

Abstract
Objective
Platelet endothelial cell adhesion molecule-1 (PECAM-1, CD31) has recently been shown to form an essential element of a mechanosensory complex that mediates endothelial responses to fluid shear stress. The aim of this study was to determine the in vivo role of PECAM-1 in atherosclerosis.
Methods and Results
We crossed C57BL/6 Pecam1−/− mice with apolipoprotein E–deficient (Apoe−/−) mice. On a Western diet, Pecam1−/−Apoe−/− mice showed reduced atherosclerotic lesion size compared to Apoe−/− mice. Striking differences were observed in the lesser curvature of the aortic arch, an area of disturbed flow, but not in the descending thoracic or abdominal aorta. Vascular cell adhesion molecule-1 (VCAM-1) expression, macrophage infiltration, and endothelial nuclear NF-κB were all reduced in Pecam1−/−Apoe−/− mice. Bone marrow transplantation suggested that endothelial PECAM-1 is the main determinant of atherosclerosis in the aortic arch, but that hematopoietic PECAM-1 promotes lesions in the abdominal aorta. In vitro data show that siRNA-based knockdown of PECAM-1 attenuates endothelial NF-κB activity and VCAM-1 expression under conditions of atheroprone flow.
Conclusion
These results indicate that endothelial PECAM-1 contributes to atherosclerotic lesion formation in regions of disturbed flow by regulating NF-κB–mediated gene expression.
Keywords: atherosclerosis, shear stress sensing, adhesion molecules, endothelium, macrophages
A therosclerosis is an inflammatory and degenerative disease of arterial walls characterized by monocyte recruitment, foam cell formation, and complex lesions with smooth muscle cell proliferation, a necrotic core, cholesterol crystals, and calcification.1,2 Apolipoprotein E–deficient (Apoe−/−) mice on a C57BL/6 background develop atherosclerotic lesions in the aorta and its major branches with a distribution similar to human atherosclerosis.3 Disease progression can be accelerated by feeding a Western diet (21% fat).4 Atherosclerosis preferentially develops in regions of disturbed flow (ie, branch points and bifurcations) that are characterized by oscillatory and low time-averaged shear stress.5 The local hemodynamic environment promotes distinct proatherosclerotic (“atheroprone”) or antiatherosclerotic (“atheroprotective”) endothelial phenotypes.6-10
A minimal complex necessary for the endothelial cell shear stress response requires platelet endothelial cell adhesion molecule-1 (PECAM-1, CD31), vascular endothelial cadherin (VE-cadherin), and vascular endothelial growth factor receptor 2 (VEGFR2).11 In this cascade, PECAM-1 senses force exerted by blood flowing across endothelial cells, leading to transactivation of endothelial VEGFR2. VEGFR2 triggers conformational activation of integrins followed by stimulation of nuclear factor of kappa light chain gene enhancer in B cells (NF-κB), a transcription factor responsible for expression of inflammatory adhesion molecules, cytokines, and chemokines. Therefore, we hypothesized that the PECAM-1–dependent mechanosensory pathway may be involved in atherogenesis.
PECAM-1 is also expressed on platelets and leukocytes12 and has been implicated in leukocyte transmigration through endothelial cell monolayers in vitro13 and in vivo.14,15 In a model of peritonitis, Pecam1−/− mice16 had no defect in leukocyte transmigration when investigated in the C57BL/6 background,17 the background used in the present study.
Materials and Methods
Mice
Pecam1−/− mice backcrossed 5 times into the C57BL/6 background (Dr S. Albelda, University of Pennsylvania)18 were crossed 3 times to C57BL/6 Apoe−/− mice. Double heterozygous offspring were used to generate Pecam1−/−Apoe−/− mice. Nine Pecam1−/−Apoe−/− and 10 Apoe−/− mice aged 10 weeks were fed a Western diet for 13 weeks (Harlan Teklad, TD88137). Pecam1−/−Apoe−/− mice were fertile and healthy under vivarium conditions. Their blood lipid profile was indistinguishable from that of Apoe−/− mice (supplemental Table I, available online at http://atvb.ahajournals.org). Anesthesia (16 μL/g, 1 part atropine sulfate 0.4 mg/mL, 1 part xylazine 20 mg/mL, 2 parts ketamine 100 mg/mL, and 16 parts 0.9% NaCl) was injected intraperitoneally before surgeries. Animal experiments and care were approved by the University of Virginia Animal Care and Use Committee according to AAALAC guidelines.
Tissue Acquisition
After carotid artery cannulation, the intact circulation of the mouse was flushed with PBS and perfusion-fixed with 4% paraformaldehyde (PFA) in PBS. The aorta was microdissected, immersed in 4% PFA/PBS for 2 days, cleaned of external fat by blunt dissection, and processed for en face preparation or paraffin embedding and sectioning.
En Face Preparation and Measurement of Atherosclerotic Lesion
Aortas were stained with oil red O and mounted en face.19 Digital microphotographs of aortas were analyzed for lesion size in specific regions (supplemental Figure I) by finding percent stained surface area using ImageJ (NIH).
Bone Marrow Transplantation
Pecam1−/−Apoe−/− mice were lethally irradiated and reconstituted with Pecam1−/−Apoe−/− or Apoe−/− bone marrow (n=3 recipients for each).20 Mice began a 16-week Western diet 6 weeks after irradiation.
