

Abstract
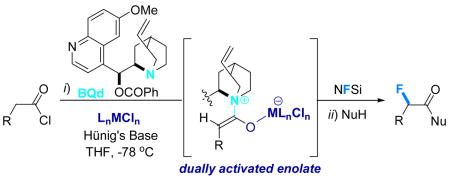
In this Communication, we disclose a catalytic, highly enantioselective (up to >99% ee) α-fluorination of acid chlorides to produce a variety of optically active carboxylic acid derivatives from readily accessible and commercially available starting materials. The reaction depends on dually activated ketene enolates generated from two discrete catalysts - a chiral nucleophile and an achiral transition metal complex working in tandem. The active, putative α-fluorobis(sulfonimide) intermediates readily transacylate in situ under mild conditions upon addition of a wide variety of nucleophiles, including complex natural products. As a consequence, the power of this method is witnessed by the broad range of α-fluorinated products that can be accessed efficiently depending on the work up conditions.
In many ways, medicinal chemistry has entered the “age of fluorine.” The number of drugs and drug candidates that contain fluorine has increased exponentially over the past few years, making its installation in a stereoselective (especially an enantioselective) manner of vital importance to the synthetic chemist.1 Along these lines, several impressive examples of catalytic, enantioselective fluorination have recently appeared in which β-keto esters, imides, and aldehydes serve as substrates to produce products in high enantioselectivity and yield.2 However, one very important complementary piece of the puzzle would be an enantioselective α-fluorination of ketene enolates that could produce simple, optically enriched α-fluorinated carboxylic acid derivatives directly.3 In this communication, we report a catalytic, highly enantioselective α-fluorination of acid chlorides to produce products of broad scope and in excellent yield. This new reaction exploits a recently developed “dual activation” strategy from our labs in which a chiral nucleophile is combined with a transition metal-based Lewis acid cocatalyst to access metal-coordinated, chiral ketene enolates.4 In this instance, these catalytically generated, dually-activated enolates are efficiently fluorinated with commercially available N-fluorodibenzene-sulfonimide (NFSi)5 to produce configurationally stable α-fluorinated carboxylic acid derivatives in high enantiomeric excess (ee) (eq 1). The power of this new method is demonstrated in the broad range of different derivatives that can be synthesized depending on the work up conditions; fluorinated carboxylic acids, amides, esters, and even peptides are all accessible depending on the nucleophile employed to quench the reaction.
 |
(1) |
Our interest in catalytic, enantioselective halogenation extends back over several years.6 We originally reported an enantioselective chlorination and bromination of acid chlorides using cinchona alkaloid derivatives as catalysts, and polyhalogenated quinones as mild sources of halogen in conjunction with a variety of stoichiometric bases. Unfortunately, we could never adequately extend this method to an analogous enantioselective fluorination. At best, only low yields of product were observed – the ketene enolate nucleophilicity was seemingly never high enough to allow a smooth reaction with standard fluorinating agents, including NFSi. We returned to the challenge after a lengthy recess upon our discovery that the reactivity of ketene enolates towards o-quinones in enantioselective oxygenation reactions could be substantially enhanced by the inclusion of a transition metal cocatalyst, namely (Ph3P)2PdCl2.4 Extensive evidence showed that this metal species binds to the ketene enolate oxygen, thereby enhancing both its reactivity and chemoselectivity. Consequently, whereas the “metal-free” reaction of p-methoxyphenylacetyl chloride (1a), Hünig’s base, 10 mol% benzoylquinidine (BQd), and 1 equiv NFSi in THF at −78 °C (followed by a methanol quench)7 produces a modest yield of the chiral, α-fluorinated methyl ester 2a, including 10 mol% trans-(Ph3P)2PdCl2 as a cocatalyst dramatically increases the yield of product 2a to 76% (98% ee). Isolation of the initially formed, putative bis(sulfonimide) intermediate (5) is difficult due to its lability; this fact necessitated a quenching reaction that turned out to be a blessing in disguise. A number of middle-transition series-based cocatalysts were then screened, with the results summarized in Table 1. For example, the metal free reaction (entry 1) optimized at 39% yield of 2a (97% ee).8 Wilkinson’s catalyst (entry 2) produced a marginal increase, whereas (PPh3)2PtCl2 (entries 3 and 4) produces a notable increase in yield. Although the yields for the metal screen vary, the ee’s for the test reaction stay consistently high. trans-(PPh3)2PdCl2 (76%) and the chelating complex (dppp)NiCl29 (83%) performed the best, producing the desired product in 98% and 99% ee, entries 6 and 8, respectively.
