

Abstract
The human malaria parasite Plasmodium vivax is responsible for 25-40% of the ~515 million annual cases of malaria worldwide. Although seldom fatal, the parasite elicits severe and incapacitating clinical symptoms and often relapses months after a primary infection has cleared. Despite its importance as a major human pathogen, P. vivax is little studied because it cannot be propagated in the laboratory except in non-human primates. We determined the genome sequence of P. vivax in order to shed light on its distinctive biologic features, and as a means to drive development of new drugs and vaccines. Here we describe the synteny and isochore structure of P. vivax chromosomes, and show that the parasite resembles other malaria parasites in gene content and metabolic potential, but possesses novel gene families and potential alternate invasion pathways not recognized previously. Completion of the P. vivax genome provides the scientific community with a valuable resource that can be used to advance scientific investigation into this neglected species.
Plasmodium vivax is the major cause of malaria outside Africa, mainly afflicting Asia and the Americas 1. A disease of poor people living on the margins of developing economies, vivax malaria traps many societies in a relentless cycle of poverty. Intermittent transmission makes protective immunity rare, and the disease strikes all ages. Repeated acute febrile episodes of debilitating intensity can occur for months. In children this can lead to life-long learning impairment, while incapacitation of adults has tremendous direct economic consequences through lost productivity and depletion of meagre financial reserves. Drug resistance in P. vivax is spreading, hindering management of clinical cases, and reports of severe pathology, including respiratory distress and coma, are challenging the description of vivax malaria as ‘benign’1.
Several biological characteristics underlie the distinct pathogenic and epidemiologic nature of vivax malaria. In contrast to P. falciparum, P. vivax is only capable of infecting reticulocytes, causing severe anaemia by dyserythropoiesis and destruction of infected and uninfected erythrocytes despite much lower parasitemias. P. vivax cannot infect Duffy blood group-negative reticulocytes (a trait shared with the closely-related monkey malaria parasite P. knowlesi), and is thus absent from West Africa where Duffy negativity predominates 2. Differences in Anopheles mosquito dynamics allow P. vivax transmission in temperate climates not tolerated by P. falciparum. In such regions P. vivax infects hepatocytes but may persist as dormant hypnozoites for months or years before initiating blood-stage infections (relapses) during another transmission season.
Since P. vivax kills infrequently and is not amenable to continuous in vitro culture, it has been relatively little studied in comparison to P. falciparum. The P. vivax genome sequence we report here, and comparative analyses with sequenced malaria parasites P. falciparum 3, the rodent parasite P. yoelii yoelii 4,5, and the primate parasite P. knowlesi 6 (an excellent model for in vivo studies of human malaria), provide significant insights into the biology of this neglected parasite.
Genome sequencing and characteristics
The ~26.8 Mb nuclear genome sequence of P. vivax (Salvador I) was sequenced by whole genome shotgun methods to 10-fold coverage, targeted gap closure and finishing, and manual curation of automated annotation. Details of these and other methods are given in Supplementary Information linked to this paper’s online version (www.nature.com/nature). Large contigs totalling ~22.6 Mb were assigned to the 14 P. vivax chromosomes; ~4.3 Mb of small subtelomeric contigs remain unassigned due to their repetitive nature (Supplementary Table 1). P. vivax chromosomes are unique among human Plasmodium species in exhibiting a form of isochore structure 7, with subtelomeric regions of low G+C content and chromosome internal regions of significantly higher G+C content. We finished the subtelomeric ends of several P. vivax chromosomes, allowing us to define their isochore boundaries (Plate 1).
Plate 1. Synteny maps showing the comparative organization of Plasmodium chromosomes.

Putative orthologs were computed between P. falciparum (Pf), P. vivax (Pv), P. knowlesi (Pk) and P. y. yoelii (Py) proteomes and used to define blocks of synteny (shaded regions) between Py - Pk, Pk - Pv, and Pv – Pf chromosomes. Genes on contigs that could not be assigned to chromosomes are not shown (see Supplementary Information). The composite rodent malaria parasite (cRMP) chromosomes generated in Ref. 18 are shown. Plots below the Pv chromosomes display the following: MS, the position of polymorphic microsatellites; G+C-skew, the base composition within each strand; %G+C, the percent G+C; and for Pv-Pk, two evolutionary parameters dS and ω. Inset: Distribution of selective constraints (ω) for Biological Process (A), Molecular Function (B) and Cellular Component (C) Gene Ontology classifications. Selective constraint is also shown for several motifs (D): proteins containing predicted transmembrane domains (TMM) and/or signal peptides (SP); GPI-anchored proteins; proteins predicted to be exported from the cell (exportome). Each grey box represents the interquartile range (IQR), which contains the sample’s 25% to 75% range (quartiles Q1 to Q3, respectively), and the median is indicated (black tick within IQR). Horizontal tick marks outside IQR show the range of all elements within Q1-1.5*IQR and Q3+1.5*IQR (~99.3% interval of a normal distribution).
In many aspects, the genomes of mammalian Plasmodium species (P. falciparum, P. knowlesi, P. vivax, P. yoelii) are remarkably uniform, ranging from 23-27 Mb across 14 chromosomes, and comprising ~5,500 genes, most of which (~51%) contain at least one intron (Table 1). However, differences in nucleotide bias can be extreme (e.g. P. vivax and P. falciparum average percent G+C ~42.3 and ~19.4, respectively), and a large gene family found in P. y. yoelii raised its gene count to ~5,880 4. A remarkable 77% of genes are orthologous between the four species (Supplementary Fig. 1); almost half of these encode conserved hypothetical proteins of unknown function. In P. falciparum, the high incidence of tandem repeats and low complexity regions (LCRs) in proteins, especially antigens, has led researchers to propose that LCRs are involved in immune evasion mechanisms, such as antigen diversification 8 and reducing the host’s antibody response to critical epitopes by acting as a ‘smokescreen’ 9. We found that LCRs tend to constitute a smaller proportion of P. vivax proteins on average (39%) than P. falciparum proteins (60%; Supplementary Fig. 2), and that LCR expansion partly accounts for the slightly larger size of P. falciparum proteins (Supplementary Table 2), but how this relates to differences in immune evasion mechanisms between P. vivax and P. falciparum is unclear.
Table 1.
