

Abstract
Introduction
Cerebral vasospasm after subarachnoid hemorrhage (SAH) is a serious complication resulting in delayed neurological deficit, increased morbidity, mortality, longer hospital stays, and rehabilitation time. It afflicts approximately 35 per 100,000 Americans per year, and there is currently no effective therapy. We present in vitro data suggesting that increasing intrinsic nitric oxide relaxation pathways in vascular smooth muscle via dopaminergic agonism ameliorates cerebral vasospasm after SAH.
Methods
Cerebrospinal fluid (CSF) from patients with cerebral vasospasm after SAH (CSFV) was used to induce vasospasm in porcine carotid artery in vitro. Dopamine was added to test its ability to reverse spasm, and specific dopamine receptor antagonists were used to determine which receptor mediated the protection. Immunohistochemical techniques confirmed the presence of dopamine receptor subtypes and the involvement of NOS in the mechanism of dopamine protection.
Results
Dopamine receptor 1, 2, and 3 subtypes are all present in porcine carotid artery. Dopamine significantly reversed spasm in vitro (67% relaxation), and this relaxation was prevented by Haloperidol, a D2R antagonist (10% relaxation, P < 0.05), but not by D1 or D3-receptor antagonism. Both eNOS and iNOS expression were increased significantly in response to CSFV alone, and this was significantly enhanced by addition of dopamine, and blocked by Haloperidol.
Conclusion
Cerebral vasospasm is significantly reversed in a functional measure of vasospasm in vitro by dopamine, via a D2R-mediated pathway. The increase in NOS protein seen in both the endothelium and vascular smooth muscle in response to CSFV is enhanced by dopamine, also in a D2R-dependent mechanism.
Keywords: Vasospasm, Intracranial, Subarachnoid hemorrhage (SAH), Dopamine, Nitric oxide, Nitric oxide synthase
Introduction
Delayed cerebral vasospasm is a significant cause of morbidity and mortality in patients surviving an aneurysmal subarachnoid hemorrhage (SAH). Cerebral vasospasm occurs in 30–50% of all surviving SAH patients, leading to further stroke, poor recovery, neurological deficit, and often death. Interestingly, there is a delay of 3–10 days between the SAH and the onset of cerebral vasospasm [1]. Despite extensive research on the etiology of cerebral vasospasm after SAH [2–5], the molecule(s) or mechanism(s) responsible for this essentially irreversible constriction of cerebral vessels have not yet been definitively identified, and there are currently no effective treatments available. Cerebral vasospasm is currently managed pharmacologically using calcium antagonists such as Nimodipine® (Bayer) [6] with minimal effects on vessel diameter. Another form of management is “Triple-H” therapy [7]. This is the combined therapeutic approach of hypertension (achieved using phenylephrine HCl or some other pressor), hypervolemia, and hemodilution (achieved using normal saline intravenously, with or without a bolus of albumin to aid liquid retention). The aim of this aggressive prophylactic therapy is to counteract the hemodynamic consequences of cerebral vasospasm after SAH, which include decreased cerebral blood flow, and loss of brain vessel auto-regulation. Although Triple-H therapy is widely used in the neurocritical care community and is the best treatment available to date, there are significant complications and limitations to this method including pulmonary edema, myocardial ischemia, hyponatremia, cerebral edema, as well as the complications associated with indwelling catheters [7]. Cerebral vasospasm also encompasses neurological deficit independent of vessel narrowing, and the etiology of this aspect of the disease is not yet understood. In this study, we focus on the vascular aspect of cerebral vasospasm, which is a major contributor to the syndrome.
One of the many therapies postulated for cerebral vasospasm involves nitric oxide (NO) [8]. NO produced by nitric oxide synthase (NOS) is a potent vasorelaxant [9], and disruption of this endogenous relaxation pathway has been postulated as a cause for cerebral vasospasm after SAH [10]. To this end, a number of animal studies have been conducted with limited success in demonstrating lasting vasorelaxation in vivo [11, 12]. The main limitation of infusions of NO and NO-donors is that they are short-lived in the circulation such that they have to be continuously infused into the cranial circulation, or even the subarachnoid space, to be effective.
Evidence exists for communication between dopamine and neuronal NOS (nNOS) in neural tissue [13–15], and in this study, we have seen that this also occurs with D2R agonism and vascular inducible NOS (iNOS) and endothelial NOS (eNOS) in vitro. Upregulation of an intrinsic relaxation pathway via intravenous administration of dopamine could leads to development of an effective therapeutic strategy for cerebral vasospasm after SAH.