Histopathology and Immunoperoxidase
Paraffin-embedded aortas were sectioned from aortic valve to descending thoracic aorta and stained with the Modified Russell-Movat Pentachrome Method (Armed Forces Institute of Pathology) or antibodies against Mac2 (clone M3/38, Accurate Chemicals),21 P-selectin (rabbit polyclonal, Dr S. Green, University of Virginia),22 VCAM-1 (SC-1504, Santa Cruz Biotech), intercellular adhesion molecule-1 (ICAM-1) (ICAM-1, SC-1511), CD3 (SC-1127), or CD20 (SC-7735) (Santa Cruz Biotech).23,24 Microwave antigen retrieval, Vectastain Elite Kit (Vector Labs), and diaminobenzidine (Dako Corp.) were used for localization of antigens.23 Sections were counterstained with Harris hematoxylin (Richard-Allen Scientific).
En Face Immunofluorescence
Aortic rings 1 to 2 mm thick, with anatomic location and orientation noted, were each placed in 5 to 10 mL of an antigen retrieval solution (Antigen Unmasking Solution H-3300, Vector Labs) and processed per manufacturer instructions. Rings were permeabilized with 0.2% Triton X-100 for 5 minutes, washed twice with 1% IgG-free BSA solution, and incubated with Alexa-546 –preconjugated (Molecular Probes A20183) antip65 antibody (1 μg/100 μL, mouse monoclonal 3026, Chemicon) in 1% IgG-free BSA at 4°C with TOTO-3 (Molecular Probes). An Alexa-546 –preconjugated IgG3 corresponding to antip65 antibody was used as a control (data not shown). Rings were washed, opened with the endothelium exposed on a glass slide, and mounted with antifading mounting gel (GelMount, Fisher).
Immunofluorescence on Cross-Sections
Paraffin-embedded aortic sections were mounted on glass slides, deparaffinized, rehydrated, processed for antigen retrieval, blocked with 10% goat or donkey serum in PBS/FSGO, and incubated overnight at 4°C with Alexa-546-preconjugated antip65 or anti-VCAM-1 (above), respectively. Donkey antigoat Alexa-546 was added for VCAM-1 preparations. Sections were stained with TOTO-3 and mounted with antifading mounting gel.
Confocal Imaging
Images of en face and paraffin-embedded sections were interrogated for each protein (Nikon C1 confocal microscope). All images were acquired at the same gain, aperture, and exposure settings.
Quantification of NF-κB
Intensity of nuclear NF-κB p65 was assessed by importing confocal images of stained aortic sections into MetaMorph Imaging (Molecular Devices). Average nuclear NF-κB intensity was measured for each TOTO-3-positive nucleus.
Western Blot
Dermal microvascular endothelial cells were harvested and plated for 48 hours in M199, 10% FBS, 1:250 endothelial cell growth supplement (ECGS, Sigma), and heparin (Sigma). Atheroprone waveform25 was applied by a cone-and-plate viscometer for 16 hours with MCDB-131 (Gibco), 2% FBS, 1:1000 ECGS/heparin, and 4% dextran (Sigma). Samples were collected, run on 10% SDS-PAGE, transferred to polyvinylidene fluoride (PVDF) membrane, and blotted for phospho-p65 (rabbit polyclonal 3031, Cell Signaling) and total p65 (rabbit polyclonal 3034).
In Vitro Flow Experiment, siRNA, and Luciferase Reporter Transfection
Passage two human umbilical vein endothelial cells (HUVECs) were isolated and plated at 50% confluence on 1% gelatin in M199 growth media (Biowhitaker), 10% FBS (Gibco), 5 μg/mL ECGS (Biomedical Technologies), 10 μg/mL heparin (Sigma), 2 mmol/L L-glutamine (Gibco), and 100U penicillin/streptomycin (Invitrogen). HUVECs were treated with 330pmol of control (D-001810, Dharmacon) or human PECAM-1 siRNA (L-017029—00) and 19.8 μL oligofectamine (Invitrogen) in 3 mL of OptiMEM-I media for 5 hours. High growth media (20% FBS) was used for 24 hours to grow to confluence. Cells were infected with 5 MOI adenovirus containing NF-κB-luciferase reporter (Vector Labs) for 16 hours. Cells were washed in DPBS and placed in reduced serum media (M199 supplemented with 2% FBS, 5 μg/mL ECGS, 10 μg/mL heparin, 2 mmol/L L-glutamine, 100U penicillin/streptomycin, and 2% dextran by weight to increase viscosity). Atheroprone or atheroprotective shear stress was applied for 24 hours using a cone-and-plate viscometer. Samples were collected in passive lysis buffer (Promega) for measurement of luciferase luminescence or SDS-MAPK sample buffer (Cell Signaling) for Western blot of human PECAM-1 and VCAM-1 (R&D Systems).
Statistics
Data are represented as mean±SEM and analyzed using a 2-tailed heteroscedastic Student t test or nonparametric Wilcoxon–Mann test. P<0.05 was considered significant.
Results
En Face Analysis of Atherosclerotic Lesions
We measured atherosclerotic lesion formation in specific aortic regions (supplemental Figure I) from Pecam1−/−Apoe−/− and matched Apoe−/− mice on Western diet for 13 weeks. Oil red O staining revealed robust lesions at branch points of major and minor arteries from both groups and along the lesser curvature of the aortic arch of Apoe−/− mice (Figure 1A). Lesion size was reduced in Pecam1−/−Apoe−/− mice by 26% in whole aortas (P<0.05), 28% in aortic arches (P<0.02), and 42% in lesser curvatures (P<0.005). Differences in the thoracic and abdominal aortas were not significant (Figure 1B). These findings suggest that PECAM-1 promotes atherosclerotic lesion development in a flow-dependent manner.