Table 1.
Metal Complexes Tested for Dual System
 | |||
|---|---|---|---|
| entry | metal complex | % yielda | % ee |
| 1 | no metal | 39 | 97 |
| 2 | (PPh3)3RhCl | 42 | 97 |
| 3 | trans-(PPh3)2PtCl2b | 60 | 98 |
| 4 | cis-(PPh3)2PtCl2 | 62 | 98 |
| 5 | cis-(PPh3)2PdCl2 | 72 | 97 |
| 6 | trans-(PPh3)2PdCl2b | 76 | 98 |
| 7 | (PPh3)2NiCl2 | 77 | 97 |
| 8 | (dppp)NiCl2c | 83 | 99 |
reactions run with 1 equiv acid chloride, base, NFSi, and cataylsts: 10 mol% BQd, and 10 mol% metal complex, except where noted; reactions quenched with methanol after 8 h; yield for pure product.
used 1 mol%;
used 3 mol%.
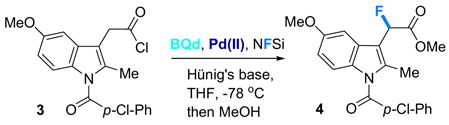
Accordingly, both achiral Pd- and Ni-based phosphine complexes were chosen (interchangeably) for further screening on a range of acid chlorides, as presented in Table 2. Acid halides containing aromatic as well as heterocyclic substituents proved to be good substrates, producing products in very high ee and good to excellent yields.10 Protected aminoalkyl-substituted acid chlorides also work well, leading to α-fluorinated β-amino acid derivatives. Another salient example is shown in the fluorination of readily available acid chloride 3 to provide fluorinated product 4 (84% yield and 95% ee), a derivative of the anti-inflammatory drug indomethacin11 (eq 2). Other examples, such as the fluorination of an aldol adduct to form 2l, show that the reaction is also compatible with isomerizable carbon-carbon double bonds.
Table 2.
Asymmetric, Bifunctional Catalytic α-Fluorination
 | ||||||
|---|---|---|---|---|---|---|
| entry | R | cocatalyst [M]a | NuH | product | % yielda | % ee [de]g |
| 1 | p-MeOPh | Ni(II) | MeOH | 2a | 83 | 99 |
| 2 | p-MeOPh | Pd(II) | L-NH2-Phe-OEtb | 2b* | 68 | >99g |
| 3 | p-MeOPh | Pd(II) | PhSHb | 2c | 67 | 98 |
| 4 | p-MeOPh | Ni(II) | N-Boc-L-prolinolb | 2d | 90 | >99g |
| 5 | Ph | Ni(II) | MeOH | 2e | 61 | 99d |
| 6 | Ph | Ni(II) | H2O | 2f* | 60 | 99d |
| 7 | 1-Np | Ni(II) | MeOH | 2g | 68 | 98 |
| 8 | 1-Np | Ni(II) | N-Boc-L-Cys-OMeb | 2h* | 80 | >99g |
| 9 | 2-Np | Pd(II) | MeOH | 2i | 63 | >99 |
| 10 | 2-thiophene | Pd(II) | MeOH | 2j | 69 | 99 |
| 11 | 3-(N-benzoylindolyl) | Pd(II) | MeOH | 2k* | 58 | 94 |
| 12 | 2-(3-Ph-(ethylcinnamate)) | Ni(II) | MeOH | 2l* | 71 | 99 |
| 13 | phthalimido-CH2 | Pd(II) | MeOH | 2m | 72 | >99 |
| 14 | phthalimido-CH2e | Pd(II) | MeOH | 2n* | 74 | 99 |
| 15 | phthalimido-CH2 | Pd(II) | NH(CH2)5b | 2o* | 79 | >99 |
| 16 | indof | Pd(II) | MeOH | 4* | 84 | 95 |
| 17 | phthalimido-CH2 | Pd(II) | (+)-emetinec | 7* | 91 | >99g |

| ||||||
reactions run with 1 equiv acid chloride, Hünig’s base, NFSi, and catalysts: 10 mol% BQd, and 3 mol% (1,3-dppp)NiCl2 or trans-(PPh3)2PdCl2 in THF at −78 °C, and were quenched with nucleophile after 6 - 15 h; yield for pure product based on limiting reagent. An excess of NuH was used except:
1.1 equiv NuH, or
0.8 equiv NuH.