Comparison of nuclear genome features between four Plasmodium species.
| Feature | P. vivax | P. knowlesi | P. falciparum | P. y. yoelii |
|---|---|---|---|---|
| Genome | ||||
| Size (Mb) | 26.8 | 23.5 | 23.3 | 23.1 |
| No. of chromosomes | 14 | 14 | 14 | 14 |
| Coverage (fold) | 10 | 8 | 14.5 | 5 |
| (G+C) content (%) | 42.3 | 37.5 | 19.4 | 22.6 |
| Genes | ||||
| No. genes | 5433* | 5188* | 5403* | 5878§ |
| Mean gene length‡ | 2164† | 2180†† | 2283 | 1298§ |
| Gene density (bp/gene) | 4462.9† | 4593†† | 4312 | 2566§ |
| Percent coding‡ | 48.5† | 47.4†† | 52.6 | 50.6§ |
| Genes with introns (%) | 52.1† | 51.6†† | 53.9 | 54.2§ |
| Exons | ||||
| Mean no. per gene | 2.5† | 2.6†† | 2.4 | 2.0§ |
| (G+C) content (%) | 46.5† | 40.2†† | 23.7 | 24.8§ |
| Mean length (bp) | 957† | 836.8†† | 935 | 641§ |
| Introns | ||||
| (G+C) content (%) | 49.8† | 38.6†† | 13.6 | 21.1§ |
| Mean length (bp) | 192† | 224.4†† | 179 | 209§ |
| Intergenic regions | ||||
| (G+C) content (%) | 42.5† | 34.56†† | 13.7 | 20.7§ |
| Mean length (bp) | 1994† | 2049.4†† | 1745 | 859§ |
| RNAs | ||||
| No. of tRNA genes | 44 | 41 | 43 | 39 |
| No. 5S rRNA genes | 3 | 0** | 3 | 3 |
| No. 5.8S/18S/28S rRNA units | 7 | 5 | 7 | 4 |
Including pseudogenes and partial genes, excluding non-coding RNA genes
Excluding genes from 2,745 small A+T-rich contigs
Excluding introns
Excluding partial genes
not present in P. knowlesi assembly version 4.0
Excluding genes from 511 small contigs
Notwithstanding the recent functional characterization of the Apicomplexan AP2 family of transcriptional regulators in Plasmodium 10, the parasite appears to lack most of the standard eukaryotic transcriptional machinery, such as transcription-associated proteins (TAPs)11, but is rich in regulatory sequences 12, fostering the idea that gene expression regulation in Plasmodium is complex and unusual. Our initial studies found no significant differences in the TAP repertoire between P. falciparum, P. vivax and P. knowlesi, indicating that transcriptional mechanisms are similar in all three species (Supplementary Table 3). Genes encoding mRNA stability proteins containing a CCCH-zinc finger were abundant in all three species, affirming the importance of post-transcriptional regulation in the control of gene expression across Plasmodium. A genome scan of P. vivax for known core promoter elements such as TATA and CAAT boxes identified some candidates, but many of them lacked positional specificity. Similarly, a search for novel promoter elements in regions upstream of ~1,800 mapped transcription start sites (5’ UTRs), and for RNA binding proteins in ~1,300 3’ UTRs, also failed to produce convincing candidates (data not shown). To determine whether binding sites are conserved between P. falciparum and other primate Plasmodium species, we searched for over-represented nucleotide ‘words’ in regions upstream of clusters of potentially co-regulated genes conserved in P.vivax, P. falciparum, P. knowlesi and P. y. yoelii (Supplementary Information). Seven putative novel regulatory binding sites conserved across at least two species were identified (Supplementary Table 4). These binding sites were associated with core eukaryotic processes such as dephosphorylation and with parasite-specific functions such as cell invasion. Independent support for two of our predicted sites comes from a recent report of the sporozoite-associated motif 5’-TGCATGCA-3’ and the merozoite invasion-related 5’-GTGTGCACAC-3’ 13 motif. In our analysis these two sites, together with the dephosphorylation-associated motif 5’-GCACGCGTGC-3’, were conserved across the four Plasmodium species.
Examination of parasite population structure in the field is key to understanding transmission dynamics, the spread of drug resistance, and to design and test malaria control efforts. Many population studies have exploited the abundant polymorphic microsatellites in the P. falciparum genome, primarily simple sequence repeats such as [TA] dinucleotide and polyA/polyT 14. We screened the P. vivax genome for microsatellites, identifying ~160 that are polymorphic between eight P. vivax laboratory lines (Supplementary Table 5; Plate 1). P. vivax microsatellites average 27.5% G+C, with an average repeat unit length of 3.1 nucleotides and an average copy number of 19.1. We found fewer microsatellites in P. vivax than in P. falciparum (as noted previously 15), likely due to the more conventional nucleotide composition of the former. Even so, these genome-wide polymorphic markers are already facilitating studies of P. vivax population structure and genetic diversity 16,17.
Chromosome synteny and genome evolution
Previous studies have indicated significant conservation of gene synteny between Plasmodium parasites 4 in direct proportion to their genetic distance. We generated a synteny map of P. vivax, P. knowlesi, P. falciparum, and the rodent malaria parasites P. yoelii, P. berghei and P. chabaudi (considered as a single lineage 18 due to their virtually complete synteny; Plate 1). The P. vivax and P. knowlesi chromosomes are highly syntenic except for microsyntenic breaks at species-specific genes (in particular the P. knowlesi kir and SICAvar genes; see ref. 6); a previous study identified such breaks as foci for the evolution of host-parasite interaction genes18. The karyotypes of P. vivax and P. knowlesi correspond to the most parsimonious reconstruction of the ancestral form of the six species; the karyotypes of P. falciparum and the rodent malaria parasites can be reconstructed from this form through nine and six chromosomal rearrangements, respectively (Supplementary Fig. 3). No ‘hot-spots’ of synteny breakage were identified, indicating that intersyntenic breakpoints were not ‘reused’ during the divergence of the species, and no obvious motifs except for AT-rich regions and LCRs were identified in regions of the P. vivax genome predicted to have recombined to give single P. falciparum chromosomes. Of the 3,336 orthologs between all six species, 3,305 (99%) were found to be positionally conserved (Supplementary Table 6).
