

Abstract
We have shown previously that a potent synthetic antagonist of Grb2 SH2 domain binding (1) blocks growth factor stimulated motility, invasion, and angiogenesis in cultured cell models, as well as tumor metastasis in animals. To characterize the selectivity of 1 for the SH2 domain of Grb2 over other proteins containing similar structural binding motifs, we synthesized a biotinylated derivative (3) that retained high affinity Grb2 SH2 domain binding and potent biological activity. To investigate the selectivity of 1 and 3 for Grb2, the biotinylated antagonist 3 was used to immobilize target proteins from cell extracts for subsequent identification by mass spectrometry. Non-specific binding was identified in parallel using a biotinylated analog that lacked a single critical binding determinant. The mechanism of action of the antagonist was further characterized by immunoprecipitation, immunoblotting and light microscopy. This approach to defining protein binding antagonist selectivity and molecular basis of action should be widely applicable in drug development.
Introduction
Antagonists of protein-protein interactions have significant therapeutic potential in a broad range of indications, including cancer. However, defining target selectivity and the molecular basis of therapeutic effects are important challenges to their development and use. For this class of drug in particular, systematic exploration of these subjects at the earliest stages of development can profoundly improve prediction, measurement and avoidance of drug toxicity, identification of appropriate patient populations and drug efficacy assessment. In the long term, systematically defining selectivity and mechanism of action may also accelerate target validation and facilitate the design of improved therapeutic and diagnostic agents. The modification of investigational compounds with chemical tags such as biotin is among the most direct strategies yet devised to address these questions and this approach was utilized in our present investigation.
Ligand binding, receptor overexpression and/or oncogenic mutations can induce receptor tyrosine kinase (RTK) activation and autophosphorylation of specific tyrosine residues within RTK intracellular domains. A prominent consequence of tyrosine phosphorylation is the formation of docking sites for proteins containing Src homology 2 (SH2) domains.1 One of the best characterized proteins of this class is growth factor receptor bound protein 2 (Grb2), an adapter that acts as a critical downstream intermediary in several oncogenic signaling pathways. Originally isolated through screening for epidermal growth factor receptor (EGFR) interacting proteins2, Grb2 associates with several signaling and regulatory proteins. Through its SH2 domain, which is a conserved sequence of approximately 100 amino acids, Grb2 can interact directly with RTKs (e.g. hepatocyte growth factor (HGF) receptor, platelet-derived growth factor receptor) and non-receptor tyrosine kinases (e.g. focal adhesion kinase (FAK) and Bcr/Abl) by preferential binding to phosphopeptide motifs of the form pYXNX, where pY represent phosphotyrosine, N is asparagine and X is any residue.3 The amino- and carboxyl-terminal Src homology 3 (SH3) domains of Grb2, which have a conserved sequence of around 50 amino acids, bind proline-rich regions within additional interacting proteins. Through these two SH3 domains, Grb2 links activated RTKs with several key intracellular regulatory networks, including the Ras/Erk pathway controlling cell cycle progression, and the p21-activating kinase (PAK1) and Arp2/3/WASp pathways regulating the actin filament system, cell shape change and motility.4,5 Grb2 is also a key intermediate of integrin signaling through its interaction with activated FAK at pY925, which resides within a canonical recognition motif (pYXNX) for the Grb2 SH2 domain.6 Grb2-FAK binding triggers a signaling sequence involved in angiogenesis and epithelial-mesenchymal transition (EMT), both of which are important contributors to tumor progression.7,8
The critical roles served by Grb2 in cell motility and angiogenesis make it a logical therapeutic target for pathological processes leading to the spread of solid tumors through local invasion and metastasis.9 Potent, synthetic, low molecular weight antagonists of Grb2 SH2 domain binding have been developed that block RTK-Grb2 interaction and growth factor-stimulated motility and matrix invasion by several tumor cell lines.10 We have previously reported that the Grb2 SH2 domain binding antagonist 1 (Figure 1) can inhibit cell motility,11 angiogenesis12 and tumor metastasis in vivo.13 To better understand the SH2 domain selectivity and mechanism of action of this class of compounds, we designed and synthesized biotinylated derivatives of two biologically active structures and established that high affinity Grb2 binding was maintained. The synthesis and binding evaluation of a macrocyclic Grb2 SH2 domain binding antagonist that bears the biotin functionality at a carboxyl-terminal position was described in a prior report.14 In the current work, we present the synthesis of a hydroxylated variant of 1 (2) as well as an open-chain biotinylated version of 1 (3) (Figure 1). We use 2 and 3 as tools to explore ligand selectivity through immobilization by streptavidin-coated beads and subsequent mass spectrometry analysis. We further evaluated the biological mechanism of action of the compound in intact cells and report herein the inhibition of lamellipodia formation by 1 as a molecular basis for its anti-motility activity.
Figure 1.

Chemical structures of Grb2-SH2 domain binding antagonists and analogues.
Results and Discussion
Synthetic
Syntheses of 2 and the biotinylated conjugates 3 and 4 were similar to the previously reported procedures14 and differed mainly in the preparation of segment 9. Starting from 1,5-dihydroxyl naphthalene 5, the monohydroxyl group was protected as the triflate using N-phenyltrifluoromethanesulfonimide and 2,4,6-collidine (Scheme 1).15 Subsequent Heck reaction proved troublesome because the C-O bond is more stable as compared to the C-halide bond and requires higher cleavage energy. Thus, no reaction was observed under the commonly used Heck reaction conditions of palladium (II) acetate and tri-O-tolylphosphine. However, with the more active phosphine ligand 1,3-bis(diphenylphosphine)propane (DPPP),16 two coupling products were obtained as the expected adduct 7 and its reductive product 8 in combined 81% yield. After reduction of both adducts with lithium aluminum hydride, the primary amine 9 was obtained in 54% yield. Using standard coupling procedures, asparagine and 1-amino-cyclohexenecarboxylic acid were added to yield the dipeptide 10.14
Scheme 1.

Synthesis of the upper segment 10a
aReagents, conditions and yields: (a) Tf2NPh, 2,4,6-collidine, DMAP, DMF-CH2Cl2, reflux, 12 h, 47%; (b) Pd(OAc)2, DPPP, Et3N, reflux, 14 h, 81%; (c) LiAlH4, THF, rt., overnight, 54%; (d) (i) Boc-Asn-OH, DIPCDI, HOBt, DMF, rt, 12 h 87%; (ii) TFA, CH2Cl2, rt, 1 h, 79%; (iii) Fmoc-1-amino-cyclohexenecarboxylic acid, EDC, HOBt, DMF, rt, 12 h, 79%; (iv) piperidine, CH3CN, rt, 2 h, 86%.
Coupling of the upper peptide segment 10 and the bottom residues 11a, b (11a was prepared as described17; 11b was available from Novobiochem Corp.) was performed in the presence of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC) and hydroxybenzotriazole (HOBt) to give 12a and 12b in 79% yield and 74% yield respectively (Scheme 2). Piperidine-mediated cleavage of N-Fmoc followed by protection of the resulting free amino group with tert-butyloxalyl chloride produced 13a and 13b in 43% yield and 80% yield respectively. Of note, the 5-phenol remained unaffected by the coupling reaction conditions and was subsequently coupled to the biotinylated linker in the next step. Global deprotection of tert-butyl ester of 13a gave final product 2. Coupling of 13a and 13b to the biotinylated linker was performed in the presence of EDC and 4-dimethylaminopyridine (DMAP) to provide the desired product 14a and 14b in 78% and 42% yield respectively. These were converted to the corresponding free acids 3 and 4 after cleavage of the tert-butyl esters.
Scheme 2.

Synthesis of inhibitor 2 and biotinylated ligands 3 and 4a
aReagents, conditions and yields: (a) EDC, HOBt, DMF, rt, 12 h (79% for 12a; 74% for 12b); (b) (i) piperidine, CH3CN, rt, 2 h; (ii) tbutyloxalyl chloride, iPr2NEt, DMF, rt, 12 h, (two steps, 43% for 13a; 80% for 13b). (c) TFA-TES-H2O, rt, 1 h 42%; (d) EDC, DMAP, DMF, rt, 12 h (78% for 14a; 42% for 14b); (e) TFA-HS(CH2)2SH-H2O, rt, 1 h (41% for 3; 56% for 4).
To characterize the selectivity of the Grb2 SH2 domain antagonist compound 1 for the SH2 domain of Grb2 over other proteins containing similar motifs, analogues 2 and 3 were prepared, the latter bearing biotin functionality at a carboxyl-terminal position. The compound was biotinylated by attaching the biotin to the naphthyl ring as described above. Biotin is commonly attached to biologically active molecules by means of a 6-aminocaproic acid ester spacer without unduly affecting their biological activities; the spacer minimizes potential steric interactions between avidin and the Grb2 SH2 domain-ligand complex. A biotinylated variant of 1 that had very poor Grb2 SH2 domain binding affinity (4) was prepared in parallel and used as a negative control throughout the course of these studies. Analogue 4 is similar to 3, except that 4 does not contain a phosphoryl mimetic at the 4-position of the tyrosyl mimetic aryl ring, which is a critical requirement for high affinity SH2 domain binding.