correlation confirmed sense of induction, see S.I.
BQ was used instead of BQd and yields the (S)-enantiomer.
3-[N-(p-Cl-benzoyl)-(5-MeO-2-Me-indol)].
diastereomeric excess (de) is measured.
Product is depicted.
Additionally, simply by modifying the quench conditions, an array of chiral, α-fluorinated carboxylic acid derivatives can be produced. Along with an alcohol quench that provides esters, a water work up affords α-fluoro carboxylic acids, compounds that should be of potentially broad utility. An amine-based work up affords amides; accordingly, thioesters can be readily accessed. In a representative example, work up with L-phenylalanine ethyl ester produces the fluorinated peptide 2b in 68% yield (>99% diastereomeric excess [de]).
 |
(2) |
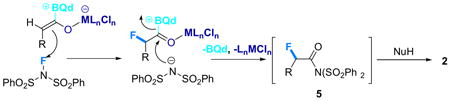
From a mechanistic standpoint, the absence of observable concentrations of free, protonated dibenzenesulfonimide during the course of the reaction suggests that it indeed reacts to form intermediate 5. This leads us to propose a mechanism (eq 3), which is also based on our previous data on metal-bound zwitterionic ketene enolates.4 Fluorination of the dually-activated
 |
(3) |
enolate leads to an acyl ammonium salt that reacts with the liberated dibenzenesulfonimide anion to form the active amide intermediate 5. As established, this species effects a transacylation with added nucleophiles to generate the final products in high ee and excellent yields.
Finally, one of the prime advantages of generating chiral, α-fluorinated reactive intermediates “in a flask” is the ability to quench them with drugs, natural products, and other exotic nucleophiles to produce interesting and potentially useful derivatives. For example, work up of a standard fluorination of 3-phthalimidopropionyl chloride (1b) with the antiprotozoal isoquinoline alkaloid12 natural product (+)-emetine produces the diastereomerically pure fluorinated derivative 7 in 91% yield (eq 4).
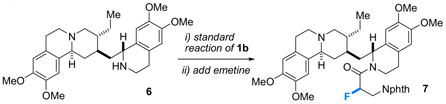 |
(4) |
Along these lines, one can imagine a wide range of fluorinated intermediates coupled with a vast array of natural nucleophiles to produce a virtually limitless number of derivatives. Future work on enantioselective fluorination will concentrate on the synthesis of other medicinally significant intermediates and on a detailed mechanistic investigation of this new method.
Supplementary Material
Procedures and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
T.L. thanks the NIH (GM064559) and the John Simon Guggenheim Memorial Foundation for support. D.H.P. thanks Johns Hopkins for a Zeltmann Fellowship.