Methods
Physiological Saline Solution
Physiological saline solution (PSS) was used in the force measurement experiments, and as a diluent for the drugs. It comprised the following (in mmol/l): 118 NaCl, 25 NaHCO3, 5.7 KCl, 1.2 MgCl2, 2.4 CaCl2, 0.75 NaH2PO4, and 11 glucose. KCl PSS (80 mmol/l KCl) was used in the force measurement experiments to achieve maximal tension, where NaCl was replaced isoosmotically with KCl [16].
Porcine Carotid Artery Isometric Force Measurements
Porcine carotid arteries (extra-dural) were obtained from Large White pigs (Stehlin’s Meat, Cincinnati, OH 45251), which weighed between 80 and 100 kg at time of slaughter. Since the excised tissue was brought in from outside, an IACUC protocol was not required. The carotid artery was removed from the pig within 10 min of sacrifice and placed in ice-cold PSS. The adventitia was carefully removed, and the tissue cut into rings of 5 mm in length. These rings were mounted onto a pair of wire triangles, one of which was fixed, and the other was attached by means of a non-compressible wire to a Harvard Instruments® force transducer. The tissues were immersed in PSS in a water-jacketed organ bath (Radnoti Glass®, equilibrated at 37°C and with 95%/5% O2/CO2. Prior to addition of cerebrospinal fluid (CSF) or pharmacological agents, the tissues were stretched manually to their optimum length, and the viability of the tissues was ascertained by addition of PSS containing 80 mmol/l KCl, giving a maximal physiological contraction.
In Vitro Model of Cerebral Vasospasm After SAH
In this laboratory, we use an in vitro model of cerebral vasospasm after SAH, which is described fully in the literature [2, 17]. Briefly, CSF from patients who have suffered a SAH was collected and used to elicit contraction in porcine carotid smooth muscle. CSF is drained from these patients as standard of care, and local IRB permission was obtained to collect store and analyze this fluid. We have found that only CSF from patients with angiographically visible cerebral vasospasm elicits a significant, sustained, increase in contraction in our model [18]. As such, our model allows for the in vivo formation of vasospastic CSF (CSFV) in human patients, thus eliminating the discrepancy in mechanism of hemorrhage that may occur in animal models. Porcine carotid arteries in vitro were bathed in undiluted CSFV in order to model cerebral vasospasm after SAH. Agents were added before or after addition of CSF in order to assess their effectiveness at preventing/reversing cerebral vasospasm in vitro.
Materials
High-grade chemicals were obtained from Acros Organics (Morris Plains, NJ) unless otherwise stated. Dopamine, L-stepholidine (LSPD, a D1-receptor antagonist), Haloperiodol (a D2-receptor antagonist), and U991914A (a D3-receptor antagonist) were obtained from Alexis Corporation San Diego, CA, and added to the tissue in the organ bath to a final concentration of 10 μmol/l for dopamine and 1 μmol/l for the antagonists.
Immunohistochemistry
Porcine carotid arteries were exposed to agents while mounted on the force measurement apparatus and fixed in situ with 4% paraformaldehyde. Tissues were exposed to CSF for 1 h, and the agent of choice for 30 min. Where a combination of agonist/antagonist is added, the additions are made simultaneously. The fixed tissues are mounted on a freezing microtome with the lumen parallel to the blade angle. Transverse 50-μm sections were cut and collected into PBS. The sections were incubated with antibodies to iNOS, eNOS, D1R, D2R, or D3R (US Biologicals, Swampscott, MA). After incubation with secondary antibodies conjugated to horseradish peroxidase, the Vectastain HRP development system (Burlingame, CA) was used to visualize specific binding of the primary antibodies. The stained sections were mounted onto slides for analysis with a Zeiss (Hallbergmoos, Germany) Axio-Cam system. Staining intensity was quantified using ImageQuant (Sunnyvale, CA) software.
Statistics
For comparison between groups and their controls, unpaired t-tests were used. Significance for multiple groups was determined using ANOVA. A significant difference was determined when P < 0.05. Errors are reported as standard deviation. Minitab Statistical Analysis software (State College, PA) was used for these analyses.