Binding Affinities of Grb2 SH2 Domain Antagonists
The Grb2 SH2 domain binding affinities of 2 and its biotin-containing counterpart 3 were determined by surface plasmon resonance (SPR). Binding constants for SH2 domain-small molecule interactions typically have been determined indirectly, by measuring the ability of a small molecule to compete with sensor-bound reference pTyr-containing peptide for the binding of SH2 domain-containing protein in the mobile phase. Significant improvements in the sensitivity and operation of SPR instrumentation now permit accurate direct binding measurements of small compounds such as 2 and 3 to SH2 domain proteins immobilized on the sensor chip. Data were analyzed to a one-site binding model using Scrubber software (Experimental Section). As shown in Table 1, compound 2 bound to amine-coupled Grb2 SH2 domain with a steady state affinity of 132 nM, while compound 3 displayed three-fold lower binding affinity (405 nM), indicating only modest perturbation of antagonist-SH2 domain interaction by the added biotin functionality.
Table 1.
Grb2 and Shc SH2 domain steady state binding affinities determined by surface plasmon resonance spectrometry (Biacore).
| Compound | SH2 Domain | Coupling* | Affinity (nM) | Ratio |
|---|---|---|---|---|
| 2 | Grb2 | amine | 132 | 1 |
| 3 | 405 | 3 | ||
| 1 | Biotin-Grb2 | SA | 370 | 1 |
| 1 | Biotin-Shc | 24000 | 65 | |
| 2 | Biotin-Grb2 | SA | 93 | 1 |
| 2 | Biotin-Shc | 9400 | 101 |
Coupling to the chip surface via amine linkage or streptavidin (SA) capture of biotinylated recombinant proteins.
As part of our target selectivity assessment of these SH2 antagonists, we compared their binding to the SH2 domains of Grb2 and Shc by SPR. The Shc SH2 domain recognizes the same general sequence motif as all known SH2 domains (pYXXX), but unlike Grb2 does not require asparagine (N) at the pY+2 position (pYXNX). While prior studies indicate that compounds such as 3 are unlikely to bind Src-like SH2 domains with high affinity, the potential for Shc SH2 domain binding is not easily predicted because its three dimensional structure differs significantly from those of the Src-like kinases and Grb2.18 For these SPR studies, the Grb2 and Shc SH2 domains were recombinantly engineered to contain a biotin tag at the encoded 5-prime end.19 Immobilization of these constructs on sensor chips coated with streptavidin (SA) provides uniform orientation on the chip surface and thereby less variability in ligand binding. The binding affinity of 2 for SA-immobilized biotin-Grb2 was in good agreement with the value obtained when Grb2 was immobilized by amine coupling (93 vs. 132 nM, respectively, Table 1). Of note, compound 2 displayed 100-fold lower binding affinity for biotin-Shc than for biotin-Grb2 (93 vs. 9400 nM, Table 1), indicating a high degree of Grb2 selectivity. Consistent with its overall similarity to compound 2, compound 1 also displayed dramatically lower binding affinity for the SH2 domain of Shc relative to that of Grb2 (370 vs. 24000 nM, Table 1).
Biotinylated 3 Retains Anti-motility Activity and is Highly Selective for Grb2
We have shown previously that the SH2 domain binding antagonist 1 blocks cell motility in a dose dependent fashion.11, 13 To confirm that biotin-containing 3 retains the biological activity of the parent compound 1 in intact cells, we performed cell migration studies using modified Boyden chambers in the presence or absence of the protein motility factor HGF (50 ng/ml). As shown in Figure 2A, 3 has anti-migratory activity comparable to the parent compound 1, resulting in significant and potent inhibition of migration by the prostate tumor-derived cell line PC3M. Detailed dose-response analysis of inhibition of migration by 3 yielded an estimated IC50 value of 14 nM (Figure 2B; R2=0.9, P < 0.01). No inhibition of cell migration was observed in response to treatment with the negative control analogue 4 at any concentration tested (data not shown).
Figure 2.

Inhibition of cell migration by 1 and 3. (A) Inhibition of HGF-stimulated (50 ng/mL; white bar), or unstimulated (black bar) PC3M cell migration by 1 and 3. Values represent mean number of migrated cells per 10× microscopic field, expressed as percent maximum, +/- standard deviation. (B) Dose-response analysis of inhibition of PC3M cell migration by 3.
In order to verify that 3 could efficiently capture Grb2 from detergent extracts of cultured cells, SA-coated beads were treated with biotin-containing 3 or 4 under conditions designed to saturate available SA sites. The compound-conjugated SA beads were then incubated with non-ionic detergent extracts prepared from cultured tumor cells and washed with buffers of increasing ionic strength (150 - 500 mM NaCl) to remove loosely-bound protein (monitored by silver staining; data not shown). The washes were analyzed for retention of Grb2 by SDS-PAGE and immunoblotting (Figure 3A). Grb2 was clearly retained on compound 3-conjugated SA beads, but in contrast, ligand 4 did not capture Grb2 under any conditions tested. We also confirmed the ability of 3 to capture proteins that are known to bind to the SH3 domains of Grb2: SDS-PAGE and immunoblot analysis of proteins captured by SA-bound 3 revealed the presence of Nck1, Nck2, Gab1 and Sos1 (Figure 3B).
Figure 3.
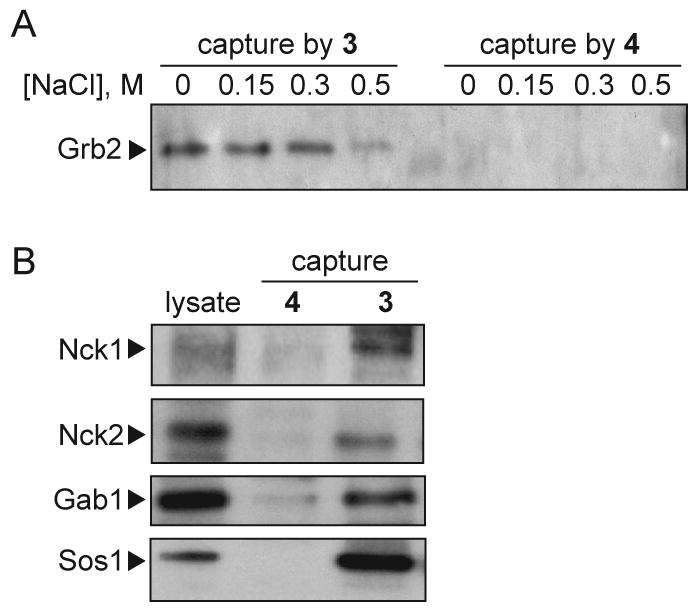
Protein capture by 3 and 4. (A) SDS PAGE and anti-Grb2 immunoblot analysis of lysates prepared from the human leiomyosarcoma cell line SK-LMS-1. Following target protein capture using 3 (left) or 4 (right), SA-coated beads were washed with buffer containing NaCl at the concentrations (M) indicated above each lane. (B) SDS-PAGE and immunoblot analysis of SK LMS-1 lysates (left lane) for known Grb2-associated proteins (top to bottom) Nck1, Nck2, Gab1 and Sos1 captured using 4 (middle lane) or 3 (right lane).
Proteins captured from cell extracts using compound 3-conjugated SA beads under conditions where Grb2 binding was detected by immunoblotting were subjected to exhaustive trypsin digestion; the resulting peptides were then separated chromatographically by HPLC and analyzed by tandem mass spectrometry (ESI-MS/MS) on a linear ion trap mass spectrometer. Representative ESI-MS/MS spectra of two [M+2H]2+ tryptic peptides from Grb2 (NH2-GACHGQTGMFPR-COOH and NH2-ATADDELSFK-COOH; NCBI sequence accession NM_203506.2) that were captured by compound 3-conjugated SA beads, but not by compound 4-conjugated beads, and which represent 10.1% sequence coverage, are shown in Figure 4. The b ion series is shown in red and the y ion series in blue. Statistical analysis using the Sequest algorithm yielded peptide correlation (Xcorr) scores of 3.707 (p=1.9e-9) (Figure 4A) and 2.98 (p=1.4e-5) (Figure 4B) for the two peptides. The difference in correlation between the top two peptide matches (ΔCn) were 0.44 and 0.28 for the spectra shown in Figure 4A and 4B, respectively; together with the peptide correlation scores, these values confirm robust protein identification. Importantly, Grb2 was the only SH2-domain containing protein detected by ESI-MS/MS among the proteins captured from cell lysates by 3 or the negative control 4. As shown in Table 2, five other proteins were captured differentially by 3 with Grb2; further research will be needed to determine whether their capture was mediated by Grb2 or by direct interaction with 3. Overall, these results confirm and extend our prior findings that 1 and closely related structures selectively block the Grb2 SH2 domain and not structurally distinct SH2 domains such as those present in Shc or phosphatidylinositol-3 kinase.11,13
Figure 4.