We used 3,322 high-quality P. vivax / P. knowlesi orthologs to obtain maximum likelihood estimates of the rate of substitution at synonymous (dS) and non-synonymous (dN) sites, as well as ω (dN/dS; Supplementary Table 7 and Plate 1). P. vivax chromosomes differ significantly in their average values for both dS and dN, but the two variables are strongly correlated within and between chromosomes (Supplementary Fig. 4). The chromosomes also differ significantly in average %GC4, the G+C content in third codon positions of four-fold degenerate amino acids. This variable is positively correlated with average dS and inversely correlated with chromosome length, such that synonymous sites in genes on the smallest chromosomes (~1 Mb) evolve ~1.5 times faster than genes on the two largest (~3 Mb) chromosomes (Supplementary Fig. 5). These observations strongly suggest the existence of heterogeneous mutation rates across the genome. It is unclear if this is due to cytosine-to-thymine deamination, which is more likely in G+C-rich regions, since it is not known whether DNA methylation occurs in P. vivax. The degree of selective constraint (ω) also varies across classes of genes. Genes encoding glycosylphosphatidylinositol (GPI) anchored proteins, cell adhesion proteins, exportome proteins, and proteins with transmembrane or signal peptide motifs, all of which are at least partly extra-cellular, were found to evolve significantly faster than genes involved in, for example, carbohydrate metabolism, enzyme regulation and cell structure (Supplementary Table 8, Plate 1 inset). The host immune system, by targeting extracellular peptides, appears to have strongly influenced evolutionary rate variation between gene classes in Plasmodium.
A highly-conserved Plasmodium metabolome
We found that key metabolic pathways, housekeeping functions, and the repertoire of predicted membrane transporters are highly conserved between the P. vivax and P. falciparum 3 proteomes (Supplementary Table 9), suggesting that the two species have much the same metabolic potential. Conservation of metabolic processes also extends to the apicoplast, an Apicomplexan plastid secondarily acquired from an ancient cyanobacterium. The apicoplast has lost photosynthetic function, but is essential to the parasite’s metabolism, hosting nuclear-encoded proteins that are targeted to the apicoplast lumen by a conserved bipartite N-terminal presequence. The complete genome sequence of P. vivax offers an opportunity to update and improve the apicoplast proteome that was predicted in silico 3. Apicoplast-targeted proteins conserved in P. vivax participate in major metabolic processes previously recognized in P. falciparum 19, such as complete Type II fatty acid synthesis (FASII), isopentenyl diphosphate (IPP) and iron sulfur cluster assembly pathways, and a fragmented haem synthesis pathway distributed between the apicoplast and mitochondria. Conservation of these pathways in P. vivax is important because synthetic pathways for FASII and IPPs are targets for antimalarial chemotherapeutics 20. The revised Plasmodium apicoplast proteome (Supplementary Table 10) also clarifies the localisation of two important processes. We show thiamine pyrophosphate biosynthesis, previously hypothesised to take place in the apicoplast 19, to be cytosolic. Conversely, we confirm a glyoxalate pathway in the apicoplast, with glyoxalase I and glyoxalase II enzymes being targeted there 21; both enzymes are potential drug targets. Thus, comparison of overall apicoplast metabolic capabilities shows very few differences between P. vivax and P. falciparum.
P. vivax can form hypnozoites, a latent hepatic stage responsible for patent parasitemia relapses months or even years after an initial mosquito-induced infection 22. Hypnozoites survive most drugs that kill blood stage parasites; complete elimination of P. vivax infections (radical cure) requires primaquine, the only licensed drug that can kill hypnozoite stages. However, resistance to the drug is spreading 23, and its use is contraindicated in pregnant women or patients with glucose-6-phosphate dehydrogenase deficiency, which is common in malaria-endemic regions. After an initial examination of P. vivax-specific proteins failed to identify leads (Supplementary Table 11), we hypothesized that the genetic switch for hypnozoite formation may involve P. vivax homologs of dormancy genes. Analysis of the predicted P. vivax proteome revealed some candidates (Supplementary Table 12). However such an association remains speculative, and investigation of hypnozoite formation and activation will require continued development of in vitro systems for culturing P. vivax liver stages 24.
Gene families shape Plasmodium biology
Plasmodium lineages display differential gene family expansion (Table 2) that has shaped the specific biology of each species. Phenotypes illustrating this include parasite invasion of red blood cells, and antigenic variation. Invasion of erythrocytes by extracellular Plasmodium merozoites, crucial to the development of malaria in an infected individual, depends upon specific interactions between merozoite ligands and erythrocyte surface receptors (Fig. 1). Plasmodium species-specific mechanisms act mostly during the preliminary phases of invasion (e.g., merozoite attachment and orientation). In P. vivax, but not P. falciparum, invasion is restricted to Duffy-positive reticulocytes 2. P. vivax Duffy-binding protein (DBP 25) and reticulocyte-binding proteins (RBPs 26) are the archetypes of two distinct Plasmodium families of cell-binding proteins involved in erythrocyte selection (referred to as the Duffy-binding-like ‘DBL’, and reticulocyte-binding-like ‘RBL’ families, respectively). Homologs of rbp1 and rbp2, two genes originally identified in P. vivax, include the P. falciparum rh/nbp genes (reviewed in ref. 27) and the Py235 family in P. yoelii (reviewed in ref. 28). Unexpectedly, we identified additional rbp genes in the P. vivax genome (Supplementary Table 13), including multiple rbp2 genes, which could provide P. vivax with a diversity of invasion mechanisms comparable to that of P. falciparum. This finding dispels a view that P. vivax has a relatively uncomplicated erythrocyte invasion mechanism. Instead, P. vivax likely has alternate invasion pathways, since differential expression of rbp homologs in P. falciparum 29 and P. yoelii 30 is closely linked to switching of invasion pathways (Fig. 1). All rbp2 loci occur in the subtelomeric regions of P. vivax chromosomes - non-syntenic, dynamic regions of the genome in which species-specific genes are generated (Supplementary Fig. 6).
Table 2.