Results
We first sought to show that the three most common receptor subtypes, the D1-like receptor, D1R, and the D2-like receptors, D2R and D3R, are present in vascular smooth muscle. Figure 1 shows that the porcine carotid artery does indeed contain all three receptor types, in both the endothelium and vascular layers.
Fig. 1.
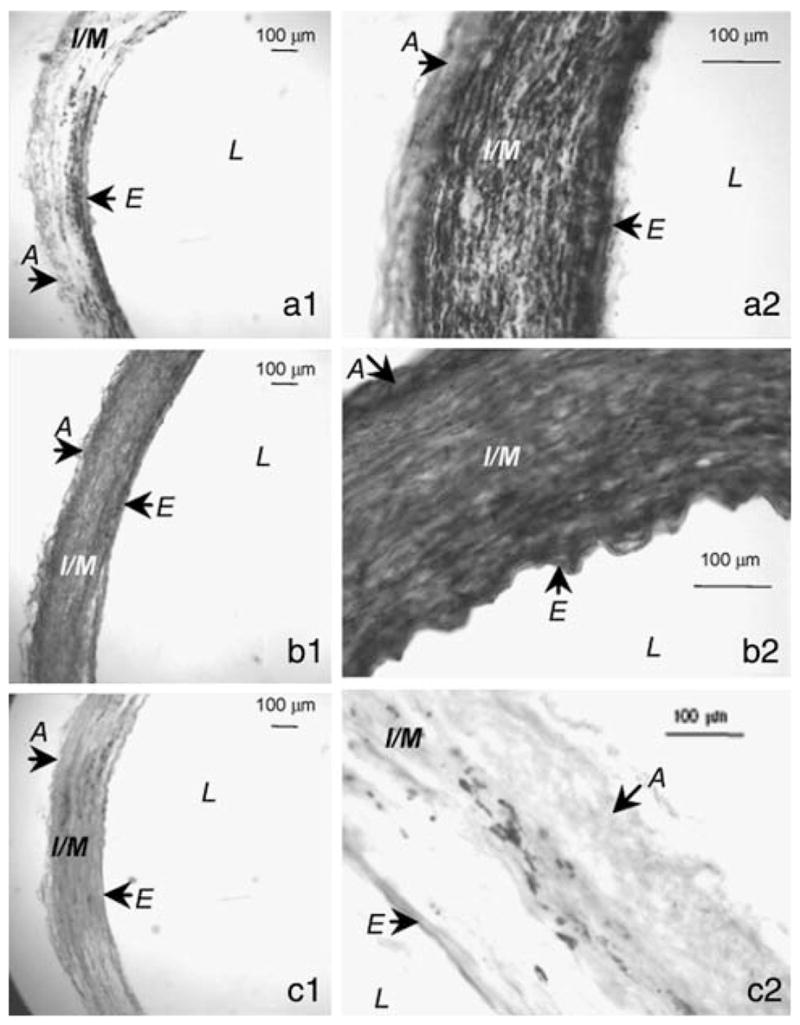
Porcine carotid artery sections subjected to immunohistochemical staining using antibodies specific for dopamine receptors (DR). D1R (a1 and a2), D2R (panel b) and D3R (c). Two different magnifications of the same section are shown for each (1 = 2X, 2 = 80X). From these representative images, it can be seen that D1 (a), D2 (b), and D3 (c) receptors are all expressed in porcine carotid arteries. D3R are the least abundant, and D2R are the most abundant, which supports our hypothesis that up-regulation of NOS in this vessel is mediated by D2R. Images are representative of 5 vessels (from different pigs) for each receptor staining. Structures: A, adventitia; E, endothelium; I/M, intima/media; L, lumen
Figure 2a shows the protective effect of dopamine against cerebral vasospasm in our in vitro model (n = 6 different pig tissues). Dopamine alone did not cause significant contraction, whereas CSFV alone elicited a pathological contraction more than twice that of the physiological maximum (70 mmol/l KCl). This CSFV-induced contraction was significantly lessened with 10 μmol/l dopamine (P < 0.05, t-test), and this protection was afforded whether the dopamine is added before or after the CSFV. In fact, relaxation is greater when dopamine is added following the CSFV. In order to determine which receptor the dopamine might be acting through, we utilized inhibitors. L-SPD (l-stepholidine) was used to inhibit D1R, Haloperidol for D2R and U99194A inhibits D3R. Figure 2b shows that only D2R antagonism prevented the protection afforded by dopamine (n = 4 different pig tissues). This strongly suggests that dopamine-mediated protection against cerebral vasospasm after SAH in vitro is D2R-mediated. Significance was determined using the t-test between tissues with and without dopamine for each antagonist and CSFV.