Representative MS/MS spectra of two [M+2H]2+ tryptic peptides from Grb2, which represent 10.1% sequence coverage, captured by 3-conjugated beads but not 4-conjugated beads. The b ion series is shown in red and the y ion series in blue. Sequest statistics: (A) Xcorr score 3.707, ΔCn=0.44 and p=1.9e-9; (B) Xcorr score = 2.98, ΔCn=0.28 and p=1.4e-5, where Xcorr is the peptide correlation score and ΔCn is the difference in correlation between the top two peptide matches.
Table 2.
Peptides identified by mass spectrometry analysis of samples captured from cell lysates by streptavidin-immobilized 3, but not by streptavidin-immobilized 4.
| Peptide Sequences Observed | NCBI Accession | Protein Name | Probability | XCorr score |
|---|---|---|---|---|
| K.LLDFGSLSNLQVTQPTVGMNFK.T | 4506693 | ribosomal protein S17 | 9.53E-12 | 48.31 |
| R.VCEEIAIIPSKK.L | ||||
| R.VIIEKYYTR.L | ||||
| K.IAGYVTHLMKR.I | ||||
| R.LGNDFHTNK.R | ||||
| K.IYVDDGLISLQVK.Q | 33286418, 33286420 | pyruvate kinase 3a | 1.10E-06 | 40.24 |
| R.NTGIICTIGPASR.S | ||||
| R.GDLGIEIPAEK.V | ||||
| R.VNFAMNVGK.A | ||||
| K.ATADDELSFK.R | 45359859 | Grb2 | 2.17E-06 | 20.83 |
| K.GACHGQTGMFPR.N | ||||
| K.AMGIMNSFVNDIFER.I | 4504257, 4504271 | histone H2B familyb | 6.51E-06 | 20.23 |
| R.KESYSIYVYK.V | ||||
| K.VANVSLLALYK.G | 4506701 | ribosomal protein S23 | 2.12E-07 | 20.20 |
| K.AHLGTALK.A | ||||
| K.SAEFLLHMLK.N | 4506617 | ribosomal protein L17 | 5.16E-05 | 10.16 |
| K.GLDVDSLVIEHIQVNK.A | ||||
Notes: Only those proteins for which two or more unique peptides were observed are shown. Probability is the p-value for the identification of the protein context of the peptide. XCorr score is the calculated correlation value for the protein based on the combined XCorr values of the peptides, obtained using the Sequest algorithm.
Unable to distinguish isoforms.
Unable to distinguish H2B family members.
Molecular and Cellular Mechanism of Action of 1
Having established the selectivity of 1 for the SH2 domain of Grb2, we further investigated its molecular mechanism of action by determining its effect on the activation state and subcellular localization of oncogenically relevant downstream signaling proteins. We focused on proteins most likely to be involved in the activities blocked by 1, namely, cell adhesion and motility. The p21-activated kinase 1 (PAK1) is an effector of Rac/Cdc42 GTPases that regulates cytoskeletal dynamics and is essential for cell migration. PAK1 binds directly to Grb2 SH3 domains, and its linkage to Grb2 is critical for PAK1 activation, autophosphorylation and lamellipodia formation downstream of growth factor-stimulated RTKs.20 We theorized that disrupting the SH2-mediated link between Grb2 and initiators of motility signaling, such as activated RTKs and integrin receptors, might also block PAK1 activation and autophosphorylation without affecting the SH3 domain-mediated PAK1-Grb2 association. We investigated the effect of 1 on growth factor-stimulated PAK1 activation by treating intact PC3M cells with 1 (1 uM) for 16 h (vs leaving them untreated), then stimulating the cells briefly with HGF and monitoring PAK1 activation by immunoblotting with a specific anti-phospho-PAK1 antibody (Figure 5A). Treatment with 1 had little effect on the steady state level of PAK1 autophosphorylation, which is not associated with cell shape change or motility (Figure 5A, left 2 lanes). In the absence of 1, HGF stimulation results in an initial decrease then increase in PAK1 autophosphorylation after 30 to 60 min, which is correlated with cell shape change and lamellipodia extension prior to increased motility (Figure 5A, middle lanes). This latter process of HGF-stimulated PAK1 activation, which is critical to motility, was completely inhibited by treatment with 1 (Figure 5A, right lanes).
Figure 5.
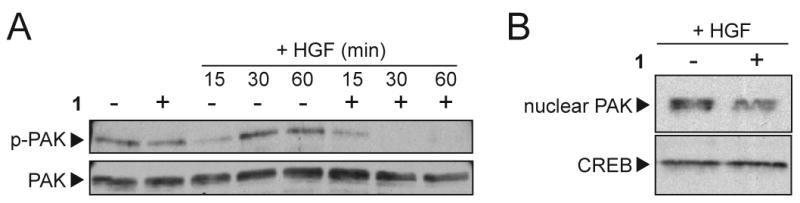
Compound 1 inhibits PAK1 activation and nuclear translocation. (A) PC3M cells were serum-deprived and treated with 1 (+;1 uM), or left untreated (-), as indicated for 16 h, then left without further treatment (left lanes) or stimulated briefly with HGF (50 ng/mL; right lanes) for the times indicated. Cell lysates were analyzed by SDS-PAGE and immunoblot analysis with anti-phospho-PAK1 antibody (upper panel), or anti-PAK1 antibody to control for protein loading (lower panel). (B) PC3M cells were treated with compound 1 for 16 h and nuclear extracts were prepared using high-salt buffer. Samples were analyzed by SDS-PAGE and immunoblot analysis with anti-PAK1 antibody (upper panel), or anti-CREB antibody (lower panel).
Growth factor-stimulated activation of PAK1 has been associated with nuclear translocation, histone protein phosphorylation and modulation of expression of several gene transcripts.21 Increased nuclear PAK1 activity has been reported in human renal cell carcinoma cells22 and in estrogen receptor-positive breast cancer 23, where it was correlated with tamoxifen resistance. We found that treatment of intact cells with 1 decreased PAK1 nuclear translocation (Figure 5B, upper panel) relative to nuclear cAMP response element-binding (CREB) protein (Figure 5B, lower panel), which is not modulated by growth factor stimulation.
Reorganization of the actin cytoskeleton is an early and critical step in lamellipodia formation prior to cell migration. We examined whether antagonism of Grb2 SH2 domain binding affected growth-factor stimulated actin reorganization and lamellipodia formation by staining cells with fluorescently labeled phalloidin, a toxin that interacts specifically with polymerized F-actin bundles. Lamellipodia formation is conveniently analyzed in monolayer cell cultures by directing their formation at an artificially created wound edge. To this end, confluent monolayers were scratched with a pipet tip, stimulated with HGF in the presence or absence of 1 for 6 h, then stained with fluorescently-tagged phalloidin. Because lamellipodia formation is cell-type dependent, we examined this process in the prostate tumor-derived cell line, PC3M and in the highly organized, normal epithelial cell line MDCK (Figure 6). HGF-stimulated PC3M cells displayed abundant pointed edges of advancing plasma membrane in the direction of cell migration (Figure 6, top left panel), while treatment with 1 markedly reduced phalloidin staining at the leading edge, indicative of a reduced actin bundle reorganization and membrane extension (Figure 6, bottom left panel). In contrast to PC3M cells, the actin cytoskeletal structure of MDCK cells is more uniformly organized (Figure 5, right panels). HGF stimulation produced the formation of well-structured advancing lamellipodia (Figure 6 top center and right panels). Treatment with 1 significantly reduced the area and distance that lamellipodia extended into the wound, consistent with inhibition of directional cell migration (Figure 6, bottom center and right panels).11, 13
Figure 6.
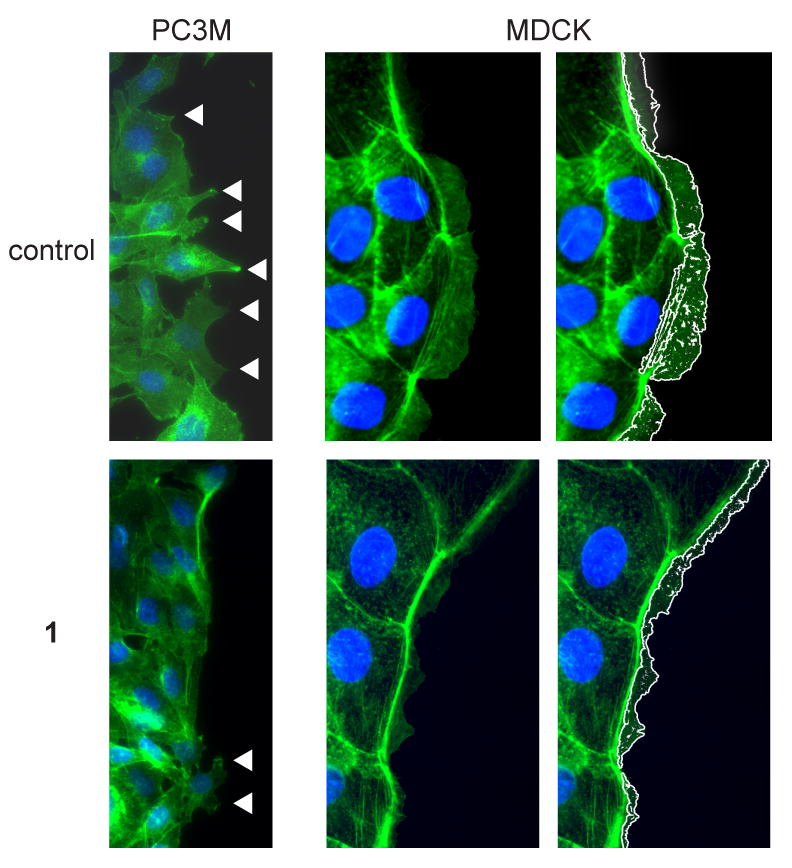
Compound 1 inhibits lamellipodia formation. Alexa 488-phalloidin (green) and DAPI staining (blue) of PC3M cells (left panels) and MDCK cells (center and right panels) treated with HGF alone (top panels, control) or HGF + 1 (1 uM; lower panels). For PC3M cells, arrowheads indicate regions of cellular extension into the artificial wound. For MDCK cells, advancing lamellipodia observed in center panels are outlined in white in the right panels for clarity.