A comparison of the sequences and antimalarial binding sites of P. vivax and P. falciparum orthologs predicted to be involved in artemisinin and atovaquone drug interactions. References and further details are given in the Supplementary Information.
| Antimalarial drug | Gene | Gene PID* | Protein PID* | X-ray (X) structure or Homology (H) model | Drug binding site residues (red: polymorphism between P. vivax and P. falciparum) | Polymorphisms associated or suspected with resistance (blue: unique to P. falciparum) | Conclusions |
|---|---|---|---|---|---|---|---|
| Artemisinin derivatives | P. vivax atpase6 | 60.7 | 70.0 | H* | (I261 A2631† F264 Q267 L268 I271 I275 A313 I942 I946 N949 I950 V953 A954 F957 S1008 L1009 I1010 L1015 Y1018 I1019)* | None reported | P. vivax predicted to be more resistant to artemisinin than P. falciparum |
| P. falciparum atpase6 | H* | (I261 L2631 F264 Q267 L268 I271 I275 A313 I973 I977 N980 I981 V984 A985 F988 N1039 L1044 I1045 L1050 Y1053 I1054)* | (E432K A623E S769N**)2 | ||||
| Atovaquone | P. vivax cyt b | 86.0 | 89.5 | H* | (I119 F123 Y126 M133 V140 I141 L144 I258 P260 F264 F267 Y268 L271 V284 L285 L288)* | None reported | Resistance mutations in P. falciparum likely to affect the same residues in P. vivax |
| P. falciparum cyt b | H3 | (I119 F123 Y126 M133 V140 I141 L144 I258 P260 F264 F267 Y268†† L271 V284 L285 L288)3 | M133I3‡ T142I3‡ L144F3‡ I258M3‡ F267I3‡ Y268S3/N4 L271V3‡ K272R3‡ P275T3‡ G280D3‡ V284K3‡ |
Determined during this study
Identified from in vitro drug selection studies
Predicted to be located in a drug active site and predicted to cause resistance
Predicted to be located in a drug active site and mutations known to cause resistance
Association with resistance not conclusive
Abbreviations: PID, percent identity; atpase6, adenosine triphosphatase-6; cyt b, cytochrome b.
Figure 1. Predicted erythrocyte invasion pathways and dominant ligands of Plasmodium species.
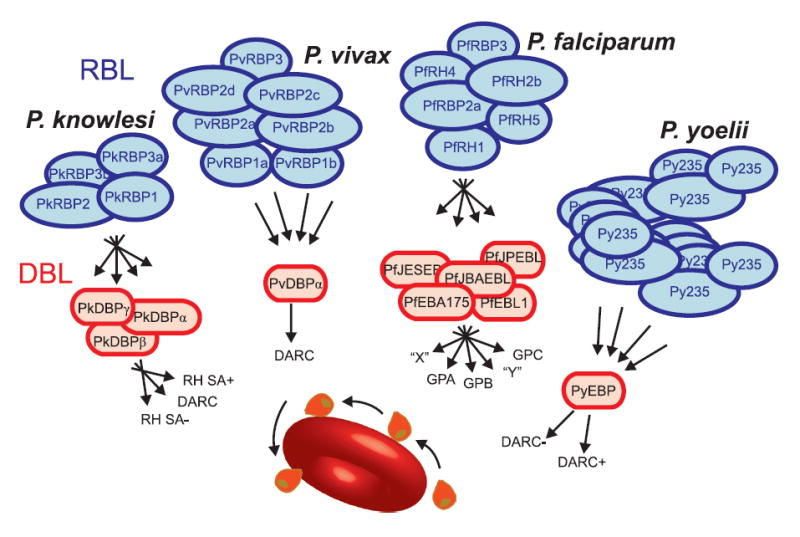
RBL and DBL invasion families predicted from several Plasmodium proteomes are shown above a Plasmodium merozoite colliding and reorientating on the red blood cell surface. Species-specific RBL families interact with an array of species-specific DBL proteins that utilize both alternative (crossed arrows) and fixed (straight arrows) pathways with known or predicted receptors on the surface of erythrocytes. Blocking these receptor-ligand interactions offers a potential mechanism to prevent clinical malaria. GPA/B/C: P. falciparum glycophorin A/B/C receptors; “X”, “Y”: predicted receptors; RH SA+/-: Rhesus sialic acid dependent (+) and independent (-) pathways; DARC: Duffy antigen receptor for chemokines dependent (+) and independent (-) pathways; * presence of this pathway is controversial.
The final phase of invasion, merozoite entry into an intraerythrocytic vacuole, uses an intracytoplasmic molecular motor (components of which are highly conserved between Plasmodium species) coupled to simultaneous shedding of crucial merozoite surface proteins (MSPs). There are at least ten distinct MSPs (Supplementary Table 14), and P. vivax genome analysis reveals two particularly interesting MSP families, MSP3 and MSP7. Eleven members of the msp3 gene family occur in tandem on a ~60 kb region of P. vivax chromosome 10 (Supplementary Fig. 7), and show weak similarity to four msp3 gene family members on P. falciparum chromosome 10 and to two P. knowlesi msp3 genes located on different chromosomes. Thus, there has been a significant expansion of the msp3 gene family in P. vivax, perhaps as a means to enhance immune evasion, as P. falciparum and P. vivax msp3 gene family members have been shown to be antigenic and to partially immunize non-human primates against blood stage parasites 31. In P. falciparum, MSP6 (a member of the MSP3 family that lacks heptad repeats) non-covalently binds with MSP1, but there is no counterpart to MSP6 in P. vivax. MSP7, another P. falciparum antigen that binds to MSP1 on the surface of merozoites, has also been expanded in P. vivax, with eleven copies on chromosome 12, compared to six and three members in P. falciparum and P. y. yoelii respectively; it is not known if any P. vivax MSP7 proteins bind to MSP1. The surface coats of merozoites and extracellular forms of Plasmodium parasites are composed largely of GPI-anchored proteins, many of which are important targets of protective immune responses and thus constitute promising vaccine candidates. When we predicted the GPI-anchored proteome of P. vivax and compared it to validated P. falciparum GPI-anchored proteins 32, 29 of the 30 GPI-anchored proteins identified in P. falciparum had counterparts in P. vivax (Supplementary Fig. 8), an extraordinary level of conservation. MSP2 (the second most abundant merozoite surface protein in P. falciparum) is absent in the P. vivax genome, and P. vivax contains one additional GPI-anchored protein that appears to be a member of the ‘six cysteine’ apicomplexan-specific gene family 33. Both the P. vivax and P. knowlesi genomes encode an apparently paralogous gene next to msp1, which is the largest and most abundant protein on the P. falciparum merozoite surface. P. vivax MAP1 is not closely related to MSP1 (11% identity, 22% similarity) although their sizes, a predicted GPI-attachment site, and structural features such as a C-terminal double EGF module, are similar.