Fig. 2.

(a) Porcine carotid artery contracts to CSF from patients with vasospasm (CSFV) to greater than that of 70 mM potassium chloride, the physiological maximum Ca2+-dependent contraction. Dopamine (DA) alone had little effect, but in combination with CSFV, DA prevented the majority of the contraction. n = 6, errors are SD. (b) In the porcine carotid artery in vitro, the DA protection is abolished by the specific D2R antagonist Haloperidol, but not by LSPD (L-stepholidine, D1R antagonist) or U99194A (D3R antagonist). This suggests the protection is mediated through D2R. n = 4, errors are SD. * Significant differences determined by ANOVA, P < 0.05
Figure 3a shows quantification of endothelial eNOS by immunohistochemistry. There is significant increase in eNOS elicited by CSFV alone, but this is, again significantly, enhanced by the presence of 10 μmol/l dopamine (P < 0.01). Taking the data in Fig. 2 implicating the involvement of D2R into consideration, we used the D2R antagonist, Haloperidol, to show that blocking D2R prevents increased eNOS expression in dopamine-treated vasospastic vessels in vitro. Haloperidol significantly reduced staining for both eNOS and iNOS compared to dopamine + CSFV. From each of 5 different pieces of tissue (from 5 different pigs), 6 tissue sections were analyzed and all the data averaged. Haloperidol also prevented dopamine-dependent increase in iNOS and eNOS staining in the absence of CSFV (data not shown).
Fig. 3.

(a) Quantification of endothelial nitric oxide synthase in the endothelium. ImageQuant® was used to determine the pixel count/area for the endothelium. Control was set at 100% and values are expressed as percent increase from control (untreated tissue). (b) Similar quantification of inducible nitric oxide synthase in the vascular smooth muscle layers. n = 5, errors are SD
A similar pattern is seen in Fig. 3b, which illustrates iNOS expression in the vascular smooth muscle. There was a significant increase with CSFV alone, which was enhanced by dopamine. This increase due to dopamine was ablated by Haloperidol (P < 0.01). From each of 5 different pieces of tissue (from 5 different pigs), 6 tissue sections were analyzed and all the data averaged.
Discussion
Nitric oxide can lead to relaxation of smooth muscle by reducing the intracellular calcium concentration in several ways (see Fig. 4). However, the widespread use of a calcium channel blocker Nimodipine® (Bayer) in SAH patients to prevent cerebral vasospasm is based on this premise also and no vasodilatation is seen in the spastic vessels; rather, improved neurological outcome associated with use of Nimodipine is likely due to prevention of calcium overload in the damaged neurons [6, 19–21]. So, perhaps the reduction in intracellular calcium in the vascular smooth muscle is not enough to reverse vasospasm [22]. Protein kinase G (cGMP-dependent protein kinase), which is activated as a result of NO pathway activation, is also known to phosphorylate and activate myosin light chain phosphatase. This, in turn, dephosphorylates smooth muscle myosin light chains and leads to smooth muscle relaxation. Figure 4 illustrates the multiple ways in which NO can lead to vasodilatation.
Fig. 4.