Conclusions
We describe herein the synthesis and biochemical characterization of a biotinylated derivative of the Grb2-SH2 domain inhibitor 1 and used this as a tool to assess its SH2 domain binding selectivity. It is estimated that more than 100 different SH2 domains are present in the human genome 24; each of which recognizes phosphotyrosine (pTyr) in the context of 3-5 additional residues, usually carboxyl-terminal to pTyr 25. Compound 1 is a peptidomimetic that was originally designed to mimic the sequence Asn-pTyr-Val-Asn-Ile-Ile-Glu, where pTyr has been substituted with a hydrolytically stable 4-(2′-malonyl)-phenylalanine residue.17,26 Several studies have revealed the importance of Asn at the position Y+2 within this sequence for selective recognition by the SH2 domain of Grb2. In addition to design criteria, it is essential to identify bona fide intracellular targets for synthetic antagonists of protein-protein interactions. Prior co-immunoprecipitation experiments provided indirect evidence that Grb2 is the primary target of 1 and that 1 was able to disrupt Grb2-EGFR and Grb2-c-Met interactions.11, 27
As a more efficient and direct alternative to screening 1 for binding against all known SH2 domain containing proteins, we devised a capture strategy coupled with mass spectral analysis using a biotinylated analog of the Grb2 SH2 domain binding antagonist 1. This approach may be broadly useful to confirm the selectivity of small molecule protein binding within the growing field of targeted drug development. Biotin/streptavidin based capture combined with proteomic analysis should be a powerful tool for identifying adapter proteins along with their associated proteins.
The findings that Grb2 SH2 domain binding antagonists cause significant remodeling of the actin cytoskeleton and inhibit PAK1 further confirm the importance of Grb2 as a regulator of actin-based cell motility. Coincident with these effects, we have previously shown that treatment of cells with 1 disrupts HGF-stimulated physical association between Grb2 and focal adhesion kinase (FAK), thereby inhibiting focal adhesion formation.13 Together these results suggest that selective disruption of Grb2 SH2 domain-mediated signaling by a single agent may be a viable strategy to target several molecular events required for cell migration, which is critical to pathologic processes such as tumor invasion, tumor angiogenesis, and metastasis.
Experimental Section
Surface Plasmon Resonance Analysis of Drug-Protein Binding
Grb2 SH2 domain-binding experiments were performed on a Biacore T100 instrument (Biacore Inc., Piscataway NJ) using methods reported previously.28 All biotinylated Grb2 SH2 domain proteins were expressed and purified by the Protein Expression Laboratory and Protein Chemistry Laboratory, SAIC-Frederick. For the T100 analysis using biotin-capture surfaces, streptavidin (Neutravidin, Pierce catalog number 31000) dissolved in 10 mM sodium acetate, pH 4.5, was immobilized onto carboxymethyl 5′ dextran surface (CM5 sensor chip, Biacore Inc.) by amine coupling using the immobilization wizard supplied by the Biacore T100 software with a 5000 RU target on each of four flow cells. Lyophilized biotinylated Grb2 SH2 domain protein was reconstituted in 50% DMSO in H2O to make a stock solution of 1 mg/mL and stored at -80°C. A 20 ug aliquot of this solution was used for immobilization by diluting in 1XPBS (phosphate-buffered saline, pH 7.4), which was also used as the running buffer for Grb2 SH2 domain capture. A manual method for building a capture surface was used with a flow rate of 20 uL/min and a capture target of 2500 RU for flow cells 2 and 4. Synthetic inhibitors were serially diluted in running buffer to concentrations ranging from 1.25 nM to 1500 nM and serially injected at 25°C at a flow rate of 30 uL/min for 2 min. Each concentration was followed by two blank buffer injections, and every injection was performed in duplicate within each experiment. To subtract background noise from each data set, all samples were also run over Neutravidin reference surfaces to allow double referencing. Data were fit to a simple 1:1 interaction model using the global data analysis program Scrubber (http://www.biologic.com.au/).
Protein Capture and Immunoblotting
SK-LMS-1 cells were serum-deprived for 16 h, stimulated with recombinant human HGF (25 ng/ml; R&D Systems, Minneapolis, MN) for 20 min and lysed in cold non-ionic detergent containing buffer. Compound 3 (3 uM) or 4 (3 uM) was added and the lysate was incubated on ice for 1 h. SA-coated beads (Streptavidin on UltraLink Resin, Pierce Biotechnology, Rockford, IL) were added and lysates were mixed at 4°C for 1 h. SA-coated beads were then washed 5 times in lysis buffer containing increasing concentrations of NaCl (150 mM to 500 mM), then stored at -80°C in PBS. Samples were analyzed by SDS-PAGE and immunoblotting, or, in parallel, subjected to exhaustive tryptic digestion for mass spectrometry analysis. Immunoblotting was performed as described using an anti-Grb2 antibody (Santa Cruz Biotech).13
Mass Spectrometry
Proteins captured on SA-coated beads were washed three times with 100 mM ammonium bicarbonate to exchange the buffer, reduced with 10 mM DTT for 1 h at 51°C and alkylated with 55 mM iodoacetamide for 45 min at 25°C. The proteins were then digested with trypsin for 6 h at 25°C with agitation. The beads were pelleted and the supernatant was removed. Peptides in the supernatant were acidified by adding 0.1% formic acid. Separation of the peptides was performed at 500 nL/min and was coupled to online analysis by tandem mass spectrometry (nLC-ESI-MS/MS) on an LTQ ion trap mass spectrometer (ThermoElectron, San Jose, CA) equipped with a nanospray ion source. From the eluted peptides, 10 uL were loaded onto a 0.1 × 150 mm Magic C18AQ column (Michrom Bioresources, Inc, Auburn, CA) inline after a nanotrap column using the Paradigm MS4 MDLC (Michrom Bioresources, Inc., Auburn, CA). Elution of the peptides into the mass spectrometer was performed with a linear gradient from 95% mobile phase A (2% acetonitrile, 0.1% formic acid, 97.9% water) to 65% mobile phase B (10% water, 0.1% acetic acid, 89.9% acetonitrile) over 45 min and then to 95% mobile phase B in 5 min. The peptides were detected in positive ion mode using a data-dependent method in which the top seven ions detected in an initial survey scan were selected for MS/MS analysis. The MS/MS spectra were searched against the human NCBI database using TurboSEQUEST in BioWorks v. 3.2 (ThermoElectron, San Jose, CA).
Motility Assay
Cell migration was measured using modified Boyden chambers as described.11 PC3M cells were seeded at 200,000 cells per chamber after treatment with the respective compounds for 24 h. Cell migration was stimulated using 50 ng/ml recombinant human HGF (R&D Systems, Minneapolis, MN). Mean values from four fields observed by light microscopy (1 × 1.4 mm) were calculated for each of triplicate wells per condition.
Immunofluorescence Videomicroscopy
PC3M and MDCK cells plated in chamber slides were serum-deprived in the presence or absence of 1 (1 uM) for 24 h. Cells were then removed from a small portion of the monolayer using a pipet tip to simulate wounding and cells were incubated in the presence or absence of HGF (50 ng/ml) to stimulate motility prior to fixation and permeabilization. Slides were blocked with 5% bovine serum albumin in PBS for 1 h and then incubated overnight at 4°C with Alexa488-conjugated phalloidin (Molecular Probes). Slides were washed with PBS, nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI; Molecular Probes) and digital epifluorescence images were acquired using an inverted Olympus videomicroscope and IPLab software (Scanalytics).
Synthesis
Trifluoro-methanesulfonic acid 5-hydroxy-naphthalen-1-yl ester (6)
A solution of 5-dihydroxynaphthalene (10.18 g, 63.6 mmol), N-phenyltrifluoromethanesulfonimide (22.7 g, 63.6 mmol), 2,4,6-collidine (8.4 mL, 63.6 mmol) and DMAP (930 mg, 7.62 mmol) in CH2Cl2/DMF (240 mL, 19:5 v/v) was refluxed for 12 h. The solvent was evaporated in vacuo and the residue was dissolved in EtOAc, washed with 5% HCl solution, H2O, brine, and dried over Na2SO4. After evaporation of solvent, the remaining residue was purified by silica gel flash chromatography to give 6 as brown solid. (8.70 g, 47% yield). 1H NMR (DMSO-d6) δ 10.70 (s, 1H), 8.27 (d, 1H, J = 8.4 Hz), 7.63 (d, 1H, J = 7.2 Hz), 7.59-7.54 (m, 2H), 7.35 (d, 1H, J = 8.4 Hz), 7.06 (d, 1H, J = 7.4 Hz). MS (FAB) m/z 291 [M-H]-.