A second notable parasite phenotype is antigenic variation: the ability to vary surface proteins during the course of an infection in order to evade the host’s immune response. In P. falciparum, antigenic variation is mediated by species-specific gene families such as var, members of which are expressed clonally and regulated epigenetically 34. In P. vivax, the largest multigene family vir, part of the pir (Plasmodium interspersed repeats) superfamily found in several Plasmodium species 5, has been implicated in antigenic variation; 35 gene copies were previously identified 35. We identified 346 vir genes in the P. vivax genome located within AT-rich subtelomeric regions of P. vivax chromosomes (Plate 1). Structurally, vir genes vary greatly, ranging from 156-2,316 bp in length and containing 1-5 exons. VIR proteins were previously classified into six subfamilies (A-F) based upon sequence similarity 35, and representatives of these subfamilies were identified in patient isolates 36. Clustering the VIRs in the Salvador I genome yielded six new subfamilies (G-L) and confirmed gene expression for several of these in natural infections (Supplementary Table 15). Motif analysis of the total VIR repertoire (Fig. 2) showed that approximately half (171) contain a transmembrane domain, and half (160) contain a motif similar to the PEXEL/VSP sequence linked to export of parasite proteins 37,38. Introns from 25 vir genes contain a conserved motif proximal to the donor splice site, suggesting possible functionality of the sequence in the control of vir gene expression, as has been shown for P. falciparum var introns 39. Motif-shuffling among the sequences is apparent, particularly among large VIR proteins that have undergone an expansion of some motifs at the amino terminus. Similarly to P. falciparum var genes, in situ hybridization analysis has shown that P. vivax chromosome ends localize to the nuclear periphery 40, where ectopic recombination favors the generation of variants and gene expansion. Although the repeat structure of P. vivax subtelomeric regions is not as extensive as that seen in P. falciparum 6, P. vivax likely uses chromosomal exchange as a mechanism for generating antigenic diversity. VIR proteins represent an extremely diverse family, members of which currently appear more divergent than members of other partially characterized PIR families such as the P. chabaudi CIR (135 members) and the P. berghei BIR (245 members) families (Supplementary Fig. 9). Shared structural characteristics have been shown between VIR subfamily D proteins and the P. falciparum Pfmc-2tm family located at Maurer’s clefts, and VIR subfamily A proteins and the P. falciparum SURFIN family found on the surface of infected erythrocytes 41. We speculate that the extreme diversity and sub-structuring of VIR proteins indicate members different subcellular localizations and functions, including immune evasion.
Figure 2. VIR protein motifs and organization.
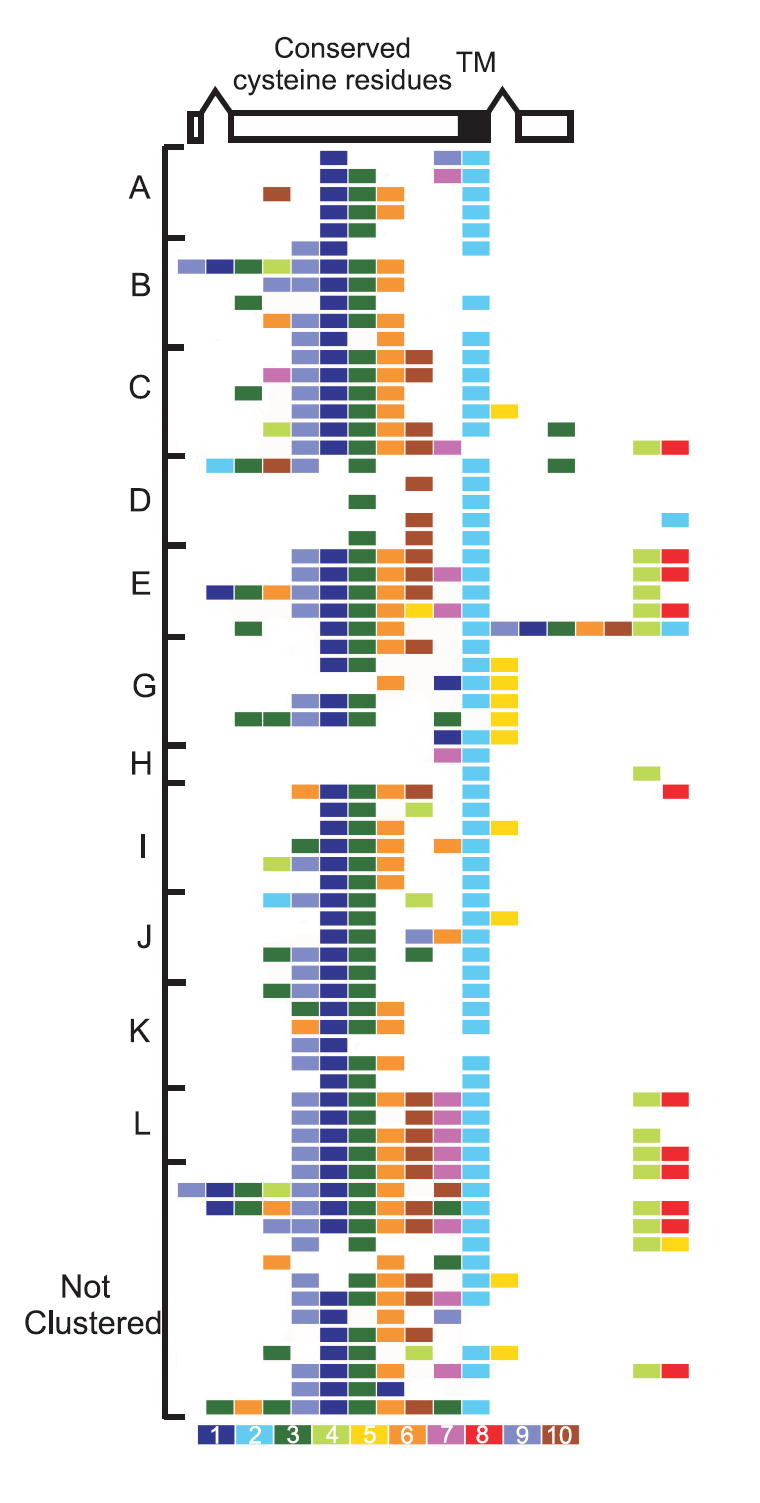
The structure of an archetypal vir gene is shown at top, followed by VIR subfamily motifs arranged from the N-terminus (left) to the C-terminus (right). Consensus motif sequences numbered in decreasing order of statistical significance are shown color coded below the figure. Motif 2: transmembrane (TM) domain; motif 3: PEXEL/VSP-like motif; all remaining motifs are predicted exposed globular domains. The overall organization and order of the motifs is maintained, with the central core motifs 9, 1, 3, 6 and 10 followed by C-terminus motifs 7, 2, 4, 8 and 5 embedded in a variant-sized portion of the molecule. Motifs are listed in the Supplementary Information.