An illustration of the pathways proposed to be involved in DA activation of NOS and the subsequent mechanisms by which NOS effects vasodilatation [12]. DA (dopamine) binds to the D2R (D2-dopamine receptor) at the plasma membrane, thus activating AC (adenylate cyclase). This leads to cAMP (3′–5′ cyclic adenosine monophosphate) production which is a second messenger that activates PKA (cyclic AMP-dependent protein kinase). PKA phosphorylates and activates NOS (nitric oxide synthase), resulting in a positive feedback loop and increased expression of both iNOS and eNOS. NO is produced which then activates sGC (soluble Guanylyl cyclase) which converts guanosine triphosphate (GTP) into 3′–5′ cyclic guanosine monophosphate (cGMP). cGMP acts as a second messenger and activates cGMP-dependent protein kinase (PKG′). This kinase acts on multiple target proteins to the same end: vasodilatation. (i) Phosphorylation of membrane ion channels leads to influx of potassium and prevention of calcium influx. Hyperpolarization ensues and calcium entry is further retarded. (ii) Phosphorylation of IP3 receptor-associated cGMP kinase substrate (IRAG) prevents release of calcium from intracellular stores. (iii) Phosphorylation of phospholamban and consequent relief of its inhibition of sarco-endoplasmic reticulum Ca2+ ATPase (SERCA) results in increased sequestration of cytosolic calcium into the sarcoplasmic reticulum. Pathways (i)–(iii) result in decreased intracellular calcium concentrations and therefore less smooth muscle contraction. (iv) PKG phosphorylates and activates myosin light chain phosphatase (MLCP). This dephosphorylates the myosin light chains, allowing dissociation of actomyosin, and relaxation. (v) This whole process can be blocked with Haloperidol, a D2R antagonist
Although the use of exogenous NO or NO donors as a relaxant to treat cerebral vasospasm after SAH has been investigated before [23], here we show that dopamine applied to the vasculature can raise expression of both iNOS and eNOS. Cross-talk between dopamine and NOS has been sparsely reported. von Essen et al. [24] showed that dopamine induces increased cerebral blood flow in dogs, and that this increase is blocked by haloperidol. In a subsequent publication [25], the same authors showed that this dopamine-induced increase in CBF is increased when a NO donor (nitroglycerine) was infused simultaneously. However, the authors do not postulate any relationship between dopamine and NOS. In patients with cerebral vasospasm after SAH, combinatorial therapy with aminophylline, nitroprusside (an NO donor), and dopamine seemed to act synergistically and improve patient outcome, but this was a series of case studies, and no published clinical trial data are available. In renal arteries, dopamine led to induction of iNOS in the vascular tissue, and this was blocked with haloperidol [26]. Another isoenzyme of NOS that we did not investigate here, mitochondrial NOS (mtNOS) [27], may also be upregulated by DR agonism. Czerniczyniac et al. found that dopamine at 1 and 15 mM significantly increased NO production in isolated mouse sub-mitochondrial synaptosome-free membrane preparations, and that this NO production was abolished by L-NNA or L-NAME [28]. Dopamine also affected other mitochondrial functional parameters thought to be regulated in part by mtNOS [29]. The concentrations of dopamine seen to have an effect in this study are supra-physiological, but could be seen locally as a result of pathology in dopaminergic regions. The authors suggest that this response to dopamine could be related to that of the response of nNOS to dopamine [15, 30], because mtNOS is an isoenzyme of nNOS [29].
So how does binding of dopamine to a D2R lead to increased NOS expression? There is very little reported work in this area. Butt et al. postulated that NOS activation may be via cAMP signaling. NOS can be phosphorylated (and consequently, activated) by protein kinase A and protein kinase GII. When phosphorylated, the enzyme becomes Ca2+-independent [31]. Dopamine can increase cAMP levels via D2R [32], so this is a possible mechanism. It is worth mentioning again that this study focused on the vascular aspects of cerebral vasospasm, and we cannot address the nonvascular-related neuro-worsening seen in some patients after SAH with our in vitro model. While we have used dopamine for these in vitro studies, the pressor effects of dopamine in patients have undesirable side-effects. Many neurocritical care units have switched to norepinephrine or phenylephrine as pressor of choice in triple-H therapy [33]. Bromocriptine is used in the Neurointensive care setting to arouse patients, usually those with traumatic brain injury. This is a D2/3R dopaminergic and does not have pressor effects [34]. This may be a viable option for moving these studies into an in vivo model.
Conclusion
We have shown that there is a D2R-mediated relationship between dopamine and eNOS and iNOS upregulation in vascular smooth muscle and endothelium (further investigations are required to assess the involvement of mtNOS) which leads to relaxation of vessels in a pathological state of constriction in vitro. Medications such as amantadine (MAO-A inhibitor), bromocriptine (D2R agonist), or other dopamine agonists, Sinemet® (combination of levodopa and carbidopa, both converted to dopamine in the brain), and Modafinil ([2-[(diphenylmethyl)sulfinyl]acetamide], an atypical wake-promoting mixed agonist dopaminergic), have been reported to improve alertness and cognition after TBI and SAH [35–39] and are often used to promote arousal in comatose recovering traumatic brain injury patients in the neurocritical care setting. That these dopaminergic agonists currently are used safely in this patient population is encouraging and may provide an avenue for pharmacologic therapy to prevent or even reverse vasospasm in the SAH patient.