Figure 1.
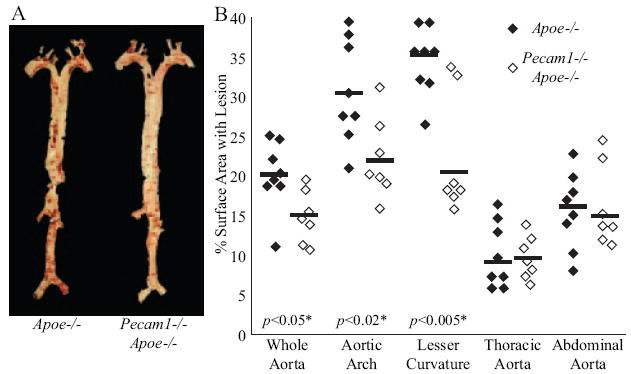
PECAM-1 promotes atherosclerosis in regions of disturbed flow. A, Representative oil red O–stained aortas from Apoe−/− and Pecam1−/−Apoe−/− mice. B, Pecam1−/−Apoe−/− mice (2 females, 5 males) develop less lesion than Apoe−/− mice (1 female, 7 males) on the whole aorta, aortic arch, and lesser curvature, but not the thoracic and abdominal aorta.
Bone Marrow Transplantation
To test whether PECAM-1 on endothelial or hematopoietic cells determines atherosclerotic lesion formation, Pecam1−/−Apoe−/− mice were lethally irradiated, reconstituted with either Apoe−/− or Pecam1−/−Apoe−/− bone marrow, and fed a Western diet for 16 weeks. Pecam1−/−Apoe−/− mice reconstituted with Apoe−/− bone marrow developed more than twice as much lesion in the abdominal aorta compared to mice receiving Pecam1−/−Apoe−/− bone marrow (129% increase, P<0.05, supplemental Figure II). This suggests that PECAM-1 on leukocytes and platelets promotes atherosclerosis in the abdominal aorta. Differences in the aortic arch were not significant between mice receiving Apoe−/− (22±1%) and Pecam1−/−Apoe−/− bone marrow (25±3%, supplemental Figure II), suggesting that endothelial PECAM-1 determines atherosclerotic lesions in the aortic arch, an area of disturbed flow.
Involvement of NF-κB Activation
To investigate the mechanism by which PECAM-1 promotes atherosclerosis at sites of disturbed flow, we stained aortas en face for NF-κB p65. Endothelial cells lining the lesser curvature of Apoe−/− but not Pecam1−/−Apoe−/− mice demonstrated robust nuclear p65 localization (Figure 2A). This suggests that PECAM-1 absence reduces nuclear NF-κB. We also compared p65-stained aortic arch cross-sections from Apoe−/− and Pecam1−/−Apoe−/− mice (Figure 2B). Pecam1−/−Apoe−/− mice demonstrated a 77% and 59% reduction in NF-κB staining in the lesser and greater curvatures, respectively, compared to Apoe−/− controls (P<0.01, Figure 2C).
Figure 2.
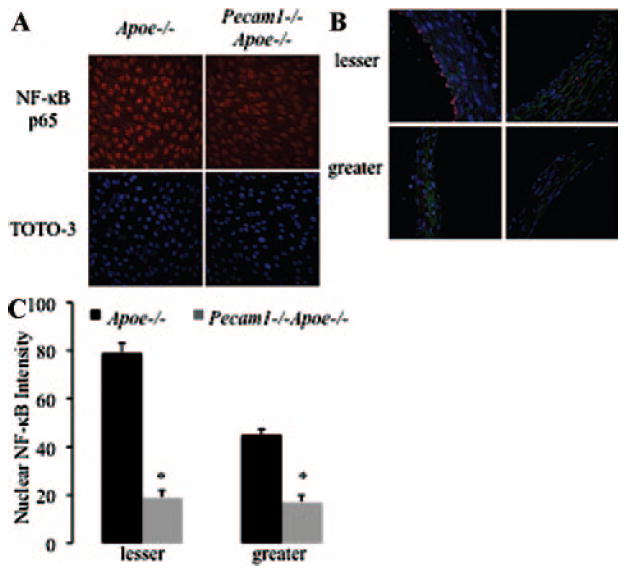
Nuclear NF-κB translocation is PECAM-1–dependent. A, En face aortic arches stained for NF-κB p65 and TOTO-3 (nuclear stain). B, Aortic arch cross-sections stained for NF-κB p65 and TOTO-3. C, Quantification of nuclear NF-κB staining in the lesser and greater curvatures (n=44 to 84 nuclei, 11 sections, 2 mice per group, *P<0.01).
To determine whether regional flow patterns affect PECAM-1 regulation of NF-κB activity, we cultured human umbilical vein endothelial cells (HUVECs) under atheroprone or atheroprotective flow patterns derived from the human circulation25 or static conditions. Atheroprone flow increased VCAM-1 expression, which was reduced by siRNA targeting PECAM-1 (P<0.001, Figure 3A, supplemental Figure III). NF-κB activity measured using a luciferase reporter increased 3.6-fold in HUVECs treated with control siRNA under atheroprone compared to atheroprotective flow (P<0.001, Figure 3B). NF-κB activity under atheroprone flow decreased by nearly 50% after siRNA-based PECAM-1 knockdown to 20±13% of control levels (P<0.001). siRNA treatment did not alter NF-κB activity under static conditions or atheroprotective flow. PECAM-1 knockdown also attenuated the VCAM-1 protein expression under atheroprone flow (P<0.001, supplemental Figure III). Collectively, these data indicate that PECAM-1 is necessary for NF-κB activation and VCAM-1 expression under atheroprone flow.