We identified eight novel gene families (Pv-fam-a to -e and -g to -i; Supplementary Table 16) in the P. vivax genome, most of which are located in subtelomeric regions (Plate 1). Of particular interest are (1) the PvTRAG (Pv-fam-a) gene family (36 genes), one member of which was previously identified; it encodes a protein localised to the caveola-vesicle complex of infected erythrocytes, and has been shown to elicit a humoral immune response during the course of natural infections 42; and (2) the Pv-fam-e family (Supplementary Fig. 10), 36 copies of which are found in two loci on either side of the predicted centromere on chromosome 5, with one 10-gene locus present in a 47% GC region, and a second 26-gene locus present in a 36% GC region. While P. vivax proteins have a fairly balanced codon composition, using all 61 sense codons almost equally (effective number of codons, Nc = 54.2), their orthologs in P. falciparum are more biased (Nc = 37.5), with G- and C-ending codons nearly absent from four-fold degenerate amino acids (Supplementary Table 17). However, P. vivax gene families, which are predominantly located in AT-rich regions, have a codon composition of Nc = 47. This pattern suggests a strong influence of local mutation pattern on the nucleotide composition of genes and indicates a potential for differential gene expression.
Plasmodium drug interaction genes
The sexual stages of P. vivax are produced before the onset of clinical symptoms, permitting mosquito transmission early in an infection. Such early parasite transmission may delay development of resistance to many of the antimalarial drugs used to treat vivax malaria, despite the extensive long-term use of these drugs in regions endemic for both P. vivax and P. falciparum 43. Nevertheless, P. vivax can develop resistance to most of the current antimalarial drugs. To understand the interactions between antimalarial drugs and the parasite proteins implicated in drug binding and resistance, we examined crystal structures and developed homology models for several P. vivax proteins in the predicted proteome, and compared the predicted binding sites and reported mutations with those of their P. falciparum orthologs (Table 2).
Currently, the most efficacious novel antimalarial drugs are derivatives of artemisinin (qinghaosu) and atovaquone, used predominantly in combination therapies. Arteminsinin derivatives, the most potent drugs recommended for treatment, may target a sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA)-type protein, ATPase6 44. We constructed homology models of P. vivax and P. falciparum ATPase6 and identified two residues in the putative active sites for artemisinin that differ between the two species (P. vivax A263 and S1008, equivalent to L263 and N1039 in P. falciparum). A change in residue 263 from leucine to alanine results in a three-fold decrease in susceptibility of P. falciparum to artemisinin 44, and indeed the IC50 (inhibitory concentration at which fifty percent of parasites die) for some P. vivax field isolates appears higher than the IC50 for P. falciparum 45, although it should be noted that clinical resistance of any human Plasmodium species to artemisinin derivatives has yet to be documented. Atovaquone, used in combination with the antifolate proguanil, selectively inhibits mitochondrial electron transport at the cytochrome bc(1) complex; mutations in the cytochrome b (cytb) gene can interfere with this inhibition, causing resistance. We constructed a homology model of P. vivax CYTB and compared it to the P. falciparum CYTB homology model 46, revealing almost identical structures, including the predicted atovaquone active sites. Although there are no reports of P. vivax treatment failures, our studies indicate that should resistance arise, the same sites in P. vivax CYTB may be implicated.
Towards a policy shift for vivax malaria
Despite the insights into parasite biology provided by the P. vivax genome, many important questions remain that can only be addressed by functional studies. For example, we were unable to find differences in the predicted P. vivax proteome that might explain the rheological behaviour of P. vivax-infected erythrocytes, which remain flexible and can repeatedly pass through the spleen, unlike P. falciparum-infected erythrocytes, whose rigidity facilitates cytoadherence and avoidance of splenic clearance 47. Studies of the hypnozoite transcriptome, although technically challenging, would radically increase our inadequate knowledge of the biology of this dormant form. Studies are currently underway to develop new in vitro culture systems 48, which could provide badly-needed biological material for such functional studies.
The malaria research and control communities were challenged recently to once again establish the eradication of malaria as a policy goal 49. Given the significant contribution of P. vivax to the global malaria situation 1,43, it is imperative that these efforts include elimination of P. vivax as well as P. falciparum. Elimination of P. vivax presents special challenges, in particular the parasite’s production of dormant hypnozoites that enables relapses long after the initial parasitemia has cleared. Indeed, an important aspect of P. vivax eradication will be the development of new drugs to replace primaquine for radical cure. Although the development of new drugs targeting P. vivax liver stages is a formidable task, recent developments offer hope that this goal can be accomplished 50.
Methods summary
Genome sequencing, assembly, mapping and annotation
Saimiri boliviensis boliviensis monkeys were infected with the Salvador I strain of P. vivax isolated from a patient from El Salvador. Extracted parasite DNA was used to make genomic DNA libraries for shotgun sequencing. Reads were assembled into scaffolds, inter-scaffold gaps closed, and scaffolds assigned to P. vivax chromosomes through hybridization of scaffold-specific probes to pulsed-field gel separated chromosomes. Gene prediction algorithms were used to predict gene models, and each model was manually checked for structural inconsistencies. Gene function was assigned using an automated annotation pipeline with subsequent manual curation.
Genome analysis
Methods for the in silco analysis of the genome sequence are described in the Supplementary Information.
Studies requiring laboratory experimentation
Polymorphic microsatellite identification
Primers flanking 333 microsatellites identified from the genome sequence and designed for field studies where access to capillary electrophoresis equipment may not be possible, were used to amplify the loci from eight world-wide P. vivax laboratory strains adapted to growth in monkeys (Brazil I, Miami II, Pakchong, Panama I, Nica, Thai II, Vietnam IV and Indonesia XIX). Amplicons were separated by electrophoresis on agarose gels and scored for size differences.
Vir gene expression
cDNA was generated from total RNA extracted from the Salvador I isolate and from three patient isolates from Brazil. Primers were designed to eight vir gene subfamilies and used to amplify the loci.
Supplementary Material
Full methods and associated references are available in the online version of the paper at www.nature.com/nature.