References
- 1.(a) Kirk KL. J Fluorine Chem. 2006;127:1013–1029. [Google Scholar]; (b) Ismail FMD. J Fluorine Chem. 2002;118:27–33. [Google Scholar]; (c) Maienfisch P, Hall RG. Chimia. 2004;58:93–99. [Google Scholar]
- 2.Representative examples: Reddy DS, Shibata N, Nagai J, Nakamura S, Toru T, Kanemasa S. Angew Chem Int Ed. 2008;47:164–168. doi: 10.1002/anie.200704093.Suzuki T, Hamashima Y, Sodeoka M. Angew Chem Int Ed. 2007;46:5435–5439. doi: 10.1002/anie.200701071.Perseghini M, Massaccesi M, Liu Y, Togni A. Tetrahedron. 2006;62:7180–7190.Beeson TD, MacMillan DWC. J Am Chem Soc. 2005;127:8826–8828. doi: 10.1021/ja051805f.Shibata N, Kohno J, Takai K, Ishimaru T, Nakamura S, Toru T, Kanemasa S. Angew Chem Int Ed. 2005;44:4204–4207. doi: 10.1002/anie.200501041.Pihko PM. Angew Chem Int Ed. 2006;45:544–547. doi: 10.1002/anie.200502425.Steiner DD, Mase N, Barbas CF., III Angew Chem Int Ed. 2005;44:3706–3710. doi: 10.1002/anie.200500571.Hamashima Y, Suzuki T, Takano H, Shimura Y, Sodeoka M. J Am Chem Soc. 2005;127:10164–10165. doi: 10.1021/ja0513077.Ma JA, Cahard D. Tetrahedron: Asymmetry. 2004;15:1007–1011.Kim DY, Park EJ. Org Lett. 2002;4:545–547. doi: 10.1021/ol010281v.Hamashima Y, Yagi K, Takano H, Tamas L, Sodeoka M. J Am Chem Soc. 2002;124:14530–14531. doi: 10.1021/ja028464f.Hamashima Y, Takano H, Hotta D, Sodeoka M. Org Lett. 2003;5:3225–3228. doi: 10.1021/ol035053a.Hintermann L, Togni A. Angew Chem Int Ed. 2000;39:4359–4362. doi: 10.1002/1521-3773(20001201)39:23<4359::AID-ANIE4359>3.0.CO;2-P.Marigo M, Feilenbach D, Braunton A, Kjaersgaard A, Jorgensen KA. Angew Chem Int Ed. 2005;44:3703–3706. doi: 10.1002/anie.200500395.
- 3.In a pioneering example, Sodeoka et al. form derivatizable, optically enriched α-fluoroarylacetic acid imides using a chiral, Ni(II)-based catalyst system, see reference [2b].
- 4.Abraham CJ, Paull DH, Bekele T, Scerba MT, Lectka T. J Am Chem Soc. 2008 doi: 10.1021/ja806818a. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.For a brief review on the chemistry of NFSi, see Rostami A. Synlett. 2007;18:2924–2925.
- 6.(a) France S, Weatherwax A, Lectka T. Eur J Org Chem. 2005:475–479. [Google Scholar]; (b) France S, Wack H, Taggi AE, Hafez AM, Wagerle TR, Shah MH, Dusich CL, Lectka T. J Am Chem Soc. 2004;126:4245–4255. doi: 10.1021/ja039046t. [DOI] [PubMed] [Google Scholar]; (c) Hafez AM, Taggi AE, Wack H, Esterbrook J, Lectka T. Org Lett. 2001;3:2049–2051. doi: 10.1021/ol0160147. [DOI] [PubMed] [Google Scholar]; (d) Wack H, Taggi AE, Hafez AM, Drury WJ, III, Lectka T. J Am Chem Soc. 2001;123:1531–1532. doi: 10.1021/ja005791j. [DOI] [PubMed] [Google Scholar]
- 7.Evidence suggests that N-acyl-N, N-bis(sulfonyl)amines are strong, but moisture sensitive, acylating agents, see: Blaschette A, Safari F. Chemiker-Zeitung. 1988;112:313–315.
- 8.Screening of an early transition metal salt, Sc(OTf)3, produced only a marginal increase in yield over the base reaction and lower ee (96%).
- 9.“Dppp” is an abbreviation for 1,3-bis(diphenylphosphino)propane.
- 10.Under standard reaction conditions, simple aliphatic acid halides react very slowly; further studies are addressing this low reactivity.
- 11.Sheng H, Shao J, Kirkland SC, Isakson P, Coffey RJ, Morrow J, Beauchamp RD, DuBois RN. J Clin Invest. 1997;99:2254–2259. doi: 10.1172/JCI119400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.For a recent review of isoquinoline alkaloid chemistry, see: Chrzanowska M, Rozwadowska MD. Chem Rev. 2004;104:3341–3370. doi: 10.1021/cr030692k.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Procedures and compound characterization. This material is available free of charge via the Internet at http://pubs.acs.org.