Figure 3.
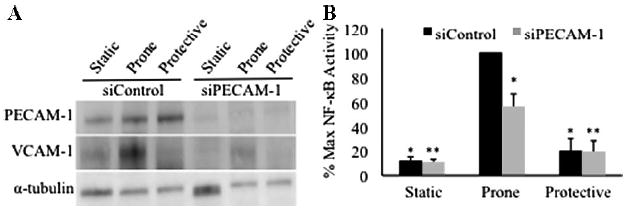
PECAM-1–dependent activation of NF-κB under atheroprone shear stress. Ad-NF-κ B-Luc–transfected endothelial cells cotransfected with control siRNA (siControl) or siRNA to PECAM-1 (siPECAM-1) were exposed to static conditions, atheroprone (“prone”) flow, and atheroprotective (“protective”) flow. A, siPECAM-1 attenuates PECAM-1 and VCAM-1 expression. B, NF-κB activity increases under atheroprone flow in the presence of PECAM-1. (*P<0.001 vs atheroprone siControl, ** P<0.001 vs atheroprone siPECAM-1, n=5).
Cross-Sectional Analysis of Atherosclerotic Lesions
We serially sectioned the entire aortic arch from aortic valve to descending thoracic aorta (Figure 4A). Foam cell-containing and complex lesions were seen in the aortic root of Apoe−/− and Pecam1−/−Apoe−/− mice (supplemental Figure IV). Lesions of the lesser curvature were strikingly absent or diminished in thickness and size in Pecam1−/−Apoe−/− mice (Figure 4B). VCAM-1 was expressed in atherosclerotic endothelial and smooth muscle cells of Apoe−/− mice,27 but this was visibly reduced in Pecam1−/−Apoe−/− mice (Figure 4C). Pecam1−/−Apoe−/− mice expressed VCAM-1 on 19±5% of the endothelial circumference of aortic cross-sections compared to 51±6% in Apoe−/− mice (n=13 to 15 cross-sections, 3 mice per group). This difference was significant in the ascending aorta (P<0.05) but much less pronounced in the more distal segments of the aortic arch and the descending aorta (data not shown). VCAM-1 is relatively specific for supporting the adhesion of mononuclear leukocytes. Its expression correlates well with the occurrence of atherosclerotic lesions.31
Figure 4.
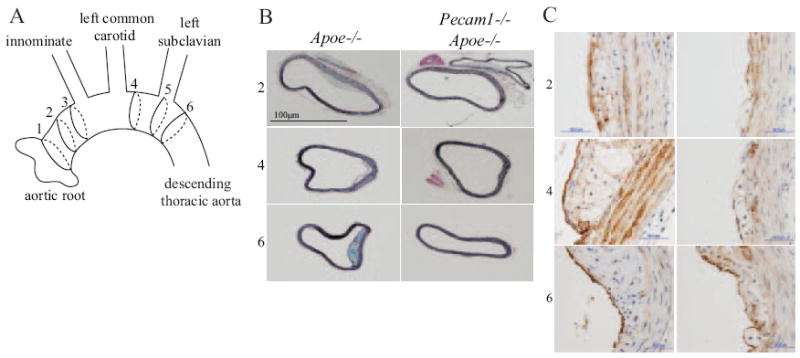
PECAM-1–deficient aortas have decreased lesion and VCAM-1. A, Aorta was interrogated at various positions: (1) root, (2–3) proximal to innominate artery, (4) between innominate and left subclavian arteries, (5) at left subclavian artery, (6) descending thoracic aorta. B, Movat stain, greater and lesser curvatures are on the left and right of each image, respectively. C, VCAM-1 protein expression in Apoe−/− (left) and Pecam1−/−Apoe−/− (right) mice.
Macrophage presence by Mac2 staining was also dramatically reduced in Pecam1−/−Apoe−/− mice (supplemental Figure V). Foam cells were present on 41±1% of the endothelial circumference of Movat-stained aortic cross-sections in Apoe−/− mice compared to 10±1% in Pecam1−/−Apoe−/− mice (P<0.00001, n=18 to 19 cross-sections, 3 mice per group). Maximum thickness of foam cell regions reached 116±4 μm in Apoe−/− mice, but only 45±3 μm in Pecam1−/−Apoe−/− mice (P<0.0003). Differences in ICAM-1, P-selectin, (supplemental Figure V), T cells (CD3) and B cells (CD20, data not shown) were not observed.
Discussion
Endothelial cells have long been known to respond to steady32,33 and transient shear stress.34,35 The latter has been hypothesized to determine the localization of atherosclerotic lesions in vivo.36 PECAM-1 plays a key role in the flow-dependent mechanotransduction events leading to atherosclerosis,11 but this was not found in all experimental systems37 and has not been directly investigated in vivo. Oscillatory or low time-averaged shear stress promotes the development of atherosclerotic lesions, whereas areas with high shear stress are protected.38 Cheng et al manipulated the shear stress pattern using a circumferential cuff,38 which also alters local blood pressure and may disturb the adventitia, an area that contains inflammatory cells and participates in the process of atherosclerosis.24 The data from Pecam1−/−Apoe−/− mice described here suggest that PECAM-1 on the endothelium is critically important for atherosclerotic lesion development in the aortic arch of Apoe−/− mice. This may be attributable to the involvement of PECAM-1 in the mechanosensory complex containing PECAM-1–VE-cadherin–VEGFR2.11,39 Our data suggest that causative factors for atherosclerotic lesion development may vary for different locations in the vasculature.