Acknowledgments
We are indebted to the P. vivax research community for their support, and in particular to M. Gottlieb and V. McGovern for facilitating financial support. Funding came from the following sources: P. vivax sequencing, assembly and closure, US Department of Defense and National Institute of Allergy and Infectious Diseases; genome mapping, Burroughs Wellcome Fund; and selective constraint analysis, National Institute of General Medical Sciences. We wish to thank TIGR’s SeqCore, Closure and IFX core facilities, E. Lee, J. Sundaram, J. Orvis, B. Haas and T. Creasy for engineering support, R. K. Smith Jr. for annotation support, E. Lyons and H. Zhang for technical assistance, H. Potts for statistical analysis, T. McCutchan for rDNA sequence annotation and S. Perkins for the Plasmodium phylogeny.
Footnotes
Competing interests statement The authors declare that they have no competing financial interests.
Sequence and annotation data for the genome were deposited in GenBank under the project accession number AAKM00000000 and are also available at the Plasmodium genome sequence database PlasmoDB at http://plasmodb.org. A minimal tiling path of clones covering each chromosome is available through the malaria repository MR4 www.mr4.org, and a long-oligo array through the Pathogen Functional Genomics Resource Center http://pfgrc.jcvi.org
References
- 1.Price RN, et al. Vivax Malaria: Neglected and Not Benign. Am J Trop Med Hyg. 2007;77(Suppl 6):79–87. [PMC free article] [PubMed] [Google Scholar]
- 2.Miller LH, Mason SJ, Clyde DF, McGinniss MH. The resistance factor to Plasmodium vivax in blacks. The Duffy-blood-group genotype, FyFy. N Engl J Med. 1976;295:302–304. doi: 10.1056/NEJM197608052950602. [DOI] [PubMed] [Google Scholar]
- 3.Gardner MJ, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carlton JM, et al. Genome sequence and comparative analysis of the model rodent malaria parasite Plasmodium yoelii yoelii. Nature. 2002;419:512–519. doi: 10.1038/nature01099. [DOI] [PubMed] [Google Scholar]
- 5.Hall N, et al. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005;307:82–86. doi: 10.1126/science.1103717. [DOI] [PubMed] [Google Scholar]
- 6.Pain A, et al. The genome sequence of Plasmodium knowlesi: the fifth human malaria parasite. Nature. 2008 this issue. [Google Scholar]
- 7.McCutchan TF, Dame JB, Miller LH, Barnwell J. Evolutionary relatedness of Plasmodium species as determined by the structure of DNA. Science. 1984;225:808–811. doi: 10.1126/science.6382604. [DOI] [PubMed] [Google Scholar]
- 8.Hughes AL. The evolution of amino acid repeat arrays in Plasmodium and other organisms. J Mol Evol. 2004;59:528–535. doi: 10.1007/s00239-004-2645-4. [DOI] [PubMed] [Google Scholar]
- 9.Anders RF. Multiple cross-reactivities amongst antigens of Plasmodium falciparum impair the development of protective immunity against malaria. Parasite Immunol. 1986;8:529–539. doi: 10.1111/j.1365-3024.1986.tb00867.x. [DOI] [PubMed] [Google Scholar]
- 10.De Silva EK, et al. Specific DNA-binding by Apicomplexan AP2 transcription factors. Proc Natl Acad Sci. 2008;17:8393–8398. doi: 10.1073/pnas.0801993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coulson RM, Hall N, Ouzounis CA. Comparative genomics of transcriptional control in the human malaria parasite Plasmodium falciparum. Genome Res. 2004;14:1548–1554. doi: 10.1101/gr.2218604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Noort V, Huynen MA. Combinatorial gene regulation in Plasmodium falciparum. Trends Genet. 2006;22:73–78. doi: 10.1016/j.tig.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 13.Young JA, et al. In silico discovery of transcription regulatory elements in Plasmodium falciparum. BMC Genomics. 2008;9:70. doi: 10.1186/1471-2164-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferdig MT, Su XZ. Microsatellite markers and genetic mapping in Plasmodium falciparum. Parasitol Today. 2000;16:307–312. doi: 10.1016/s0169-4758(00)01676-8. [DOI] [PubMed] [Google Scholar]
- 15.Feng X, et al. Single-nucleotide polymorphisms and genome diversity in Plasmodium vivax. Proc Natl Acad Sci USA. 2003;100:8502–8507. doi: 10.1073/pnas.1232502100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Imwong M, et al. Relapses of Plasmodium vivax infection usually result from activation of heterologous hypnozoites. J Infect Dis. 2007;195:927–933. doi: 10.1086/512241. [DOI] [PubMed] [Google Scholar]
- 17.Joy DA, et al. Local adaptation and vector-mediated population structure in Plasmodium vivax malaria. Mol Bio Evo. 2008;25:1245–1252. doi: 10.1093/molbev/msn073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kooij TW, et al. A Plasmodium whole-genome synteny map: indels and synteny breakpoints as foci for species-specific genes. PLoS Pathog. 2005;1:e44. doi: 10.1371/journal.ppat.0010044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ralph SA, et al. Tropical infectious diseases: Metabolic maps and functions of the Plasmodium falciparum apicoplast. Nat Rev Microbiol. 2004;2:203–216. doi: 10.1038/nrmicro843. [DOI] [PubMed] [Google Scholar]
- 20.Sato S, Wilson RJ. The plastid of Plasmodium spp.: a target for inhibitors. Curr Top Microbiol Immunol. 2005;295:251–273. doi: 10.1007/3-540-29088-5_10. [DOI] [PubMed] [Google Scholar]
- 21.Akoachere M, et al. Characterization of the glyoxalases of the malarial parasite Plasmodium falciparum and comparison with their human counterparts. Biol Chem. 2005;386:41–52. doi: 10.1515/BC.2005.006. [DOI] [PubMed] [Google Scholar]
- 22.Krotoski WA. The hypnozoite and malarial relapse. Prog Clin Parasitol. 1989;1:1–19. [PubMed] [Google Scholar]
- 23.Baird JK, Hoffman SL. Primaquine therapy for malaria. Clin Infect Dis. 2004;39:1336–1345. doi: 10.1086/424663. [DOI] [PubMed] [Google Scholar]
- 24.Sattabongkot J, et al. Establishment of a human hepatocyte line that supports in vitro development of the exo-erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax. Am J Trop Med Hyg. 2006;74:708–715. [PubMed] [Google Scholar]
