameters:
Before discussing our choice of the hyperparameters  and
and  we discuss the improper Jeffreys prior on the variances,
we discuss the improper Jeffreys prior on the variances,  , which is equivalent to the uniform distribution on
, which is equivalent to the uniform distribution on  and is the limiting case of the scaled inverse-
and is the limiting case of the scaled inverse- density with
density with 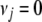 . The Jeffreys prior yields an improper posterior on each
. The Jeffreys prior yields an improper posterior on each 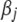 with an infinite mode at zero. As
with an infinite mode at zero. As  increases, however, a second finite local mode appears away from zero (e.g., ter Braak et al. 2005; Griffin and Brown 2007). The property of a model with the Jeffreys prior may make it problematic to fully explore the posterior, but still formally allows the analysis of posterior modes (see Griffin and Brown 2007). Much work has shown that the Jeffreys prior leads to good performance in MCMC and mode-finding analyses, yielding strong shrinkage for very small effects but weak shrinkage for large effects (Figueiredo 2003; Kiiveri 2003; Xu 2003; Bao and Mallick 2004; Griffin and Brown 2007).
increases, however, a second finite local mode appears away from zero (e.g., ter Braak et al. 2005; Griffin and Brown 2007). The property of a model with the Jeffreys prior may make it problematic to fully explore the posterior, but still formally allows the analysis of posterior modes (see Griffin and Brown 2007). Much work has shown that the Jeffreys prior leads to good performance in MCMC and mode-finding analyses, yielding strong shrinkage for very small effects but weak shrinkage for large effects (Figueiredo 2003; Kiiveri 2003; Xu 2003; Bao and Mallick 2004; Griffin and Brown 2007).
We choose 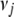 and
and 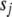 to retain the good features of the Jeffreys prior while avoiding the drawback of impropriety of both prior and posterior. This can be achieved by setting
to retain the good features of the Jeffreys prior while avoiding the drawback of impropriety of both prior and posterior. This can be achieved by setting  and
and  to be small enough so that the variances
to be small enough so that the variances 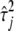 obtained by Equation 8 are close to zero for near-zero coefficients (
obtained by Equation 8 are close to zero for near-zero coefficients ( ≈ 0), but
≈ 0), but 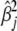 for large coefficients (
for large coefficients (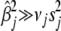 ). We set
). We set 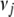 = 0.01 and
= 0.01 and 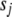 = 10−4 in our application. This prior works well and stably. However, we find that small changes from this default value (e.g.,
= 10−4 in our application. This prior works well and stably. However, we find that small changes from this default value (e.g.,  from 0 to 0.1,
from 0 to 0.1, 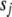 from 0 to 10−3) do not affect the results. The small positive value of
from 0 to 10−3) do not affect the results. The small positive value of  and
and  yields a proper prior on
yields a proper prior on 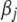 sharply peaked near zero but approximately uniform for the range away from zero and thus leads to strong sparseness in the fitted model while allowing for occasional large coefficients. Finally, our scaled inverse-
sharply peaked near zero but approximately uniform for the range away from zero and thus leads to strong sparseness in the fitted model while allowing for occasional large coefficients. Finally, our scaled inverse-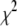 prior includes uniform priors for
prior includes uniform priors for  as a special case (
as a special case (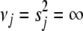 ), and thus we can perform no shrinkage for certain variables (e.g., relevant covariates).
), and thus we can perform no shrinkage for certain variables (e.g., relevant covariates).
Model search strategy:
In principle the above hierarchical models can handle any number of variables [see the augmented regression (8), where the number of data points (n + J) is larger than the number of variables J] and thus can simultaneously estimate genetic effects of a large number of markers. For biological interpretability, however, we prefer using only a subset of the genetic effects in the model. Furthermore, including all possible effects requires large memory and intensive computation. Therefore, we propose a model search strategy to build a parsimonious model, beginning with a model with no genetic effect but some relevant covariates if any, gradually adding different types of genetic effects into the model and then fitting the new model. It is expected that many variables have no effects on the phenotype and should be pruned from the model. We set a very small threshold value t1 (say 10−8) and delete genetic effects satisfying 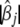 < t1 from the model.
< t1 from the model.
Our approach differs from classical forward stepwise methods by simultaneously adding many correlated variables and deleting many near-zero variables. As described below, our search strategy can take advantage of the special correlation structure in QTL data: genotypes on the same chromosomes are correlated. In summary, our approach proceeds as follows:
Searching for main effects: for each chromosome c (
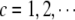 ), simultaneously add all possible main effects for markers on chromosome c into the current model, fit the model, and then delete some main effects on the basis of the above-mentioned criterion.
), simultaneously add all possible main effects for markers on chromosome c into the current model, fit the model, and then delete some main effects on the basis of the above-mentioned criterion.Searching for epistatic effects among the included main effects: simultaneously add all possible epistatic interactions among the included main effects into the current model, fit the model, and delete some epistatic effects.