Several possible explanations may account for residual atherosclerotic lesions in Pecam1−/−Apoe−/− mice. First, although shear stress–induced phosphatidylinositol-3-OH kinase p85 and AKT phosphorylation were completely absent in Pecam1−/− endothelial cells, shear stress applied to Pecam1−/− endothelial cells activated αVβ3 integrin to a small extent.11 Second, other unidentified mechanisms in addition to the PECAM-1 pathway may exist for sensing shear stress. Third, rheological factors such as increased residence time of monocytes and inflammatory cytokines at atheroprone sites may promote atherosclerosis.40,41 Fourth, prolonged permeability of inflamed endothelium in Pecam1−/− mice42 may enhance monocyte recruitment and the action of inflammatory factors. Finally, PECAM-1 may prime the endothelium in response to atheroprone flow, but its impact may diminish over time as other factors contribute.43
PECAM-1 regulates transendothelial migration of monocytes44 and neutrophils.13 In a model of acute lung inflammation, the importance of PECAM-1 for neutrophil recruitment was demonstrated in vivo.15 Intravital microscopy showed that PECAM-1–specific antibody hampered leukocyte movement across the basement membrane but not transendothelial migration.45,46 However, the role of PECAM-1 in transendothelial migration depends on the mouse strain, because C57BL/6 mice showed no discernible defect in two models of inflammation.17 Because we investigated Apoe−/− mice on a C57BL/6 background, it is unlikely that PECAM-1 absence significantly curbed atherosclerosis by inhibiting transmigration.
Bone marrow transplantation experiments performed here demonstrate that PECAM-1 expressed on leukocytes or platelets promotes atherosclerosis in the abdominal aorta, but not the aortic arch, even in the absence of PECAM-1 on the endothelium. This suggests a putative role for either homophilic binding between leukocyte and platelet PECAM-1 via immunoglobulin-like domain 1 or heterophilic binding via immunoglobulin-like domains 1, 2, and 6.44 As one possible mechanism, elevated very low density lipoprotein leads to increased human endothelial expression of CD38,47 which may be a PECAM-1 ligand.48
The present findings show that the net effect of removing Pecam1 from the mouse genome is atheroprotective in regions of the aorta exposed to atheroprone flow patterns. We believe dynamic fluid shear stress generates tension between adjacent endothelial cells and activates PECAM-1, which leads to nuclear translocation of NF-κB and expression of inflammatory and adhesive mediators, such as VCAM-1, that promote atherosclerosis. NF-κB regulation by PECAM-1 might depend on the underlying extracellular matrix on which the endothelial cells are sitting. Indeed, endothelial cells on fibronectin-rich, but not collagen-rich, matrices activate NF-κB in response to atheroprone flow.49
PECAM-1 may also regulate atherosclerosis by mechanisms not investigated here. Pecam1−/− mice have increased bleeding times,50 and altered hemostatic function may reduce atherosclerosis. Shear stress sensing is also involved in regulating nitric oxide production,51,52 preventing apoptosis, and maintaining anticoagulant properties, which might be affected following knockout of the Pecam1 gene.
Polymorphisms in the human PECAM1 gene correlate with the incidence of coronary atherosclerosis,53 coronary artery disease,54 and myocardial infarction.55 A 53G>A polymorphism in the 5′ untranslated region of PECAM1 reduced shear stress response in vitro. Compared with 53G homozygotes, carriers of the 53A allele showed less focal progression of disease of coronary atherosclerosis in the LOCAT study and a similar trend in diffuse progression of disease in the REGRESS study.53 Another study reports that a 373C>G polymorphism, which may affect homophilic interactions of PECAM-1 via a Leu125Val exchange in the first immunoglobulin-like domain,56 is associated with coronary artery disease in Asian Indians.55 Polymorphisms in the sixth immunoglobulin-like domain (Asn563Ser), which is important for heterophilic interactions with integrins,57 and the cytoplasmic domain (Gly670Arg), which participates in signal transduction,58 have been recognized as risk factors for myocardial infarction in the Japanese.55
In conclusion, the present data identify the PECAM-1–dependent endothelial shear stress response as a key factor in atherosclerotic lesion development in the aortic arch of the Apoe−/− mouse, a model that shares many similarities with the human disease.
Supplementary Material
Acknowledgments
We thank Dr William Muller (Cornell University) for critical reading of the manuscript and Drs Steven Albelda (University of Pennsylvania) and Sam Green (University of Virginia) for providing materials.
Sources of Funding This work was supported by AHA 2005 Student Scholarship in Cardiovascular Disease and Stroke to B.L.H., NIH 5T32HL007284-32 to R.E.F., NIH 5R01HL082836-02 to B.R.B., Deutsche Forschungsgemeinschaft AZ 428/2-1 to A.Z., and NIH 5R01HL058108-12 to K.L.
Footnotes
Disclosures None.