3-(5-Hydroxy-naphthalen-1-yl)-acrylonitrile (7)
To a solution of 6 (7.10 g, 24.2 mmol), acrylonitrile (3.3 mL, 50.1 mmol) and Et3N (7.5 mL, 50.1 mmol) was added Pd(OAc)2 (146 mg, 0.66 mmol) and DPPP (296 mg, 0.66 mmol). The mixture was refluxed under argon (24 h), then cooled to room temperature, diluted with EtOAc and filtered through celite. The filtrate was washed with saturated NH4Cl solution and brine, dried over Na2SO4. After evaporation of solvent, the remaining residue was purified by silica gel flash chromatography to provide 7 (2.33 g, 49% yield) as a yellow solid and 8 (1.52 g, 32% yield) as yellow oil. 1H NMR (CDCl3) δ 8.28 (d, 1H, J = 8.5 Hz), 8.13 (d, 1H, J = 16.4 Hz), 7.53 (d, 1H, J = 8.6 Hz), 7.43-7.33 (m, 3H), 6.82 (d, 1H, J = 7.5 Hz), 5.89 (d, 1H, J = 16.4 Hz). MS (FAB) m/z 195 (M)+; 196 (MH)+.
5-(3-Amino-propyl)-naphthalen-1-ol (9)
Compound 7 (2.62 g, 13.4 mmol) was dissolved in THF (300 mL). To the stirred solution was added LiAlH4 (1.09 g, 26.8 mmol) in several portions. The mixture was stirred at room temperature overnight. EtOAc (40 mL) was added followed by 10% NaOHaq (2 mL). The mixture was stirred for additional 1 h. After filtration, the filtrate was removed and the residue was purified by silica gel flash chromatography to provide 9 as a yellow oil (1.47 g, 54% yield). 1H NMR (CD3OD) δ 8.09 (dd, 1H, J = 3.2 Hz & 6.4 Hz), 7.51 (dd, 1H, J = 0.8 Hz & 8.7 Hz), 7.31-7.25 (m, 3H), 6.80 (dd, 1H, J = 0.9 Hz & 7.4 Hz), 3.05 (t, 2H, J = 7.8 Hz), 2.72 (t, 2H, J = 7.2 Hz). 1.92-1.85 (m, 2H); MS (FAB) m/z 202 (MH)+.
(S)-Tert-butyl 4-amino-1-(3-(5-hydroxynaphthalen-1-yl)propylamino)-1,4-dioxobutan-2-ylcarbamate (N-Boc Asn-9)
To a solution of 9 (874 mg, 4.34 mmol) in DMF (21 mL) was added an active ester solution prepared by the reaction of N-Boc-Asn-OH (1.00 g, 4.34 mmol), HOBt (586 mg, 4.34 mmol) and N,N′-diisopropylcarbodiimide (DIPCDI) (0.682 mL, 4.34 mmol) in DMF (14 mL) at room temperature for 10 minutes. The resulting solution was stirred at room temperature (12 h). Solvent was evaporated and the remaining residue was dissolved in EtOAc, washed with H2O, brine, dried over Na2SO4. After evaporation of solvent, the remaining residue was purified by silica gel flash chromatography to provide N-Boc Asn-9 (1.56 g, 87% yield) as yellow oil. 1H NMR (CD3OD) δ 8.08 (m, 1H), 7.52 (d, 1H, J = 8.5 Hz), 7.32-7.27 (m, 3H), 6.80 (d, 1H, J = 7.5 Hz), 4.42 (m, 1H), 3.32-3.26 (m, 2H), 3.04 (t, 2H, J = 7.8 Hz), 2.68-2.61 (m, 2H), 1.91 (t, 2H, J =7.4 Hz), 1.43 (s, 9H). MS (FAB) m/z 416 (MH)+. HRMS cacld for C22H30N3O5 (M+H)+, 416.2185; found, 416.2183.
(2S)-2-Amino-N1-[3-(5-Hydroxy-naphthalen-1-yl)-propyl]-succinamide (N-Asn-9)
N-Boc Asn-9 (1.56 g, 3.76 mmol) was dissolved in TFA-CH2Cl2 (8 mL, 1:1 v/v) and the resulting solution was stirred at room temperature for 1 h. After evaporation of solvent, the remaining residue was purified by silica gel flash chromatography to provide N-Asn-9 as a yellow oil. (936 mg, 79% yield). 1H NMR (CD3OD) δ 8.08 (dd, 1H, J = 3.2 Hz & 6.6 Hz), 7.53 (d, 1H, J = 8.5 Hz), 7.32-7.27 (m, 3H), 6.80 (dd, 1H, J = 0.6 Hz & 7.5 Hz), 3.70 (dd, 1H, J = 5.0 Hz & 8.1 Hz), 3.15-3.05 (m, 3H), 2.64 (dd, 1H, J = 5.0 Hz & 15.4 Hz), 2.48 (dd, 1H, J = 8.1 Hz & 15.4 Hz), 1.98-1.91 (m, 2H). MS (FAB) m/z 316 (MH)+. HRMS calculated for C17H22N3O3 (M+H)+, 316.1675; found, 316.1661.
(S)-(9H-Fluoren-9-yl)methyl 1-(4-amino-1-(3-(5-hydroxynaphthalen-1-yl)propylamino)-1,4-dioxobutan-2-ylcarbamoyl)cyclohexylcarbamate (N-Fmoc 10)
N-Asn-9 (901 mg, 2.86 mmol), N-Fmoc-1-amino-cyclohexenecarboxylic acid (1.043 g, 2.86 mmol), HOBt (390 mg, 2.86 mmol) and DIPCDI (0.451 mL, 2.86 mmol) were treated as the preparative procedure of N-Boc Asn-9 to provide N-Fmoc-protected 10 as a yellow solid (1.49 g, 79% yield). 1H NMR (CD3OD) δ 8.30 (d, 1 H, J = 8.1 Hz), 8.05 (dd, 1H, J = 2.0 Hz & 7.7 Hz), 7.82 (m, 1H). 7.74 (dd, 1 H, J = 3.3 & 7.5 Hz), 7.50-7.42 (m, 3H), 7.36-7.31 (m, 3 H), 7.26-7.18 (m, 5H), 6.78 (dd, 1H, J = 0.7 Hz & 7.5 Hz), 4.62 (m, 1H), 4.34 (dd, 1H, J = 6.5 Hz & 10.6 Hz), 4.22 (dd, 1H, J = 6.2 Hz & 10.7 Hz), 4.05 (t, 1H, J = 6.1 Hz), 3.27-3.21 (m, 2H), 3.00 (t, 2H, J = 7.8 Hz), 2.86 (dd, 1H, J = 6.7 Hz & 15.5 Hz), 2.71 (dd, 1H, J = 4.7 Hz & 15.7 Hz), 1.96-1.83 (m, 5H), 1.75-1.27 (m, 7H). MS (FAB) m/z 663 (MH)+. HRMS cacld for C39H42N4O6Na (M+Na)+, 685.2997; found, 685.3012.
(2S)-2-[(1-Amino-cyclohexanecarbonyl)-amino]-N1-[3-(5-Hydroxy-naphthalen-1-yl)-propyl]-succinamide (10)
To a solution of N-Fmoc-protected 10 (1.49 g, 2.25 mmol) in CH3CN (30 mL) was added piperidine (3 mL) and the resulting solution was stirred at room temperature for 2 h. After evaporation of solvent, the remaining residue was purified by silica gel flash chromatography to provide 10 as a yellow solid (847 mg, 86% yield). 1H NMR (CD3OD) δ 8.07 (dd, 1H, J = 3.1 Hz & 6.3 Hz), 7.52 (d, 1H, J = 8.6 Hz), 7.34-7.27 (m, 3 H), 6.80 (dd, 1H, J = 0.7 Hz & 7.5 Hz), 4.65 (dd, 1H, J = 5.9 Hz & 6.6 Hz), 3.30-3.27 (m, 2H), 3.07-3.03 (m, 2H), 2.73 (dd, 1H, J = 6.7 Hz & 15.4 Hz), 2.66 (dd, 1H, J = 5.9 Hz & 15.4 Hz), 1.96-1.81 (m, 4H), 1.63-1.53 (m, 5H), 1.46-1.27 (m, 3H). MS (FAB) m/z 441 (MH)+. HRMS cacld for C24H33N4O4 (M+H)+, 441.2502; found, 441.2505.