Searching for epistatic effects between the excluded and the included main effects: for chromosome c (
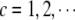 ), simultaneously add all possible epistatic interactions between the remaining main effects (not included in the current model) on chromosome c and the included main effects into the current model, fit the model, and delete some epistatic effects added in this step.
), simultaneously add all possible epistatic interactions between the remaining main effects (not included in the current model) on chromosome c and the included main effects into the current model, fit the model, and delete some epistatic effects added in this step.Searching for interactions between the covariate(s) and main effects: for chromosome c (
 ), simultaneously add all possible interactions between the covariate(s) and all possible main effects on chromosome c into the current model, fit the model, and delete some G × E interactions added in this step.
), simultaneously add all possible interactions between the covariate(s) and all possible main effects on chromosome c into the current model, fit the model, and delete some G × E interactions added in this step.
After these steps, we obtain a final model with preset covariates and genetic effects satisfying  > t1. With our shrinkage prior, the majority of genetic effects are shrunk to zero, and thus the final model includes only a limited number of variables. As shown in our simulation studies, our model search procedure picks up true effects with reasonable high probability, but can occasionally include some extraneous effects. However, inclusion of spurious effects has little effect on detecting and estimating true or strong effects, and the estimates of spurious effects (and the corresponding P-values) are smaller (larger) than those of the true effects, allowing us to easily identify strong causal effects. Although not necessary, however, it could be useful to have a formal procedure to remove extraneous effects from the final model. A simple way is to use the P-values for testing
> t1. With our shrinkage prior, the majority of genetic effects are shrunk to zero, and thus the final model includes only a limited number of variables. As shown in our simulation studies, our model search procedure picks up true effects with reasonable high probability, but can occasionally include some extraneous effects. However, inclusion of spurious effects has little effect on detecting and estimating true or strong effects, and the estimates of spurious effects (and the corresponding P-values) are smaller (larger) than those of the true effects, allowing us to easily identify strong causal effects. Although not necessary, however, it could be useful to have a formal procedure to remove extraneous effects from the final model. A simple way is to use the P-values for testing 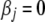 (equivalent to the likelihood-ratio test) in the final model; we set a small threshold value t2 (say 0.001) and remove genetic effects with the P-values larger than t2 from the model. As described later, a value of t2 = 0.001 can greatly reduce the rate of false positives. However, the P-values should be interpreted with caution, as they are based on the final model.
(equivalent to the likelihood-ratio test) in the final model; we set a small threshold value t2 (say 0.001) and remove genetic effects with the P-values larger than t2 from the model. As described later, a value of t2 = 0.001 can greatly reduce the rate of false positives. However, the P-values should be interpreted with caution, as they are based on the final model.
Implementation in R/qtlbim:
The freely available package R/qtlbim (www.qtlbim.org) is an extensible and interactive environment for multiple-QTL mapping in experimental crosses (Yandell et al. 2007), built on top of the widely used R/qtl (Broman et al. 2003). The previous version of R/qtlbim performs fully Bayesian analyses via MCMC algorithms to simultaneously handle covariates, main effects, epistatic and gene–environment interactions for continuous traits, and binary and ordinal traits. We have incorporated the proposed method into R/qtlbim, by creating new functions to implement our model-fitting algorithm and model search procedure. The outputs for the final model are stored as a standard glm object and thus can be summarized and displayed conventionally.
SIMULATION STUDIES
Simulation design:
Although our method can handle various types of phenotypes and models, we here evaluate the performance of the method for a normal continuous trait and a binary trait. We expect that the basic conclusions hold for other cases. In our simulations, we generated the continuous phenotypes on the basis of different multiple-QTL models and the binary trait by transforming the continuous values into two categories with the proportions of 40 and 60%, respectively. We generated a genome with 19 chromosomes, each of length 100 cM. Five percent of the marker genotypes were assumed to be randomly missing.
The first experiment was an F2 intercross composed of 400 progenies and having 11 equally spaced markers (placed 10 cM apart) on each chromosome. We considered a model with one binary covariate and five QTL with only main effects. The covariate effect explained ∼4% of the variance of the continuous phenotype. Table 1 presents the simulated positions of five QTL, their additive or dominance effects, and the approximate heritabilities (proportion of the phenotypic variation explained by an effect). As can be seen, two of five simulated QTL (Q2 and Q4) were positioned at markers, and others were in marker intervals. The second simulation was the same as the first one except that each chromosome had 51 equally spaced markers (placed 2 cM apart). Therefore, these two simulations had 209 and 969 total markers, respectively.
TABLE 1.
F2 simulation with five main-effect QTL
| QTL | Chromosome | Position (cM) | Main effect |
|---|---|---|---|
| Q1 | 1 | 12 | a = 0.5 (0.08) |
| Q2 | 1 | 60 | d = 0.7 (0.08) |
| Q3 | 2 | 23 | a = −0.5 (0.08) |
| Q4 | 2 | 50 | a = 0.5 (0.08) |
| Q5 | 8 | 24 | a = 0.4 (0.05) |
| d = −0.5 (0.04) |
The effects a and d represent additive and dominance effects, respectively.