References
- 1.Libby P. Inflammation in atherosclerosis. Nature. 2002;6917:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 2.Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature. 1993;6423:801–809. doi: 10.1038/362801a0. [DOI] [PubMed] [Google Scholar]
- 3.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;5081:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 4.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;1:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 5.Malek AM, Alper SL, Izumo S. Hemodynamic shear stress and its role in atherosclerosis. JAMA. 1999;21:2035–2042. doi: 10.1001/jama.282.21.2035. [DOI] [PubMed] [Google Scholar]
- 6.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;3:519–560. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dai G, Kaazempur-Mofrad MR, Natarajan S, Zhang Y, Vaughn S, Blackman BR, Kamm RD, Garcia-Cardena G, Gimbrone MA., Jr Distinct endothelial phenotypes evoked by arterial waveforms derived from atherosclerosis-susceptible and -resistant regions of human vasculature. Proc Natl Acad Sci U S A. 2004;41:14871–14876. doi: 10.1073/pnas.0406073101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hastings NE, Simmers MB, McDonald OG, Wamhoff BR, Blackman BR. Atherosclerosis-prone hemodynamics differentially regulates endothelial and smooth muscle cell phenotypes and promotes pro-inflammatory priming. Am J Physiol Cell Physiol. 2007;6:C1824–C1833. doi: 10.1152/ajpcell.00385.2007. [DOI] [PubMed] [Google Scholar]
- 9.Hajra L, Evans AI, Chen M, Hyduk SJ, Collins T, Cybulsky MI. The NF-kappa B signal transduction pathway in aortic endothelial cells is primed for activation in regions predisposed to atherosclerotic lesion formation. Proc Natl Acad Sci U S A. 2000;16:9052–9057. doi: 10.1073/pnas.97.16.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iiyama K, Hajra L, Iiyama M, Li H, DiChiara M, Medoff BD, Cybulsky MI. Patterns of vascular cell adhesion molecule-1 and intercellular adhesion molecule-1 expression in rabbit and mouse atherosclerotic lesions and at sites predisposed to lesion formation. Circ Res. 1999;2:199–207. doi: 10.1161/01.res.85.2.199. [DOI] [PubMed] [Google Scholar]
- 11.Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, Cao G, DeLisser H, Schwartz MA. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. 2005;7057:426–431. doi: 10.1038/nature03952. [DOI] [PubMed] [Google Scholar]
- 12.Delisser HM, Newman PJ, Albelda SM. Molecular and functional aspects of PECAM-1/CD31. Immunol Today. 1994;10:490–495. doi: 10.1016/0167-5699(94)90195-3. [DOI] [PubMed] [Google Scholar]
- 13.Muller WA, Weigl SA, Deng X, Phillips DM. PECAM-1 is required for transendothelial migration of leukocytes. J Exp Med. 1993;2:449–460. doi: 10.1084/jem.178.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bogen S, Pak J, Garifallou M, Deng X, Muller WA. Monoclonal antibody to murine PECAM-1 (CD31) blocks acute inflammation in vivo. J Exp Med. 1994;3:1059–1064. doi: 10.1084/jem.179.3.1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vaporciyan AA, Delisser HM, Yan HC, Mendiguren II, Thom SR, Jones ML, Ward PA, Albelda SM. Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science. 1993;5139:1580–1582. doi: 10.1126/science.8248808. [DOI] [PubMed] [Google Scholar]
- 16.Duncan GS, Andrew DP, Takimoto H, Kaufman SA, Yoshida H, Spellberg J, Luis de la PJ, Elia A, Wakeham A, Karan-Tamir B, Muller WA, Senaldi G, Zukowski MM, Mak TW. Genetic evidence for functional redundancy of Platelet/Endothelial cell adhesion molecule-1 (PECAM-1): CD31-deficient mice reveal PECAM-1-dependent and PECAM-1-independent functions. J Immunol. 1999;5:3022–3030. [PubMed] [Google Scholar]
- 17.Schenkel AR, Chew TW, Muller WA. Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. J Immunol. 2004;10:6403–6408. doi: 10.4049/jimmunol.173.10.6403. [DOI] [PubMed] [Google Scholar]
- 18.Albelda SM, Lau KC, Chien P, Huang ZY, Arguiris E, Bohen A, Sun J, Billet JA, Christofidou-Solomidou M, Indik ZK, Schreiber AD. Role for platelet-endothelial cell adhesion molecule-1 in macrophage Fcgamma receptor function. Am J Respir Cell Mol Biol. 2004;2:246–255. doi: 10.1165/rcmb.2003-0404OC. [DOI] [PubMed] [Google Scholar]
- 19.Nunnari JJ, Zand T, Joris I, Majno G. Quantitation of oil red O staining of the aorta in hypercholesterolemic rats. Exp Mol Pathol. 1989;1:1–8. doi: 10.1016/0014-4800(89)90002-6. [DOI] [PubMed] [Google Scholar]
- 20.Zarbock A, Singbartl K, Ley K. Complete reversal of acid-induced acute lung injury by blocking of platelet-neutrophil aggregation. J Clin Invest. 2006;12:3211–3219. doi: 10.1172/JCI29499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manka D, Forlow SB, Sanders JM, Hurwitz D, Bennett DK, Green SA, Ley K, Sarembock IJ. Critical role of platelet P-selectin in the response to arterial injury in apolipoprotein-E-deficient mice. Arterioscler Thromb Vasc Biol. 2004;6:1124–1129. doi: 10.1161/01.ATV.0000127619.04687.f4. [DOI] [PubMed] [Google Scholar]
- 22.Green SA, Setiadi H, McEver RP, Kelly RB. The cytoplasmic domain of P-selectin contains a sorting determinant that mediates rapid degradation in lysosomes. J Cell Biol. 1994;4:435–448. doi: 10.1083/jcb.124.4.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McPherson JA, Barringhaus KG, Bishop GG, Sanders JM, Rieger JM, Hesselbacher SE, Gimple LW, Powers ER, Macdonald T, Sullivan G, Linden J, Sarembock IJ. Adenosine A(2A) receptor stimulation reduces inflammation and neointimal growth in a murine carotid ligation model. Arterioscler Thromb Vasc Biol. 2001;5:791–796. doi: 10.1161/01.atv.21.5.791. [DOI] [PubMed] [Google Scholar]
- 24.Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. 2006;5:1273–1282. doi: 10.1084/jem.20052205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gelfand BD, Epstein FH, Blackman BR. Spatial and spectral heterogeneity of time-varying shear stress profiles in the carotid bifurcation by phase-contrast MRI. J Magn Reson Imaging. 2006;6:1386–1392. doi: 10.1002/jmri.20765. [DOI] [PubMed] [Google Scholar]
- 26.Deleted in proof.