Acknowledgments
The authors thank Dr. Kenneth Wagner for excellent discussions and Mrs. Shauna Beiler for porcine tissue supply. They also thank Amanda Harm and Ashlie Saffire for immunohistochemical technical assistance. This project was funded by NIH 5R0 1NS049428 (Pyne-Geithman PI).
Contributor Information
Gail J. Pyne-Geithman, Department of Neurology, University of Cincinnati, 3125 Eden Avenue, 2324 Vontz Center, Cincinnati, OH 45267-0536, USA, e-mail: pynegj@ucmail.uc.edu
Danielle N. Caudell, Department of Neurology, University of Cincinnati, 3125 Eden Avenue, 2324 Vontz Center, Cincinnati, OH 45267-0536, USA
Matthew Cooper, Department of Neurology, University of Cincinnati, 3125 Eden Avenue, 2324 Vontz Center, Cincinnati, OH 45267-0536, USA.
Joseph F. Clark, Department of Neurology, University of Cincinnati, 3125 Eden Avenue, 2324 Vontz Center, Cincinnati, OH 45267-0536, USA
Lori A. Shutter, Department of Neurology, University of Cincinnati, 3125 Eden Avenue, 2324 Vontz Center, Cincinnati, OH 45267-0536, USA Department of Neurosurgery, University of Cincinnati, Cincinnati, OH, USA; The Veterans Administration Medical Center, Cincinnati, OH, USA.
References
- 1.Weir B, Grace M, Hansen J, Rothberg C. Time course of vasospasm in man. J Neurosurg. 1978;48:173–8. doi: 10.3171/jns.1978.48.2.0173. [DOI] [PubMed] [Google Scholar]
- 2.Pyne GJ, Cadoux-Hudson TAD, Clark JF. The presence of an extractable substance in the CSF of humans with cerebral vasospasm after subarachnoid haemorrhage that correlates with phosphatase inhibition. Biochim Biophys Acta. 2000;1474:283–90. doi: 10.1016/s0304-4165(00)00030-1. [DOI] [PubMed] [Google Scholar]
- 3.Sato M, Tani E, Fujikawa H, Kaibuchi K. Involvement of Rho-kinase-mediated phosphorylation of myosin light chain in enhancement of cerebral vasospasm. Circ Res. 2000;87(3):195–200. doi: 10.1161/01.res.87.3.195. [DOI] [PubMed] [Google Scholar]
- 4.Koide M, Nishizawa S, Ohta S, Yokoyama T, Namba H. Chronological changes of the contractile mechanism in prolonged vasospasm after subarachnoid hemorrhage: from protein kinase C to protein tyrosine kinase. Neurosurgery. 2002;51(6):1468–76. doi: 10.1097/00006123-200212000-00018. [DOI] [PubMed] [Google Scholar]
- 5.Borel CO, McKee A, Parra A, et al. Possible role for vascular cell proliferation in cerebral vasospasm after subarachnoid hemorrhage. Stroke. 2003;34:427–33. doi: 10.1161/01.STR.00000538 48.06436.AB. [DOI] [PubMed] [Google Scholar]
- 6.Pickard JD, Murray GD, Illingworth R, et al. Effect of oral nimodipine on cerebral infarction and outcome after subarachnoid haemorrhage: British aneurysm nimodipine trial. BMJ. 1989;298(6674):636–42. doi: 10.1136/bmj.298.6674.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee KH, Lukowitz T, Friedman JA. “Triple-H” therapy for cerebral vasospasm following subarachnoid hemorrhage. Neurocrit Care. 2006;4:68–76. doi: 10.1385/NCC:4:1:068. [DOI] [PubMed] [Google Scholar]
- 8.Pluta RM. Delayed cerebral vasospasm and nitric oxide: review, new hypothesis and proposed treatment. Pharmacol Ther. 2005;105:23–56. doi: 10.1016/j.pharmthera.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 9.Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43(2):109–42. [PubMed] [Google Scholar]
- 10.Pluta RM, Thompson BG, Dawson TM, Snyder SH, Boock RJ, Oldfield EH. Loss of nitric oxide synthase immunoreactivity in cerebral vasospasm. J Neurosurg. 1996;84:648–54. doi: 10.3171/jns.1996.84.4.0648. [DOI] [PubMed] [Google Scholar]