Our third experiment was a backcross (BC) with 400 progenies and 11 equally spaced markers on each chromosome. We simulated one binary covariate and eight QTL with complex interactions; among the eight simulated QTL, five had main effects while other three had no main effects but interacted with other QTL or the covariate. The covariate effect explained ∼2% of the variance of the continuous phenotype. Table 2 presents the positions of simulated QTL, the genetic effects (main effects, epistasis, and G × E), and the approximate heritabilities. The fourth simulation was the same as the third except that each chromosome had 51 equally spaced markers.
TABLE 2.
Backcross simulation with eight interacting QTL
| QTL | Chromosome | Position(cM) | Main effect |
|---|---|---|---|
| Q1 | 1 | 10 | 1.0 (0.06) |
| Q2 | 1 | 60 | 1.0 (0.06) |
| Q3 | 2 | 20 | 1.0 (0.06) |
| Q4 | 2 | 40 | −1.0 (0.06) |
| Q5 | 3 | 20 | 1.0 (0.06) |
| Q6 | 3 | 80 | 0.0 (0.00) |
| Q7 | 4 | 10 | 0.0 (0.00) |
| Q8 | 5 | 10 | 0.0 (0.00) |
| Interaction pair
|
Epistasis or G × E
|
||
| Q1 | Q2 | 2.0 (0.06) | |
| Q1 | Q5 | −2.0 (0.06) | |
| Q3 | Q6 | 2.0 (0.06) | |
| Q7 | Covariate | 2.0 (0.05) | |
| Q8 | Covariate | 2.0 (0.05) | |
Effects were supplied while heritabilities in parentheses were estimated from simulated samples.
Analysis and evaluation:
Under each scenario, we carried out 200 simulation replicates. Each of the F2 simulated data sets was analyzed using two models; the first included a covariate term and main effects of all markers, and the second allowed not only the covariate and main effects but also epistatic and G × E interactions. The aim of the second analysis is to examine whether or not we detect interactions if they are not present. For each of the simulated backcross (BC) data sets, we fit a model with a covariate effect, main effects of all markers, and epistatic and G × E interactions. For all analyses, the continuous and the binary traits were analyzed using Gaussian and Probit models, respectively.
We tried several values for the hyperparameters 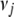 from 0 to 0.1 and
from 0 to 0.1 and 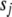 from 0 to 10−3 and got similar results. We displayed the results with (
from 0 to 10−3 and got similar results. We displayed the results with (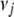 ,
, 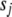 ) = (0.01, 10−4). To investigate the influence of two thresholds t1 and t2 on our model search method (see the last section), we analyzed the data sets using several values (t1 = 10−4, 10−6, 10−8, 10−10, 10−12, and t2 = 1, 0.01, 0.005, 0.001). We evaluated the performance of our method by examining the frequency of each effect included in the final model, true positives (the frequency of true effects included in the final model), and false positives (the frequency of false effects included in the final model) over the 200 simulation replicates. The inclusion frequency of an effect presents the empirical power to detect the effect. A detected main or G × E effect (epistatic effect) is considered as correct if the associated position(s) is within 10 cM of any true QTL.
) = (0.01, 10−4). To investigate the influence of two thresholds t1 and t2 on our model search method (see the last section), we analyzed the data sets using several values (t1 = 10−4, 10−6, 10−8, 10−10, 10−12, and t2 = 1, 0.01, 0.005, 0.001). We evaluated the performance of our method by examining the frequency of each effect included in the final model, true positives (the frequency of true effects included in the final model), and false positives (the frequency of false effects included in the final model) over the 200 simulation replicates. The inclusion frequency of an effect presents the empirical power to detect the effect. A detected main or G × E effect (epistatic effect) is considered as correct if the associated position(s) is within 10 cM of any true QTL.
Results:
The threshold t1 controls the minimal size of genetic effects to enter the models. We found that the final models almost picked up the same sets of effects for the above values of t1 in all the simulation experiments, and thus the choice of t1 had little influence on our model search method. Therefore, we present only the results for t1 = 10−8 below.
Figure 1 displays the genetic effects included in the final models with frequencies >0.01 for the four values of t2 in the first simulation experiment. We can see that no false effect (defined above) was included the final models with frequency >0.01. For the continuous trait, the simulated main effects located at markers, (1@60)d and (2@50)a, were detected with frequencies close to 1. As expected, the analyses of the binary trait had lower power than the continuous trait, especially for the two linked QTL on chromosome 2. For the simulated effects located within the marker intervals, our analyses picked up the corresponding effects of the flanking markers in the final models; for example, for the simulated effect (2@23)a, the final models included (2@20)a or (2@30)a. For these simulated effects, the true positives that roughly equal the sum of the inclusion frequencies of the corresponding flanking marker effects were also high. Although no interactions were present, the analyses allowing epistatic and G × E effects did not reduce the power for detecting the main effects and did not include any interactions with high frequency. Finally, it can be seen that a smaller threshold t2 slightly reduced the power. However, this threshold controls the rate of false positives, which will be illustrated shortly.