- 27.Blackman BR, Garcia-Cardena G, Gimbrone MA., Jr A new in vitro model to evaluate differential responses of endothelial cells to simulated arterial shear stress waveforms. J Biomech Eng. 2002;4:397–407. doi: 10.1115/1.1486468. [DOI] [PubMed] [Google Scholar]
- 28.Deleted in proof.
- 29.Manka DR, Wiegman P, Din S, Sanders JM, Green SA, Gimple LW, Ragosta M, Powers ER, Ley K, Sarembock IJ. Arterial injury increases expression of inflammatory adhesion molecules in the carotid arteries of apolipoprotein-E-deficient mice. J Vasc Res. 1999;5:372–378. doi: 10.1159/000025676. [DOI] [PubMed] [Google Scholar]
- 30.O’Brien KD, Allen MD, McDonald TO, Chait A, Harlan JM, Fishbein D, McCarty J, Ferguson M, Hudkins K, Benjamin CD. Vascular cell adhesion molecule-1 is expressed in human coronary atherosclerotic plaques. Implications for the mode of progression of advanced coronary atherosclerosis. J Clin Invest. 1993;2:945–951. doi: 10.1172/JCI116670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. 2001;10:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Flaherty JT, Pierce JE, Ferrans VJ, Patel DJ, Tucker WK, Fry DL. Endothelial nuclear patterns in the canine arterial tree with particular reference to hemodynamic events. Circ Res. 1972;1:23–33. doi: 10.1161/01.res.30.1.23. [DOI] [PubMed] [Google Scholar]
- 33.Dewey CF, Jr, Bussolari SR, Gimbrone MA, Jr, Davies PF. The dynamic response of vascular endothelial cells to fluid shear stress. J Biomech Eng. 1981;3:177–185. doi: 10.1115/1.3138276. [DOI] [PubMed] [Google Scholar]
- 34.Frangos JA, Eskin SG, McIntire LV, Ives CL. Flow effects on prostacyclin production by cultured human endothelial cells. Science. 1985;4693:1477–1479. doi: 10.1126/science.3883488. [DOI] [PubMed] [Google Scholar]
- 35.Helmlinger G, Geiger RV, Schreck S, Nerem RM. Effects of pulsatile flow on cultured vascular endothelial cell morphology. J Biomech Eng. 1991;2:123–131. doi: 10.1115/1.2891226. [DOI] [PubMed] [Google Scholar]
- 36.Zarins CK, Giddens DP, Bharadvaj BK, Sottiurai VS, Mabon RF, Glagov S. Carotid bifurcation atherosclerosis. Quantitative correlation of plaque localization with flow velocity profiles and wall shear stress. Circ Res. 1983;4:502–514. doi: 10.1161/01.res.53.4.502. [DOI] [PubMed] [Google Scholar]
- 37.Sumpio BE, Yun S, Cordova AC, Haga M, Zhang J, Koh Y, Madri JA. MAPKs (ERK1/2, p38)and AKT can be phosphorylated by shear stress independently of platelet endothelial cell adhesion molecule-1 (CD31) in vascular endothelial cells. J Biol Chem. 2005;12:11185–11191. doi: 10.1074/jbc.M414631200. [DOI] [PubMed] [Google Scholar]
- 38.Cheng C, Tempel D, van HR, van der BA, Grosveld F, Daemen MJ, Krams R, de Crom R. Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation. 2006;23:2744–2753. doi: 10.1161/CIRCULATIONAHA.105.590018. [DOI] [PubMed] [Google Scholar]
- 39.Shay-Salit A, Shushy M, Wolfovitz E, Yahav H, Breviario F, Dejana E, Resnick N. VEGF receptor 2 and the adherens junction as a mechanical transducer in vascular endothelial cells. Proc Natl Acad Sci U S A. 2002;14:9462–9467. doi: 10.1073/pnas.142224299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chien S, Li S, Shyy YJ. Effects of mechanical forces on signal transduction and gene expression in endothelial cells. Hypertension. 1998;1(Pt 2):162–169. doi: 10.1161/01.hyp.31.1.162. [DOI] [PubMed] [Google Scholar]
- 41.Moore JE, Jr, Ku DN, Zarins CK, Glagov S. Pulsatile flow visualization in the abdominal aorta under differing physiologic conditions: implications for increased susceptibility to atherosclerosis. J Biomech Eng. 1992;3:391–397. doi: 10.1115/1.2891400. [DOI] [PubMed] [Google Scholar]
- 42.Graesser D, Solowiej A, Bruckner M, Osterweil E, Juedes A, Davis S, Ruddle NH, Engelhardt B, Madri JA. Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest. 2002;3:383–392. doi: 10.1172/JCI13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. 2003;1:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 44.Woodfin A, Voisin MB, Nourshargh S. PECAM-1: a multi-functional molecule in inflammation and vascular biology. Arterioscler Thromb Vasc Biol. 2007;27:2514–2523. doi: 10.1161/ATVBAHA.107.151456. [DOI] [PubMed] [Google Scholar]