- 25.Fang XD, Kaslow DC, Adams JH, Miller LH. Cloning of the Plasmodium vivax Duffy receptor. Mol Biochem Parasitol. 1991;44:125–132. doi: 10.1016/0166-6851(91)90228-x. [DOI] [PubMed] [Google Scholar]
- 26.Galinski MR, Medina CC, Ingravallo P, Barnwell JW. A Reticulocyte-binding protein complex of Plasmodium vivax Merozoites. Cell. 1992;69:1213–1226. doi: 10.1016/0092-8674(92)90642-p. [DOI] [PubMed] [Google Scholar]
- 27.Cowman AF, Crabb BS. Invasion of red blood cells by malaria parasites. Cell. 2006;124:755–766. doi: 10.1016/j.cell.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Gruner AC, et al. The Py235 proteins: glimpses into the versatility of a malaria multigene family. Microbes and Infection. 2004;6:864–873. doi: 10.1016/j.micinf.2004.04.004. [DOI] [PubMed] [Google Scholar]
- 29.Duraisingh MT, et al. Phenotypic variation of Plasmodium falciparum merozoite proteins directs receptor targeting for invasion of human erythrocytes. Embo J. 2003;22:1047–1057. doi: 10.1093/emboj/cdg096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Preiser PR, Jarra W, Capiod T, Snounou G. A rhoptry-protein-associated mechanism of clonal phenotypic variation in rodent malaria. Nature. 1999;398:618–622. doi: 10.1038/19309. [DOI] [PubMed] [Google Scholar]
- 31.Roussilhon C, et al. Long-term clinical protection from falciparum malaria is strongly associated with IgG3 antibodies to merozoite surface protein 3. PLoS Medicine. 2007;4:e320. doi: 10.1371/journal.pmed.0040320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilson PR, et al. Identification and stoichiometry of glycosylphosphatidylinositol-anchored membrane proteins of the human malaria parasite Plasmodium falciparum. Mol Cell Proteomics. 2006;5:1286–1299. doi: 10.1074/mcp.M600035-MCP200. [DOI] [PubMed] [Google Scholar]
- 33.Sanders PR, et al. Distinct protein classes including novel merozoite surface antigens in Raft-like membranes of Plasmodium falciparum. J Biol Chem. 2005;280:40169–40176. doi: 10.1074/jbc.M509631200. [DOI] [PubMed] [Google Scholar]
- 34.Dzikowski R, Templeton TJ, Deitsch K. Variant antigen gene expression in malaria. Cellular Microbiol. 2006;8:1371–1381. doi: 10.1111/j.1462-5822.2006.00760.x. [DOI] [PubMed] [Google Scholar]
- 35.del Portillo HA, et al. A superfamily of variant genes encoded in the subtelomeric region of Plasmodium vivax. Nature. 2001;410:839–842. doi: 10.1038/35071118. [DOI] [PubMed] [Google Scholar]
- 36.Fernandez-Becerra C, et al. Variant proteins of Plasmodium vivax are not clonally expressed in natural infections. Mol Micro. 2005;58:648–658. doi: 10.1111/j.1365-2958.2005.04850.x. [DOI] [PubMed] [Google Scholar]
- 37.Marti M, Good RT, Rug M, Knuepfer E, Cowman AF. Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science. 2004;306:1930–1933. doi: 10.1126/science.1102452. [DOI] [PubMed] [Google Scholar]
- 38.Hiller NL, et al. A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science. 2004;306:1934–1937. doi: 10.1126/science.1102737. [DOI] [PubMed] [Google Scholar]
- 39.Frank M, et al. Strict pairing of var promoters and introns is required for var gene silencing in the malaria parasite Plasmodium falciparum. J Biol Chem. 2006;281:9942–9952. doi: 10.1074/jbc.M513067200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Scherf A, Figueiredo L, Freitas-Junior LH. Genomes and Molecular Cell Biology of Malaria Parasites. Horizon Press; Wymondham: 2004. [Google Scholar]
- 41.Merino EF, et al. Multi-character population study of the vir subtelomeric multigene superfamily of Plasmodium vivax, a major human malaria parasite. Mol Biochem Parasitol. 2006;149:10–16. doi: 10.1016/j.molbiopara.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 42.Jalah R, et al. Identification, expression, localization and serological characterization of a tryptophan-rich antigen from the human malaria parasite Plasmodium vivax. Mol Biochem Parasitol. 2005;142:158–169. doi: 10.1016/j.molbiopara.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 43.Mendis K, Sina BJ, Marchesini P, Carter R. The neglected burden of Plasmodium vivax malaria. Am J Trop Med Hyg. 2001;64:97–106. doi: 10.4269/ajtmh.2001.64.97. [DOI] [PubMed] [Google Scholar]
- 44.Uhlemann AC, et al. A single amino acid residue can determine the sensitivity of SERCAs to artemisinins. Nat Struct Mol Biol. 2005;12:628–629. doi: 10.1038/nsmb947. [DOI] [PubMed] [Google Scholar]
- 45.Russell B, et al. Determinants of in vitro drug susceptibility testing of Plasmodium vivax. Antimicrob Agents Chemother. 2008;52:1040–1045. doi: 10.1128/AAC.01334-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Korsinczky M, et al. Mutations in Plasmodium falciparum cytochrome b that are associated with atovaquone resistance are located at a putative drug-binding site. Antimicrob Agents Chemother. 2000;44:2100–2108. doi: 10.1128/aac.44.8.2100-2108.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suwanarusk R, et al. The deformability of red blood cells parasitized by Plasmodium falciparum and P. vivax. J Infect Dis. 2004;189:190–194. doi: 10.1086/380468. [DOI] [PubMed] [Google Scholar]
- 48.Udomsangpetch R, Kaneko O, Chotivanich K, Sattabongkot J. Cultivation of Plasmodium vivax. Trends Parasitol. 2008;24:85–88. doi: 10.1016/j.pt.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 49.Roberts L, Enserink M. Malaria. Did they really say … eradication? Science. 2007;318:1544–1545. doi: 10.1126/science.318.5856.1544. [DOI] [PubMed] [Google Scholar]
- 50.Carraz M, et al. A plant-derived morphinan as a novel lead compound active against malaria liver stages. PLoS Medicine. 2006;3:e513. doi: 10.1371/journal.pmed.0030513. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full methods and associated references are available in the online version of the paper at www.nature.com/nature.