Figure 1.—

F2 simulation with 19 chromosomes each having 100 cM and 11 markers: frequencies of effects included in final models over 200 replicates with the thresholds t1 = 10−8 and t2 = 1, 0.01, 0.005, and 0.001 for the continuous trait (circles) and the binary trait (triangles). Effects with frequencies <0.01 are not displayed. The right (left) panel represents analyses excluding (including) epistatic and G × E interactions. The notation for effects, e.g., (1@10)a, indicates chromosome 1, position 10 cM, and additive effect a (or dominance effect d).
Figure 2 shows the results of the analyses for the second experiment. The models including or excluding epistatic and G × E interactions produced similar results again. Our second experiment simulated 51 markers evenly distributed on each chromosome, resulting in much higher correlation among the variables. Consequently, the simulated effects were included with lower frequencies than in the first experiment. However, we were still able to identify the true effects with reasonable high power and true positives for both the continuous and the binary traits. For example, the first simulated effect (1@12)a was identified with power of ∼60% (40%) and true positives of ∼90% (80%) for the continuous (binary) trait.
Figure 2.—

F2 simulation with 19 chromosomes each having 100 cM and 51 markers: frequencies of effects included in final models over 200 replicates with the thresholds t1 = 10−8 and t2 = 1, 0.01, 0.005, and 0.001 for the continuous trait (circles) and the binary trait (triangles). Effects with frequencies <0.01 are not displayed. The right (left) panel represents analyses excluding (including) epistatic and G × E interactions. The notation for effects, e.g., (1@12)a, indicates chromosome 1, position 12 cM, and additive effect a (or dominance effect d).
The results for the BC simulations are illustrated in Figure 3. For the experiment with 11 markers on each chromosome, no false effect was included the final models with frequency >0.01. For the continuous trait, all the simulated main effects and epistatic and G × E interactions were identified in almost all 200 replicates. For all the simulated main effects and epistatic interactions, the analyses of the binary trait had lower power; the reduction of the power was larger for the main effects of the two relatively tightly linked QTL on chromosome 2 and epistasis. For the fourth experiment where 51 markers were evenly placed on each chromosome, most of the simulated effects were included in the final models with frequencies >60% for the continuous trait. Higher correlation among the variables resulted in the method occasionally picking up effects with associated positions near to the true effects. Again, the true positives for all the simulated effects were high. Finally, it was observed that the threshold t2 had little influence on the power of detecting the true effects.
Figure 3.—

Backcross simulation with 19 chromosomes each having 100 cM and 11 (right) or 51 (left) markers: frequencies of effects included in final models over 200 replicates with the thresholds t1 = 10−8 and t2 = 1, 0.01, 0.005, and 0.001 for the continuous trait (circles) and the binary trait (triangles). Effects with frequencies <0.01 are not displayed. The notation for effects, e.g., (1@10), indicates chromosome 1 and position 10 cM. The term fix.cov represents the covariate, and X1:X2 represents interaction between X1 and X2.
Figure 4 shows the false positives for the BC experiments under different P-values t2. The other experiments gave similar results. With a large value t2, the models included extraneous effects with high frequencies in the simulation replicates. Data with dense marker maps generally produced lower false positives. False effects were removed from the models rapidly as the threshold t2 decreased, indicating that false effects were associated with larger P-values. For all the cases, we observed that the estimates of the simulated effects were close to the corresponding true values. But the estimates of spurious effects were much smaller.
Figure 4.—

False positive rates for the threshold t2 = 1, 0.01, 0.005, and 0.001 for the backcross simulations. The sparse and dense maps indicate the simulations with 11 and 51 markers on each chromosome, respectively.
REAL DATA EXAMPLES
We illustrate our method with three real QTL mapping experiments. These data sets have been extensively analyzed previously and thus serve as good examples to compare our method with the existing methods and to illustrate the advantages of our method in terms of statistical and computational efficiencies and modeling flexibility. We used several values for 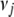 (between 0 and 0.1) and
(between 0 and 0.1) and 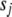 (between 0 and 10−3) and obtained identical results. We displayed the results by setting the thresholds t1 = 10−8 and t2 = 1.
(between 0 and 10−3) and obtained identical results. We displayed the results by setting the thresholds t1 = 10−8 and t2 = 1.
Barley data set:
The barley data set (Tinker et al. 1996) was reanalyzed for demonstration. The data were collected from a doubled-haploid population that contained n = 145 lines; each was grown in 25 different environments. The phenotype analyzed was the average value of kernel weight across environments. A total of 127 markers spanning seven chromosomes were genotyped. The data contained ∼5% missing marker genotypes.