- 11.Afshar JKB, Pluta RM, Boock RJ, Thompson BG, Oldfield EH. Effect of intracarotid nitric oxide on primate cerebral vasospasm after subarachnoid hemorrhage. J Neurosurg. 1995;83:118–22. doi: 10.3171/jns.1995.83.1.0118. [DOI] [PubMed] [Google Scholar]
- 12.Wolf EW, Banerjee A, Soble-Smith J, Dohan FC, White RP, Robertson JT. Reversal of cerebral vasospasm using an intrathecally administered nitric oxide donor. J Neurosurg. 1998;89:279–88. doi: 10.3171/jns.1998.89.2.0279. [DOI] [PubMed] [Google Scholar]
- 13.Choi JK, Chen YI, Hamel E, Jenkins BG. Brain hemodynamic changes mediated by dopamine receptors: role of the cerebral microvasculature in dopamine-mediated neurovascular coupling. Neuroimage. 2006;30(3):700–12. doi: 10.1016/j.neuroimage.2005.10.029. [DOI] [PubMed] [Google Scholar]
- 14.Salum C, Guimaraes FS, Brandao ML, Del Bel EA. Dopamine and nitric oxide interaction on the modulation of prepulse inhibition of the acoustic startle response in the Wistar rat. Psychopharmacology. 2006;185:133–41. doi: 10.1007/s00213-005-0277-z. [DOI] [PubMed] [Google Scholar]
- 15.Sammut S, Dec A, Mitchell D, Linardakis J, Ortiguela M, West AR. Phasic dopaminergic transmission increases NO efflux in the rat dorsal striatum via a neuronal NOS and a dopamine D1/5 receptor-dependent mechanism. Neuropsychopharmacology. 2006;31:493–505. doi: 10.1038/sj.npp.1300826. [DOI] [PubMed] [Google Scholar]
- 16.Krebs EG, Beavo JA. Phosphorylation and dephosphorylation of enzymes. Annu Rev Biochem. 1979;48:923–59. doi: 10.1146/annurev.bi.48.070179.004423. [DOI] [PubMed] [Google Scholar]
- 17.Pyne GJ, Cadoux-Hudson TAD, Clark JF. Force-function relations in vascular smooth muscle during cerebral vasospasm. Biophys J. 1998;74(2 Pt 2):A256. [Google Scholar]
- 18.Cadoux-Hudson TAD, Pyne GJ, Domingo A, Clark JF. The stimulation of vascular smooth muscle oxidative metabolism by CSF from subarachnoid haemorrhage patients. Acta Neurochir (Wien) 2001;143(1):65–72. doi: 10.1007/s007010170140. [DOI] [PubMed] [Google Scholar]
- 19.Gilsbach JM, Reulen HJ, Ljunggren B, et al. Early aneurysm and preventive therapy with intravenously administered Nimodipine. A multicentre, double-blind, dose-comparison study. Neurosurgery. 1990;26:458–64. doi: 10.1097/00006123-199003000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Ohman J, Servo A, Heiskanen O. Long term effects of Nimodipine on cerebral infarcts and outcome after aneurysmal subarachnoid haemorrhage and surgery. J Neurosurg. 1991;74:8–13. doi: 10.3171/jns.1991.74.1.0008. [DOI] [PubMed] [Google Scholar]
- 21.Whitfield PC, Pickard JD. Nimodipine. Br J Hosp Med. 1994;52(10):539–40. [PubMed] [Google Scholar]
- 22.Clark JF, Pyne GJ, Choutka OJ, et al. In vitro therapy with dobutamine, isoprenaline and sodium nitroprusside protects vascular smooth muscle metabolism from subarachnoid haemorrhage induced cerebral vasospasm. Acta Neurochir (Wien) 2001;143:721–8. doi: 10.1007/s007010170052. [DOI] [PubMed] [Google Scholar]
- 23.Pluta RM, Dejam A, Grimes G, Gladwin MT, Oldfield EH. Nitrite infusions to prevent delayed cerebral vasospasm in a primate model of subarachnoid hemorrhage. JAMA. 2005;293(12):1477–84. doi: 10.1001/jama.293.12.1477. [DOI] [PubMed] [Google Scholar]
- 24.von Essen C, Zervas NT, Brown DR, Koltun WA, Pickren KS. Local cerebral blood flow in the dog during intravenous infusion of dopamine. Surg Neurol. 1980;13:181–8. [PubMed] [Google Scholar]