2-{4-[2-(1-(1S)-{2-Carbamoyl-1- [3-(5-hydroxy-naphthalen-1-yl)-propylcarbamoyl] -ethylcarbamoyl} -cyclohexylcarbamoyl)-2-(2S)-(9H-fluoren-9-ylmethoxycarbonyl amino)-ethyl]-phenyl}-malonic acid di-tert-butyl ester (12a)
Compound 10 (603 mg, 1.37 mmol), compound 11a (826 mg, 1.37 mmol), HOBt (185 mg, 1.37 mmol) and DIPCDI (0.215 mL, 1.37 mmol) were treated as the preparative procedure of N-Boc Asn-9 to provide 12a as a yellow solid (1.058 g, 79% effective yield based on 247 mg of recovered 10). 1H NMR (CD3OD) δ 8.18 (d, 1H, J = 7.8 Hz), 8.05 (d, 1H, J = 9.1 Hz), 7.82-7.75 (m, 3H), 7.56-7.51 (m, 3H), 7.36-7.18 (m, 9H), 7.77 (d, 1H, J = 7.5 Hz), 4.52 (m, 1H), 4.45 (s, 1H), 4.42 (m, 1H), 4.32-4.21 (m, 2H), 4.08 (t, 1H, J = 6.7 Hz), 3.27-3.21 (m, 2H), 3.07-3.02 (m, 3H), 2.93-2.84 (m, 2H), 2.79 (dd, 1H, J = 5.5 Hz & 14.8 Hz), 1.99-1.92 (m, 4H), 1.77-1.70 (m, 2H), 1.44 (s, 18H), 1.42-1.24 (m, 4H). MS (FAB) m/z 1024.5 (MH)+. HRMS cacld for C59H69N5O11Na (M+Na)+, 1046.4886; found, 1046.4886.
[2-(4-tert-Butoxy-phenyl)-1-(1S)-{2-Carbamoyl-1-[3-(5-hydroxy-naphthalen-1-yl)-propylcarbamoyl]-ethylcarbamoyl}-cyclohexylcarbamoyl)-ethyl]-carbamic acid 9H-fluoren-9-ylmethyl ester (12b)
Compound 10 (185 mg, 0.42 mmol), compound 11b (230 mg, 0.50 mmol), HOBt (68 mg, 0.50 mmol) and DIPCDI (0.079 mL, 0.50 mmol) were treated as the preparative procedure of N-Boc Asn-9 to provide 12b as a yellow solid (274 mg, 74% yield). 1H NMR (CDCl3) δ 8.02-7.95 (m, 2H), 7.69-7.60 (m, 2H), 7.42-7.38 (m, 2H), 7.32-7.28 (m, 2H), 7.21-7.17 (m, 6H), 7.00 (t, 1H, J = 7.5 Hz), 6.90-6.86 (m, 2H), 6.79-6.76 (m, 2H), 6.67 (d, 1H, J = 7.3 Hz), 4.60 (m, 1H), 4.30-4.25 (m, 3H), 4.03 (m, 1H), 3.32-3.21 (m, 2H), 2.98-2.89 (m, 2H), 2.74 (m, 1H), 2.48 (m, 1H), 2.02-1.06 (m, 14H), 1.22 (s, 9H). MS (FAB) m/z 882 (MH)+.
2-{4-[2-(2S)-(tert-Butoxyoxalyl-amino)-2-(1-(1S)-{2-carbamoyl-1-[3-(5-hydroxy-naphthalen-1-yl)-propylcarbamoyl]-ethylcarbamoyl}-cyclohexylcarbamoyl)-ethyl]-phenyl}-malonic acid di-tert-butyl ester (13a)
Compound 12a (609 mg, 0.595 mmol) and piperidine (0.47 mL) were treated as preparative procedure of 10 to provide deprotected-Fmoc 12a (477 mg). To a solution of this crude material (477 mg, 0.595 mmol) in DMF (24 mL) was added iPr2NEt (0.124 mL, 0.714 mmol) and tButyloxalyl (0.091 mL, 0.714 mmol) at 0°C. The solution was stirred at room temperature overnight. After evaporation of solvent, the remaining residue was purified by silica gel flash chromatography to provide 13a as a yellow oil (240 mg, 43% yield). 1H NMR (CDCl3) δ 8.05-8.01 (m, 2H), 7.65-7.61 (m, 2H), 7.46 (d, 1H, J = 8.6 Hz), 7.27-7.25 (m, 4H), 7.08 (t, 1H, J = 8.0 Hz), 7.04-7.01 (m, 2H), 6.74 (s, 1H), 6.62 (d, 1H, J = 7.3 Hz), 6.46 (s, 1H), 5.58 (s, 1H), 4.67 (m, 1H), 4.56 (m, 1H), 4.41 (s, 1H), 3.39 (m, 1H), 3.28 (m, 1H), 3.05-2.95 (m, 4H), 2.85 (dd, 1H, J = 8.2 Hz & 14.3 Hz), 2.46 (dd, 1H, J = 4.9 Hz & 15.0 Hz), 2.00-1.62 (m, 8H), 1.47 (s, 9H), 1.46 (s, 9H), 1.45 (s, 9H), 1.44-1.24 (m, 4H). MS (FAB) m/z 930 (MH)+.
N-[2-(4-tert-Butoxy-phenyl)-(1-(1S)- {2-carbamoyl-1-[3-(5-hydroxy-naphthalen-1-yl)-propylcarbamoyl] -ethylcarbamoyl} -cyclohexylcarbamoyl)-ethyl]-oxalamic acid tert-butyl ester (13b)
Compound 12b (258 mg, 0.293 mmol) were treated as preparative procedure of 13a to provide 13b as a yellow oil (174 mg, 80% yield). 1H NMR (CD3OD) δ 8.03 (d, 1H, J = 8.2 Hz), 7.52 (d, 1H, J = 8.5 Hz), 7.32-7.21 (m, 3H), 7.05-7.02 (m, 2H), 6.85-6.81 (m, 2H), 6.75 (dd, 1H, J = 0.7 Hz & 7.5 Hz), 4.63 (dd, 1H, J = 6.9 Hz & 8.5 Hz), 4.52 (dd, 1H, J = 5.0 Hz & 7.2 Hz), 3.37-3.21 (m, 2H), 3.11-3.03 (m, 3H), 2.91 (dd, 1H, J = 8.6 Hz & 14.0 Hz), 2.86 (dd, 1H, J = 7.2 Hz & 15.4 Hz), 2.71 (dd, 1H, J = 5.0 Hz & 15.5 Hz), 1.98-1.18 (m, 12H), 1.42 (s, 9H), 1.26 (s, 9H). MS (FAB) m/z 788 (MH)+.
2-{4-[2-(1-(1S)-{2-Carbamoyl-1-[3-(5-hydroxy-naphthalen-1-yl)-propylcarbamoyl]-ethylcarbamoyl}-cyclohexylcarbamoyl)-2-(2S)-(oxalyl-amino)-ethyl]-phenyl}-malonic acid (2)
A solution of 13a (110 mg, 0.118 mmol) in a mixture of TFA-TES-H2O (2.0 mL, 3.7:0.1:0.2 v/v) was stirred at room temperature for 1 h. After evaporation of solvent, the remaining residue was purified by HPLC as follows: A linear gradient over 25 minutes of from 0.1% TFA in 5% CH3CN : 95% H2O to 0.1% TFA in 95% CH3CN : 5% H2O; Analytical retention time = 24.4 min; Preparative retention time = 14.1 min. Lyophilization provided 2 as a yellow solid (38 mg, 42% yield) in purity 97.6% as determined by HPLC. 1H NMR (d6-DMSO) δ 9.92 (s, 1H), 8.73 (d, 1H, J = 7.8 Hz), 8.22 (s, 1H), 7.93-7.89 (m, 2H), 7.43-7.40 (m, 2H), 7.29-7.15 (m, 9H), 6.83 (s, 1H), 6.77 (d, 1H, J = 7.3 Hz), 4.66 (m, 1H), 4.52 (s, 1H), 4.30 (m, 1H), 3.12-3.08 (m, 3H), 2.95-2.89 (m, 3H), 2.61 (dd, 1H, J = 6.5 Hz & 15.5 Hz), 2.43 (dd, 1H, J = 7.0 Hz & 15.5 Hz), 1.91-1.08 (m, 12H). MS (FAB) m/z 761 (M)+, 760 (M-H)-. HRMS cacld for C38H43N5O12Na (M-2Na+3H)+, 784.2800; found, 784.2810.