We used a Gaussian model to fit main effects of all markers and their epistatic interactions. The total number of effects was 8129 (127 main effects and 127 × 126/2 = 8001 epistatic effects). Our analysis took ∼0.05 min on a P4 computer. We obtained a final model with 8 main effects: 3 on chromosome 1; 2 on chromosome 2; and 1 on chromosomes 3, 4 and 7, respectively. We did not detect epistatic effects. The estimates of the main effects are displayed in the top panel of Figure 5. The P-values for these effects were 1.3e-07, 3.3e-15, 1.4e-03, 4.3e-05, 6.9e-04, 9.2e-05, 3.3e-03, and 3.1e-24, respectively. If we set t2 = 0.001, the effects 1@212.1 and 4@145.5 are removed from the final model. For comparison, we reanalyzed the data using the MCMC method (Yi et al. 2005, 2007a) implemented in R/qtlbim (Yandell et al. 2007). Our MCMC analysis also did not detect epistatic effects, but did detect multiple main effects with positions overlapping or close to those in the proposed method analysis (see the bottom panel of Figure 5).
Figure 5.—

Barley data set analyses using the proposed method (top) and the Bayesian MCMC method (bottom). (Top) The estimated genetic effects (±1 standard error) in the final model are shown. The notation for main effects, e.g., (1@0), indicates chromosome 1 and position 0 cM. The bottom panel shows profiles of Bayes factors (rescaled as 2 logeBF and negative values are truncated as zero) for main effects.
This data set has been extensively analyzed using different methods, including mode-finding algorithms (Xu 2007; Xu and Jia 2007) and shrinkage MCMC methods (Xu 2003; Yi and Xu 2008). Xu (2007) (also see Xu and Jia 2007) analyzed this data set using an empirical Bayes method and the LASSO of Tibshirani (1996). Both the analyses took ∼2 min and obtained results similar to ours. The shrinkage MCMC algorithms also gave similar results, but took a few hours (Xu 2003; Yi and Xu 2008). Therefore, our method performs comparably to the alternative sophisticated techniques and is computationally faster.
Listeria monocytogenes data set:
As a second study we illustrate the application of our method by reanalyses of the mouse data of Boyartchuk et al. (2001). This data set consisted of 116 female mice from an intercross (F2) between the BALB/cByJ and C57BL/6ByJ strains. Each animal was genotyped at 133 markers spanning 20 chromosomes. The phenotype of interest was the time to death following infection with L. monocytogenes. The animals that died from infection had a mean survival of 153.8 hr. Roughly 30% of the mice recovered from the infection and survived past the 264-hr time point. Several methods have been applied to this data set, including a two-part model (Broman 2003), a parametric proportional hazards model (Diao et al. 2004) and a unified semiparametric framework approach (Jin et al. 2007). These analyses detected significant QTL on chromosomes 1, 5, and 13 and suggestive QTL on chromosome 15.
We applied our method to the data using the two-part model of Broman (2003) rather than developing a new model for this type of phenotype. The two-part model decomposes the phenotype into a binary trait, indicating whether the subject survived, and a continuous trait for time to death for those dying within 264 hr (Broman 2003). We used a Probit regression model for the binary phenotype and a Gaussian model for the logarithm of the continuous phenotype, and fit a model with main effects of all markers and their epistatic interactions. The total number of effects was 35,378 (133 additive effects, 133 dominance effects, and 35,112 epistatic effects).
Our analyses took ∼0.02 and 0.03 min on a P4 computer, for the continuous and the binary traits, respectively. Our results were consistent with the previous ones (Figure 6). For the continuous trait, the final model included two additive effects on chromosomes 1 and 15, respectively, and one dominance effect on chromosome 13. The P-values for these effects were 3.8e-05, 1.8e-02, and 1.6e-03, respectively. For the binary trait, the final model included two additive effects on chromosomes 5 and 13, with the P-values being 1.7e-07 and 1.9e-04, respectively. We did not detect epistatic effects.
Figure 6.—

Listeria monocytogenes data set analyses. The estimated genetic effects (±1 standard error) in the final models for (top) the continuous and (bottom) the binary traits are shown. The notation for effects, e.g., (1@81.4)a, indicates chromosome 1, position 81.4 cM, and additive effect a (or dominance effect d).
We also analyzed the data using the Bayesian MCMC method (Yi et al. 2005, 2007a,b; Yandell et al. 2007). The MCMC algorithm detected additive effects on chromosomes 1 and 15 and a dominance effect on chromosome 13 for the continuous trait and additive effects on chromosomes 5 and 13 for the binary trait (not shown here). The positions of these QTL were close to the markers detected by the proposed method. In addition these main effects, the fully Bayesian analysis found evidence of epistatic effects: for the continuous trait, QTL on chromosomes 1, 5, and 9 showed evidence of epistasis, and for the binary trait, chromosomes 6 and 13 harbored epistatic QTL. However, these epistatic effects were not strong. The MCMC analyses took ∼8 and 10 min for the continuous and the binary traits, respectively.