- 25.von Essen C, Kistler P, Lees RS, Zervas NT. Cerebral blood flow and intracranial pressure in the dog during intravenous infusion of nitroglycerin alone and in combination with dopamine. Stroke. 1981;12(3):331–8. doi: 10.1161/01.str.12.3.331. [DOI] [PubMed] [Google Scholar]
- 26.Costa MA, Elesgaray R, Loria A, Balaszczuk AM, Arranz CT. Vascular and renal effects of dopamine during extracellular volume expansion: role of nitric oxide pathway. Life Sci. 2006;78:1543–9. doi: 10.1016/j.lfs.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 27.Ghafourifar P, Cadenas E. Mitochondrial nitric oxide synthase. Trends Pharmacol Sci. 2005;26(4):190–5. doi: 10.1016/j.tips.2005. 02.005. [DOI] [PubMed] [Google Scholar]
- 28.Czerniczyniec A, Bustamante J, Lores-Arnaiz S. Dopamine enhances mtNOS activity: implications in mitochondrial function. Biochim Biophys Acta. 2007;1767:1118–25. doi: 10.1016/j.bbabio.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 29.Haynes V, Elfering S, Traaseth N, Giulivi C. Mitochondrial nitric oxide synthase: enzyme expression, characterization and regulation. J Bioenerg Biomembr. 2004;36(4):341–6. doi: 10.1023/B:JOBB.0000041765.27145.08. [DOI] [PubMed] [Google Scholar]
- 30.Melis MR, Succu S, Argiolas A. Dopamine agonists increase nitric oxide production in the paraventricular nucleus of the hypothalamus; correlation with penile erection and yawning. Eur J Neurosci. 1996;8:2056–63. doi: 10.1111/j.1460-9568.1996. tb00725.x. [DOI] [PubMed] [Google Scholar]
- 31.Butt E, Bernhardt M, Smolenski A, et al. Endothelial nitric oxide synthase (type III) is activated and becomes calcium independent upon phosphorylation by cyclic nucleotide-dependent protein kinases. J Biol Chem. 2000;275(7):5179–87. doi: 10.1074/jbc. 275.7.5179. [DOI] [PubMed] [Google Scholar]
- 32.Seeman P, Van Tol HM. Dopamine receptor pharmacology. Trends Pharmacol Sci. 1994;15:264–70. doi: 10.1016/0165-6147 (94)90323-9. [DOI] [PubMed] [Google Scholar]
- 33.Miller JA, Dacey RG, Diringer MN. Safety of hypertensive hypervolemic therapy with phenylephrine in the treatment of delayed ischemic deficits after subarachnoid hemorrhage. Stroke. 1995;26:2260–6. doi: 10.1161/01.str.26.12.2260. [DOI] [PubMed] [Google Scholar]
- 34.Calne DB, Teychenne PF, Claveria LE, Eastman LE, Greenacre JK, Petrie A. Bromocriptine in Parkinsonism. BMJ. 1974;4:442–4. doi: 10.1136/bmj.4.5942.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schneider WN, Drew-Cates J, Wong TM, Dombovy ML. Cognitive and behavioural efficacy of amantidine in acute traumatic brain injury: an initial double-blind placebo-controlled study. Brain Inj. 1999;13:863–72. doi: 10.1080/026990599121061. [DOI] [PubMed] [Google Scholar]
- 36.Zafonte RD, Lexell J, Cullen N. Possible applications for dopaminergic agents following traumatic brain injury. J Head Trauma Rehabil. 2000;15:1179–82. doi: 10.1097/00001199-200010000-00014. [DOI] [PubMed] [Google Scholar]
- 37.Zafonte RD, Lexell J, Cullen N. Possible applications for dopaminergic agents following traumatic brain injury; part 2. J Head Trauma Rehabil. 2001;16:112–6. doi: 10.1097/00001199-2001 02000-00014. [DOI] [PubMed] [Google Scholar]
- 38.Teitelman E. Off-label uses of Modafinil. Am J Psychiatry. 2001;158:970–1. doi: 10.1176/ajp.158.8.1341. [DOI] [PubMed] [Google Scholar]
- 39.Powell JH, al-Adawi S, Morgan J, Greenwood RJ. Motivation deficits after brain injury: effects of bromocriptine in 11 patients. J Neurol Neurosurg Psychiatry. 1996;60:416–21. doi: 10.1136/jnnp.60.4.416. [DOI] [PMC free article] [PubMed] [Google Scholar]