- 45.Wakelin MW, Sanz MJ, Dewar A, Albelda SM, Larkin SW, Boughton-Smith N, Williams TJ, Nourshargh S. An anti-platelet-endothelial cell adhesion molecule-1 antibody inhibits leukocyte extravasation from mesenteric microvessels in vivo by blocking the passage through the basement membrane. J Exp Med. 1996;1:229–239. doi: 10.1084/jem.184.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woodfin A, Reichel CA, Khandoga A, Corada M, Voisin MB, Scheiermann C, Haskard DO, Dejana E, Krombach F, Nourshargh S. JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood. 2007;6:1848–1856. doi: 10.1182/blood-2006-09-047431. [DOI] [PubMed] [Google Scholar]
- 47.Norata GD, Pirillo A, Callegari E, Hamsten A, Catapano AL, Eriksson P. Gene expression and intracellular pathways involved in endothelial dysfunction induced by VLDL and oxidised VLDL. Cardiovasc Res. 2003;1:169–180. doi: 10.1016/s0008-6363(03)00335-3. [DOI] [PubMed] [Google Scholar]
- 48.Deaglio S, Morra M, Mallone R, Ausiello CM, Prager E, Garbarino G, Dianzani U, Stockinger H, Malavasi F. Human CD38 (ADP-ribosyl cyclase) is a counter-receptor of CD31, an Ig superfamily member. J Immunol. 1998;1:395–402. [PubMed] [Google Scholar]
- 49.Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol. 2005;1:191–202. doi: 10.1083/jcb.200410073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mahooti S, Graesser D, Patil S, Newman P, Duncan G, Mak T, Madri JA. PECAM-1 (CD31) expression modulates bleeding time in vivo. Am J Pathol. 2000;1:75–81. doi: 10.1016/S0002-9440(10)64519-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci. 2005;(Pt 18):4103–4111. doi: 10.1242/jcs.02541. [DOI] [PubMed] [Google Scholar]
- 52.Bagi Z, Frangos JA, Yeh JC, White CR, Kaley G, Koller A. PECAM-1 mediates NO-dependent dilation of arterioles to high temporal gradients of shear stress. Arterioscler Thromb Vasc Biol. 2005;8:1590–1595. doi: 10.1161/01.ATV.0000170136.71970.5f. [DOI] [PubMed] [Google Scholar]
- 53.Elrayess MA, Webb KE, Flavell DM, Syvanne M, Taskinen MR, Frick MH, Nieminen MS, Kesaniemi YA, Pasternack A, Jukema JW, Kastelein JJ, Zwinderman AH, Humphries SE. A novel functional polymorphism in the PECAM-1 gene (53G>A) is associated with progression of atherosclerosis in the LOCAT and REGRESS studies. Atherosclerosis. 2003;1:131–138. doi: 10.1016/s0021-9150(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 54.Fang L, Wei H, Chowdhury SH, Gong N, Song J, Heng CK, Sethi S, Koh TH, Chatterjee S. Association of Leu125Val polymorphism of platelet endothelial cell adhesion molecule-1 (PECAM-1) gene & soluble level of PECAM-1 with coronary artery disease in Asian Indians. Indian J Med Res. 2005;2:92–99. [PubMed] [Google Scholar]
- 55.Sasaoka T, Kimura A, Hohta SA, Fukuda N, Kurosawa T, Izumi T. Polymorphisms in the platelet-endothelial cell adhesion molecule-1 (PECAM-1) gene, Asn563Ser and Gly670Arg, associated with myocardial infarction in the Japanese. Ann N Y Acad Sci. 2001:259–269. doi: 10.1111/j.1749-6632.2001.tb03948.x. [DOI] [PubMed] [Google Scholar]
- 56.Newton JP, Buckley CD, Jones EY, Simmons DL. Residues on both faces of the first immunoglobulin fold contribute to homophilic binding sites of PECAM-1/CD31. J Biol Chem. 1997;33:20555–20563. doi: 10.1074/jbc.272.33.20555. [DOI] [PubMed] [Google Scholar]
- 57.Wong CW, Wiedle G, Ballestrem C, Wehrle-Haller B, Etteldorf S, Bruckner M, Engelhardt B, Gisler RH, Imhof BA. PECAM-1/CD31 trans-homophilic binding at the intercellular junctions is independent of its cytoplasmic domain; evidence for heterophilic interaction with integrin alphavbeta3 in Cis. Mol Biol Cell. 2000;9:3109–3121. doi: 10.1091/mbc.11.9.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newman PJ. Switched at birth: a new family for PECAM-1. J Clin Invest. 1999;1:5–9. doi: 10.1172/JCI5928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.