Obesity data set:
We also applied our method to an obesity data set of backcross mice. This data set has been extensively analyzed using Bayesian MCMC methods (Yi et al. 2005, 2006, 2007a). The cross was produced from two highly divergent strains: M16i, consisting of large and moderately obese mice, and CAST/Ei, a wild strain of small mice with lean bodies. CAST/Ei males were mated to M16i females, and F1 males were backcrossed to M16i females, resulting in 421 mice (213 males and 208 females) reaching 12 weeks of age. All mice were genotyped for 92 markers located on 19 autosomal chromosomes. We analyzed a continuous trait Fat, the sum of right gonadal and hindlimb subcutaneous fat pads. We included sex and body weight at the age of 12 weeks as covariates and permitted the inclusion of epistatic and gene–sex interactions in a Gaussian regression model. The total number of genetic effects was 4370, including 92 main effects, 4186 epistatic effects, and 92 gene–sex interactions.
Using the MCMC algorithm (which took ∼15 min), Yi et al. (2007a) detected evidence of QTL on nine chromosomes (1, 2, 6, 8, 13, 14, 15, 18, and 19). These QTL showed a complex network of interactions. The strongest main-effect QTL was found on chromosome 2, and QTL on chromosomes 8, 13, 14, 15, and 19 also had main effects. Strong epistatic interactions between chromosomes 15 and 19 and chromosomes 1 and 18 were detected. Evidence of epistasis was found between a few other chromosome pairs. The QTL on chromosome 2 was found to interact with the covariate sex.
Our hierarchical generalized linear model analysis took ∼0.15 min and obtained a final model including 12 main effects, 5 epistatic effects, and two gene–sex interactions. The estimates of the genetic effects and their P-values are displayed in Figure 7. Our results were basically consistent with the previous full Bayesian analysis; all strong effects were detected by both analyses. All the main-effect QTL detected in Yi et al. (2007a) were also identified in our reanalysis, with positions in the regions detected previously. The P-values for most of the main effects were <0.001. Main effects of two linked markers on chromosome 2 were simultaneously included in the model, which were the largest among the detected main effects. The epistatic interactions (19@38):(15@39) and (1@55):(18@73) were the strongest, which were also detected previously. Main effects on both chromosomes 1 and 18 were not included in the final model, indicating that our method can detect QTL with weak main effects but strong epistasis. Two gene–sex interactions, (2@73):SEX and (15@12):SEX, were included in the final model with P-values being 1.7e-03 and 4.4e-03, respectively.
Figure 7.—

Obesity data set analysis. The (left) estimated genetic effects (±1 standard error) in the final models and (right) their P-values are shown. The notation for additive effects, e.g., (2@73), indicates chromosome 2 and position 73 cM. The term X1:X2 represents interaction between X1 and X2.
DISCUSSION
We propose a new statistical framework for genomewide multiple QTL mapping in experimental crosses. Our method is developed on the basis of the generalized linear model framework and thus can deal with various phenotypes that the standard generalized linear models can analyze. The proposed models assume sparseness-inducing priors and thus can simultaneously fit a large number of effects, including environmental effects, main effects of numerous genomic loci, and epistatic and G × E interactions. By looking for the modes of the posterior distribution via the EM algorithm we are able to quickly identify significant effects. Experiments with extensive simulation studies and real data sets have shown good performance.
We assign independent normal distributions with unknown variances to the coefficients and devote our attention to the scaled inverse-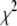 distribution for the variance parameters. Our prior includes the noninformative Jeffreys prior as a special case, which has the form of a scaled inverse-
distribution for the variance parameters. Our prior includes the noninformative Jeffreys prior as a special case, which has the form of a scaled inverse-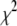 density with 0 d.f. As already shown in previous studies (Figueiredo 2003; Kiiveri 2003; Xu 2003; Bao and Mallick 2004; Griffin and Brown 2007) and in our experimental results, the Jeffreys prior usually yields good performance, inducing strong sparseness in the fitted model. However, we recommend choosing the hyperparameters in the scaled inverse-
density with 0 d.f. As already shown in previous studies (Figueiredo 2003; Kiiveri 2003; Xu 2003; Bao and Mallick 2004; Griffin and Brown 2007) and in our experimental results, the Jeffreys prior usually yields good performance, inducing strong sparseness in the fitted model. However, we recommend choosing the hyperparameters in the scaled inverse- to yield a proper prior on coefficients sharply peaked near zero but approximately uniform for the range away from zero. Our prior induces strong shrinkage for near-zero effects but weak shrinkage for large effects. Our method can be in principle extended to models with other prior distributions, for example, the exponential distributions with fixed hyperparameters. However, it would be appealing to treat the hyperparameters as unknowns and estimate them from the data so that the model can shrink the coefficients as much as can be justified by the data. Yi and Xu (2008) developed MCMC algorithms to jointly estimate all hyperparameters and model parameters and found that such procedures usually outperform those with fixed hyperparameters. Future research will extend the proposed EM algorithm to estimate hyperparameters.
to yield a proper prior on coefficients sharply peaked near zero but approximately uniform for the range away from zero. Our prior induces strong shrinkage for near-zero effects but weak shrinkage for large effects. Our method can be in principle extended to models with other prior distributions, for example, the exponential distributions with fixed hyperparameters. However, it would be appealing to treat the hyperparameters as unknowns and estimate them from the data so that the model can shrink the coefficients as much as can be justified by the data. Yi and Xu (2008) developed MCMC algorithms to jointly estimate all hyperparameters and model parameters and found that such procedures usually outperform those with fixed hyperparameters. Future research will extend the proposed EM algorithm to estimate hyperparameters.