(4R,5S,6S)-2-{4-[2-(1-{2-Carbamoyl-1-[3-(3S)-(5-{6-[5-(2-oxo-hexahydro-thieno[3,4-d]imidazol-6-yl)-pentanoylamino]-hexanoyloxy}-naphthalen-1-yl)-propylcarbamoyl}-cyclohexylcarbamoyl)-2-(2S)-(oxalyl-amino)-ethyl]-phenyl}-malonic acid tri-tert-butyl ester (14a)
To a solution of 13a (119 mg, 0.13 mmol) and 6-biotinamidocaproic acid (94 mg, 0.26 mmol) in DMF (7 mL) was added EDC (50 mg, 0.26 mmol) and DMAP (2.4 mg, 0.02 mmol) at 0°C and the resulting solution was stirred for at room temperature for 12 h. The solvent was evaporated and the residue was dissolved in EtOAc, washed with H2O, brine, and dried over Na2SO4. After evaporation of the solvent, the remaining residue was purified by silica gel flash chromatography to provide 14a as a white solid (127 mg, 78% yield). 1H NMR (CD3OD) δ 7.71 (dd, 1H, J = 1.5 Hz & 7.6 Hz), 7.50-7.39 (m, 4H), 7.29-7.19 (m, 5H), 4.72 (dd, 1H, J = 6.7 Hz & 8.5 Hz), 4.57 (dd, 1H, J = 5.0 Hz & 7.1 Hz), 4.47 (s, 1H), 4.46 (m, 1H), 4.32 (dd, 1H, J = 4.8 Hz & 7.7 Hz), 4.27 (dd, 1H, J = 4.4 Hz & 7.8 Hz), 4.13 (dd, 1H, J = 4.4 Hz & 7.9 Hz), 3.35-3.32 (m, 2H), 3.23-3.05 (m, 5H), 2.93-2.77 (m, 5H), 2.70-2.62 (m, 3H), 2.21-2.17 (m, 3H), 2.03-1.94 (m, 3H), 1.86-1.48 (m, 24H), 1.45 (s, 9H), 1.44 (s, 18H). MS (FAB) m/z 1269 (MH)+.
(4R,5S,6S)-6-[5-(2-Oxo-hexahydro-thieno[3,4-d]imidazol-6-yl)-pentanoylamino]-hexanoic acid 5-{3-{2-({1-2(tert-butoxyoxylyl-amino)-3-(4-tert-butoxy-phenyl)-propionylamino]-cyclohexylcarbonyl}-amino)-3-carbamoyl-propionylamino]-propyl}-naphthalen-1-yl ester (14b)
Compound 13b (80 mg, 0.10 mmol) were treated as preparative procedure of 14a to provide 14b as a white solid (9 mg, 42% effective yield based on 65 mg recovered 13b). 1H NMR (CD3OD) δ 8.04 (d, 1H, J = 8.6 Hz), 7.71 (dd, 1H, J = 1.7 Hz & 7.5 Hz), 7.51-7.40 (m, 3H), 7.19 (d, 1H, J = 7.0 Hz), 7.11-7.08 (m, 2H), 6.89-6.85 (m, 2H), 4.68 (dd, 1H, J = 6.8 Hz & 8.6 Hz), 4.56 (dd, 1H, J = 5.0 Hz & 7.1 Hz), 4.32 (dd, 1H, J = 4.7 Hz & 8.2 Hz), 4.13 (dd, 1H, J = 4.5 Hz & 7.8 Hz), 3.40-3.21 (m, 4H), 3.17-3.10 (m, 4H), 2.99-2.61 (m, 7H), 2.18 (t, 2H, J = 7.3 Hz), 2.02-1.24 (m, 24H), 1.45 (s, 9H), 1.30 (s, 9H). MS (FAB) m/z 1127 (MH)+.
(4R,5S,6S)-2-{4-[2-(1-{2-Carbamoyl-1-[3-(3S)-(5-{6-[5-(2-oxo-hexahydro-thieno[3,4-d]imidazol-6-yl)-pentanoylamino]-hexanoyloxy}-naphthalen-1-yl)-propylcarbamoyl}-cyclohexylcarbamoyl)-2-(2S)-(oxalyl-amino)-ethyl]-phenyl}-malonic acid (3)
A solution of 14a (127 mg, 0.1 mmol) in a mixture of TFA-ethanedithol-H2O (1.0 mL, 3.8:0.1:0.1 v/v) was stirred at room temperature for 1 h. The mixture was reduced under vacuum to a volume of 0.25 mL. Diethyl ether (10 mL) was added giving a white solid. The solid was collected and purified by HPLC as follows: A linear gradient over 25 minutes of from 0.1% TFA in 5% CH3CN : 95% H2O to 0.1% TFA in 95% CH3CN : 5% H2O; Analytical retention time = 24.6 min; Preparative retention time = 16.6 min. Liophilization provided 3 as a white solid (44.7 mg, 41% yield) in 99% purity as determined by HPLC. 1H NMR (DMSO-d6) δ 8.75 (d, 1H, J = 7.8 Hz), 8.24 (s, 1H), 7.93 (t, 2H, J = 8.2 Hz), 7.71 (t, 1H, J = 5.6 Hz), 7.63 (t, 1H, J = 4.8 Hz), 7.45-7.41 (m, 2H), 7.37-7.35 (m, 2H), 7.30 (s, 1H), 7.20-7.15 (m, 5H), 6.84 (s, 1H), 6.34 (s, 1H), 4.66 (m, 1H), 4.52 (m, 1H), 4.31 (m, 1H), 4.21 (dd, 1H, J = 5.0 Hz & 7.7 Hz), 4.04 (dd, 1H, J = 4.4 Hz & 7.7 Hz), 3.13-3.07 (m, 3H), 3.04-2.93 (m, 6H), 2.75-2.69 (m, 3H), 2.62 (dd, 1H, J = 6.2 Hz & 15.5 Hz), 2.51-2.45 (m, 2H), 1.99 (t, 2H, J = 7.4 Hz), 1.92-1.08 (m, 24H). MS (FAB) m/z 1100 (M)+, 1099 (M-H)-. HRMS cacld for C54H65N8O15Na3S (M-3Na+4H)+, 1101.4598; found, 1101.4502.
(4R,5S,6S)-6-[5-(2-Oxo-hexahydro-thieno[3,4-d]imidazol-6-yl)-pentanoylamino]-hexanoic acid 5-{3-[3-carbamoyl-2-({1-[3-(4-hydroxy-phenyl)-2-(oxylyl-amino)-propionylamino]-cyclohexylcarbonyl}-amino)-propionylamino]-propyl}-naphthalen-1-yl ester (4)
Compound 14b (9.0 mg) were treated as preparative procedure of 14a to provide 4 as a white solid. (4.5 mg, 56% yield). Analytical retention time = 26.0 min; Preparative retention time = 18.1 min (HPLC conditions as indicated for 14a). 1H NMR (DMSO-d6) δ 8.58 (d, 1H, J = 7.8 Hz), 8.19 (s, 1H), 7.95 (d, 1H, J = 8.8 Hz), 7.88 (d, 1H, J = 7.6 Hz), 7.71 (m, 1H), 7.64 (m, 1H), 7.45 (d, 2H, J = 7.3 Hz), 7.40-7.36 (m, 2H), 7.30 (s, 1H), 7.19 (d, 1H, J = 7.4 Hz), 6.95 (d, 2H, J = 7.4 Hz), 6.83 (s, 1H), 6.55 (d, 2H, J = 7.0 Hz), 6.38-6.32 (m, 2H), 4.56 (m, 1H), 4.31 (m, 1H), 4.21 (m, 1H), 4.04 (dd, 1H, J = 4.6 Hz & 7.5 Hz), 3.16-2.96 (m, 9H), 2.81-2.69 (m, 3H), 2.60 (dd, 1H, J = 6.7 Hz & 15.5 Hz), 2.61-2.56 (m, 2H), 2.01-1.10 (m, 26H). MS (FAB) m/z 1013 (M-H)-.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. This project has been funded in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract N01-CO-12400. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
List of Abbreviations
- Grb2
Growth factor receptor-bound protein 2
- SH2
Src homology 2
- RTK
Receptor tyrosine kinase
- EGFR
Epidermal growth factor receptor
- HGF
Hepatocyte growth factor
- PDGFR
Platelet-derived growth factor receptor
- FAK
Focal adhesion kinase
- Bcr
Breakpoint cluster region
- Abl
Abelson murine leukemia viral oncogene
- SH3
Src homology 3
- Ras
Rat sarcoma virus oncogene
- Erk
Extracellular signal-regulated kinase
- PAK1
p21-activating kinase
- Arp2/3
Actin related protein 2/3
- WASp
Wiskott-Aldrich syndrome protein
- EMT
Epithelial-mesenchymal transition
- SPR
Surface plasmon resonance
- SA
Streptavidin
- Nck
Non-catalytic region of tyrosine kinase adaptor protein
- Gab1
Growth factor receptor protein 2-associated protein 1
- SOS1
Son of sevenless homolog 1
- ESI-MS/MS
Electrospray ionization tandem mass spectrometry
- Rac
Ras-related C3 botulinum toxin substrate
- CDC42
Cell division cycle 42
- CREB
cAMP Response element-binding
- MDCK
Madin-Darby canine kidney cell
- DAPI
4′,6-diamidino-2-phenylindole
- DPPP
1,3-bis(diphenylphosphine)propane
- EDC
1-Ethyl-3-(3-Dimethylaminopropyl)carbodiimide
- HOBt
1-Hydroxybenzotriazole
- DMAP
4-Dimethylaminopyridine
- DMF
Dimethylformamide
- THF
Tetrahydrofuran
- DIPCDI
Diisopropylcarbodiimide
- TFA-TES
Trifluoroacetic acid-N-tris(hydroxymethyl)methyl-2-amino-ethanesulfonic acid
References
- 1.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 2.Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullrich A, Skolnik EY, Bar-Sagi D, Schlessinger J. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992;70:431–442. doi: 10.1016/0092-8674(92)90167-b. [DOI] [PubMed] [Google Scholar]