Gelman et al. (2008) proposed independent Cauchy distribution with mean 0 and scale 2.5 [i.e.,  and
and  ] for routine data analysis, and implemented a procedure to fit generalized linear models with this prior by incorporating an EM algorithm into the usual iteratively weighted least squares. However, this Cauchy prior cannot shrink small effects to zero and thus cannot be used for our high-dimensional models. Our algorithm is similar to that of Gelman et al. (2008), but differs in regarding the variances rather than the coefficients as missing data. The trick of the algorithm is to express the prior information on coefficients as additional “data points,” allowing us to take advantage of the existing iteratively weighted least-squares algorithm. Various other efficient methods have been proposed recently, but are built upon special algorithms and thus more difficult to implement (e.g., Tibshirani 1996; Efron et al. 2004; Genkin et al. 2007; Xu 2007).
] for routine data analysis, and implemented a procedure to fit generalized linear models with this prior by incorporating an EM algorithm into the usual iteratively weighted least squares. However, this Cauchy prior cannot shrink small effects to zero and thus cannot be used for our high-dimensional models. Our algorithm is similar to that of Gelman et al. (2008), but differs in regarding the variances rather than the coefficients as missing data. The trick of the algorithm is to express the prior information on coefficients as additional “data points,” allowing us to take advantage of the existing iteratively weighted least-squares algorithm. Various other efficient methods have been proposed recently, but are built upon special algorithms and thus more difficult to implement (e.g., Tibshirani 1996; Efron et al. 2004; Genkin et al. 2007; Xu 2007).
Rather than fitting all possible effects in a large model, we propose a novel model search strategy to build a parsimonious model. Our method begins with a model with no genetic effect and then adds different types of genetic effects into the model and deletes very small effects. Unlike the traditional variable selection approach, we simultaneously add or delete many variables and take care of the correlation among the variables. Kiiveri (2003) and Zhang and Xu (2005) adopted a similar criterion for pruning effects from the model, but used a backward selection procedure. Our search strategy avoids the need of large memory and computational problems when the number of effects is huge, and thus facilitates the genomewide interacting QTL analysis.
The fully Bayesian approach, employing the MCMC simulation to generate posterior samples from the joint posterior distribution of all unknowns, can provide more comprehensive posterior inferences (Gelman et al. 2003). In QTL mapping, the fully Bayesian methods can easily treat missing genotypes and positions of QTL as unknowns and use model averaging to induce robustness and stability (Yi et al. 2005, 2007a,b; Yi and Shriner 2008). We describe our method by allowing only observed markers as potential QTL and replacing missing marker genotypes by their conditional expectations. We can extend our method to detect QTL within marker intervals by inserting loci within each marker interval (pseudomarkers) as possible positions of QTL. Further investigation may be needed on how the number and the spacing of pseudomarkers affect the performance of the method. The proposed method has computational advantages over the fully Bayesian MCMC methods. High-order interactions could be implemented using our approach, but this would substantially increase the model space to be explored and thus may need further investigation. Future research will extend our method to genomewide association analysis and gene expression QTL (eQTL) mapping. Fast and efficient algorithms are an essential feature for the practical analysis of these more complicated cases.
Acknowledgments
This work was supported by National Institutes of Health grant R01 GM069430 to N.Y. and partially supported by Clinical and Translational Sciences Center grant UL1-RR024996 to S.B.
References
- Baierl, A., M. Bogdan, F. Frommlet and A. Futschik, 2006. On locating multiple interacting quantitative trait loci in intercross designs. Genetics 173 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao, K., and B. K. Mallick, 2004. Gene selection using a two-level hierarchical Bayesian model. Bioinformatics 20 3423–3430. [DOI] [PubMed] [Google Scholar]
- Bogdan, M., J. K. Ghosh and R. W. Doerge, 2004. Modifying the Schwarz Bayesian information criterion to locate multiple interacting quantitative trait loci. Genetics 167 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyartchuk, V. L., K. W. Broman, R. E. Mosher, S. E. F. D'Orazio, M. N. Starnbach et al., 2001. Multigenic control of Listeria monocytogenes susceptibility in mice. Nat. Genet. 27 259–260. [DOI] [PubMed] [Google Scholar]
- Broman, K. W., 2003. Mapping quantitative trait loci in the case of a spike in the phenotype distribution. Genetics 163 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., and T. P. Speed, 2002. A model selection approach for identification of quantitative trait loci in experimental crosses. J. R. Stat. Soc. B 64 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., H. Wu, Ś. Sen and G. A. Churchill, 2003. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19 889–890. [DOI] [PubMed] [Google Scholar]
- Carlborg, Ö., and C. Haley, 2004. Epistasis: too often neglected in complex trait studies? Nat. Rev. Genet. 5 618–625. [DOI] [PubMed] [Google Scholar]
- Diao, G., D. Y. Lin and F. Zou, 2004. Mapping quantitative trait loci with censored observations. Genetics 168 1689–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efron, B., T. Hastie, I. Johnstone and R. Tibshirani, 2004. Least angle regression. Ann. Stat. 32 407–499. [Google Scholar]
- Figueiredo, M. A. T., 2003. Adaptive sparseness for supervised learning. IEEE Trans. Patt. Anal. Mach. Intell. 25 1150–1159. [Google Scholar]
- Gelman, A., J. Carlin, H. Stern and D. Rubin, 2003. Bayesian Data Analysis. Chapman & Hall, London.
- Gelman, A., A. Jakulin, M. G. Pittau and Y. S. Su, 2008. A weakly informative default prior distribution for logistic and other regression models. Ann. Appl. Stat. (in press).
- Genkin, A., D. D. Lewis and D. Madigan, 2007. Large-scale Bayesian logistic regression for text categorization. Technometrics 49 291–304. [Google Scholar]
- Griffin, J. E., and P. J. Brown, 2007. Bayesian adaptive lassos with non-convex penalization. Technical Report, University of Warwick, Coventry, UK.
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69 315–324. [DOI] [PubMed] [Google Scholar]
- Jiang, C., and Z.-B. Zeng, 1997. Mapping quantitative trait loci with dominant and missing markers in various crosses from two inbred lines. Genetica 101 47–58. [DOI] [PubMed] [Google Scholar]
- Jin, C., J. P. Fine and B. S. Yandell, 2007. A unified semiparametric framework for quantitative trait loci analysis, with application to spike phenotypes. J. Am. Stat. Assoc. 102 56–67. [Google Scholar]
- Kao, C. H., Z-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiiveri, H., 2003. A Bayesian approach to variable selection when the number of variables is very large, pp. 127–143 in Science and Statistics: Festschrift for Terry Speed (Institute of Mathematical Statistics Lecture Notes—Monograph Series, Vol 40), edited by D. R. Goldstein. Hayward, CA.
- McCullagh, P., and J. A. Nelder, 1989. Generalized Linear Models, Ed. 2. Chapman & Hall, London.
- Meuwissen, T. H. E., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, T., and G. Casella, 2008. The Bayesian Lasso. J. Am. Stat. Assoc. 103 681–686. [Google Scholar]
- R Development Core Team, 2006. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. http://www.R-project.org.
- ter Braak, C., M. Boer and M. Bink, 2005. Extending Xu's Bayesian model for estimating polygenic effects using markers of the entire genome. Genetics 170 1435–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibshirani, R., 1996. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. B 58 267–288. [Google Scholar]
- Tinker, N. A., D. E. Mather, B. G. Rossnagel, K. J. Kasha and A. Kleinhofs, 1996. Regions of the genome that affect agronomic performance in two-row barley. Crop Sci. 36 1053–1062. [Google Scholar]
- Xu, S., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., 2007. An empirical Bayes method for estimating epistatic effects of quantitative trait loci. Biometrics 63 513–521. [DOI] [PubMed] [Google Scholar]
- Xu, S., and Z. Jia, 2007. Genome-wide analysis of epistatic effects for quantitative traits in barley. Genetics 175 1955–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell, B. S., T. Mehta, S. Banerjee, D. Shriner, R. Venkataraman et al., 2007. R/qtlbim: QTL with Bayesian interval mapping in experimental crosses. Bioinformatics 23 641–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., 2004. A unified Markov chain Monte Carlo framework for mapping multiple quantitative trait loci. Genetics 167 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., and D. Shriner, 2008. Advances in Bayesian multiple QTL mapping in experimental designs. Heredity 100 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., and S. Xu, 2008. Bayesian LASSO for quantitative trait loci mapping. Genetics 179 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., B. S. Yandell, G. A. Churchill, D. B. Allison, E. J. Eisen et al., 2005. Bayesian model selection for genome-wide epistatic QTL analysis. Genetics 170 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., D. K. Zinniel, K. Kim, E. J. Eisen, A. Bartolucci et al., 2006. Bayesian analysis of multiple epistatic QTL models for body weight and body composition in mice. Genet. Res. 87 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., D. Shriner, S. Banerjee, T. Mehta, D. Pomp et al., 2007. a An efficient Bayesian model selection approach for interacting QTL models with many effects. Genetics 176 1865–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., S. Banerjee, D. Pomp and B. S. Yandell, 2007. b Bayesian mapping of genome-wide interacting QTL for ordinal traits. Genetics 176 1855–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z.-B., T. Wang and W. Zou, 2005. Modeling quantitative trait loci and interpretation of models. Genetics 169 1711–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.-M., and S. Xu, 2005. A penalized maximum likelihood method for estimating epistatic effects of QTL. Heredity 95 96–104. [DOI] [PubMed] [Google Scholar]