- 3.Songyang Z, Shoelson SE, McGlade J, Olivier P, Pawson T, Bustelo XR, Barbacid M, Sabe H, Hanafusa H, Yi T. Specific motifs recognized by the SH2 domains of Csk, 3BP2, fps/fes, GRB-2, HCP, SHC, Syk, and Vav. Mol Cell Biol. 1994;14:2777–2785. doi: 10.1128/mcb.14.4.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tari AM, Lopez-Berestein G. GRB2: A pivotal protein in signal transduction. Seminars in Oncology. 2001;28:142–147. doi: 10.1016/s0093-7754(01)90291-x. [DOI] [PubMed] [Google Scholar]
- 5.Kumar R, Gururaj AE, Barnes CJ. p21-activated kinases in cancer. Nat Rev Cancer. 2006;6:459–471. doi: 10.1038/nrc1892. [DOI] [PubMed] [Google Scholar]
- 6.Schlaepfer DD, Hanks SK, Hunter T, Van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 7.Mitra SK, Mikolon D, Molina JE, Hsia DA, Hanson DA, Chi A, Lim ST, Bernard-Trifilo JA, Ilic D, Stupack DG, Cheresh DA, Schlaepfer DD. Intrinsic FAK activity and Y925 phosphorylation facilitate an angiogenic switch in tumors. Oncogene. 2006;25:5969–5984. doi: 10.1038/sj.onc.1209588. [DOI] [PubMed] [Google Scholar]
- 8.McLean GW, Carragher NO, Avizienyte E, Evans J, Brunton VG, Frame MC. The role of focal-adhesion kinase in cancer [mdash] a new therapeutic opportunity. Nat Rev Cancer. 2005;5:505–515. doi: 10.1038/nrc1647. [DOI] [PubMed] [Google Scholar]
- 9.Dharmawardana PG, Peruzzi B, Giubellino A, Burke J, Bottaro DP. Molecular targeting of growth factor receptor-bound 2 (Grb2) as an anti-cancer strategy. Anti-Cancer Drugs. 2006;17:13–20. doi: 10.1097/01.cad.0000185180.72604.ac. [DOI] [PubMed] [Google Scholar]
- 10.Burke TR. Development of Grb2 SH2 domain signaling antagonists: A potential new class of antiproliferative agents. International Journal of Peptide Research and Therapeutics. 2006;12:33–48. doi: 10.1007/s10989-006-9014-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atabey N, Gao Y, Yao ZJ, Breckenridge D, Soon L, Soriano JV, Burke TR, Bottaro DP. Potent Blockade of Hepatocyte Growth Factor-stimulated Cell Motility, Matrix Invasion and Branching Morphogenesis by Antagonists of Grb2 Src Homology 2 Domain Interactions. J Biol Chem. 2001;276:14308–14314. doi: 10.1074/jbc.M010202200. [DOI] [PubMed] [Google Scholar]
- 12.Soriano JV, Liu N, Gao Y, Yao ZY, Ishibashi T, Underhill C, Burke TR, Bottaro DP. Inhibition of angiogenesis by growth factor receptor bound protein 2-Src homology 2 domain binding antagonists. Molecular Cancer Therapeutics. 2004;3:1289–1299. [PubMed] [Google Scholar]
- 13.Giubellino A, Gao Y, Lee S, Lee MJ, Vasselli JR, Medepalli S, Trepel JB, Burke TR, Bottaro DP. Inhibition of tumor metastasis by a growth factor receptor bound protein 2 Src homology 2 domain-binding antagonist. Cancer Res. 2007;67:6012–6016. doi: 10.1158/0008-5472.CAN-07-0022. [DOI] [PubMed] [Google Scholar]
- 14.Shi ZD, Liu H, Zhang M, Worthy KM, Bindu L, Yang D, Fisher RJ, Burke TR. Synthesis of a C-terminally biotinylated macrocyclic peptide mimetic exhibiting high Grb2 SH2 domain-binding affinity. Bioorganic & Medicinal Chemistry. 2005;13:4200–4208. doi: 10.1016/j.bmc.2005.04.028. [DOI] [PubMed] [Google Scholar]
- 15.Sasaki H, Irie R, Hamada T, Suzuki K, Katsuki T. Rational design of Mn-salen catalyst (2): Highly enantioselective epoxidation of conjugated cis-olefins. Tetrahedron. 1994;50:11827–11838. [Google Scholar]
- 16.Cabri W, Candiani I, Bedeschi A, Santi R. 1,10-Phenanthroline derivatives: A new ligand class in the Heck reaction. Mechanistic aspects. Journal of Organic Chemistry. 1993;58:7421–7426. [Google Scholar]
- 17.Gao Y, Burke TR. Stereoselective preparation of L-4-(2′-malonyl)phenylalanine suitably protected for Fmoc-based synthesis of potent signal transduction inhibitory ligands. Synlett. 2000:134–136. [Google Scholar]
- 18.Mikol V, Baumann G, Zurini MGM, Hommel U. Crystal structure of the SH2 domain from the adaptor protein SHC: A model for peptide binding based on X-ray and NMR data. Journal of Molecular Biology. 1995;254:86–95. doi: 10.1006/jmbi.1995.0601. [DOI] [PubMed] [Google Scholar]
- 19.Oishi S, Karki RG, Shi ZD, Worthy KM, Bindu L, Chertov O, Esposito D, Frank P, Gillette WK, Maderia M, Hartley J, Nicklaus MC, Barchi J, Fisher RJ, Burke TR. Evaluation of macrocyclic Grb2 SH2 domain-binding peptide mimetics prepared by ring-closing metathesis of C-terminal allylglycines with an N-terminal beta-vinyl-substituted phosphotyrosyl mimetic. Bioorganic and Medicinal Chemistry. 2005;13:2431–2438. doi: 10.1016/j.bmc.2005.01.052. [DOI] [PubMed] [Google Scholar]
- 20.Puto LA, Pestonjamasp K, King CC, Bokoch GM. p21-activated Kinase 1 (PAK1) Interacts with the Grb2 Adapter Protein to Couple to Growth Factor Signaling. J Biol Chem. 2003;278:9388–9393. doi: 10.1074/jbc.M208414200. [DOI] [PubMed] [Google Scholar]
- 21.Singh RR, Song C, Yang Z, Kumar R. Nuclear localization and chromatin targets of p21-activated kinase 1. J Biol Chem. 2005;280:18130–18137. doi: 10.1074/jbc.M412607200. [DOI] [PubMed] [Google Scholar]
- 22.O'Sullivan GC, Tangney M, Casey G, Ambrose M, Houston A, Barry OP. Modulation of p21-activated kinase 1 alters the behavior of renal cell carcinoma. International Journal of Cancer. 2007;121:1930–1940. doi: 10.1002/ijc.22893. [DOI] [PubMed] [Google Scholar]
- 23.Rayala SK, Kumar R. Sliding p21-activated kinase 1 to nucleus impacts tamoxifen sensitivity. Biomedicine and Pharmacotherapy. 2007;61:408–411. doi: 10.1016/j.biopha.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 24.Machida K, Mayer BJ. The SH2 domain: versatile signaling module and pharmaceutical target. Biochimica et Biophysica Acta (BBA) - Proteins & Proteomics. 2005;1747:1–25. doi: 10.1016/j.bbapap.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 25.Songyang Z, Shoelson SE, Chaudhuri M, Gish G, Pawson T, Haser WG, King F, Roberts T, Ratnofsky S, Lechleider RJ, Neel BG, Birge RB, Fajardo JE, Chou MM, Hanafusa H, Schaffhausen B, Cantley LC. SH2 domains recognize specific phosphopeptide sequences. Cell. 1993;72:767–778. doi: 10.1016/0092-8674(93)90404-e. [DOI] [PubMed] [Google Scholar]
- 26.Gao Y, Luo J, Yao ZJ, Guo R, Zou H, Kelley J, Voigt JH, Yang D, Burke TR. Inhibition of Grb2 SH2 domain binding by non-phosphate-containing ligands 2. 4-(2-Malonyl)phenylalanine as a potent phosphotyrosyl mimetic. Journal of Medicinal Chemistry. 2000;43:911–920. doi: 10.1021/jm9904248. [DOI] [PubMed] [Google Scholar]
- 27.Gay B, Suarez S, Caravatti G, Furet P, Meyer T, Schoepfer J. Selective Grb2 SH2 inhibitors as anti-Ras therapy. International Journal of Cancer. 1999;83:235–241. doi: 10.1002/(sici)1097-0215(19991008)83:2<235::aid-ijc15>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 28.Oishi S, Shi ZD, Worthy KM, Bindu LK, Fisher RJ, Burke TR. Ring-closing metathesis of C-terminal allylglycine residues with an N-terminal ?-vinyl-substituted phosphotyrosyl mimetic as an approach to novel Grb2 SH2 domain-binding macrocyles. ChemBioChem. 2005;6:668–674. doi: 10.1002/cbic.200400298. [DOI] [PubMed] [Google Scholar]