

Abstract
Recent studies have shown that there is a considerable heterogeneity in the response of melanoma cell lines to MEK and BRAF inhibitors. In the current study, we address whether dysregulation of cyclin-dependent kinase 4 (CDK4) and/or cyclin D1 contribute to the BRAF inhibitor resistance of melanoma cells. Mutational screening identified a panel of melanoma cell lines that harbored both a BRAF V600E mutation and a CDK4 mutation: K22Q (1205Lu), R24C (WM39, WM46, and SK-Mel-28), and R24L (WM902B). Pharmacologic studies showed that the presence of a CDK4 mutation did not alter the sensitivity of these cell lines to the BRAF inhibitor. The only cell line with significant BRAF inhibitor resistance was found to harbor both a CDK4 mutation and a CCND1 amplification. Array comparative genomic hybridization analysis showed that CCND1 was amplified in 17% of BRAF V600E–mutated human metastatic melanoma samples, indicating the clinical relevance of this finding. As the levels of CCND1 amplification in cell lines are lower than those seen in clinical specimens, we overexpressed cyclin D1 alone and in the presence of CDK4 in a drug-sensitive melanoma line. Cyclin D1 overexpression alone increased resistance and this was enhanced when cyclin D1 and CDK4 were concurrently overexpressed. In conclusion, increased levels of cyclin D1, resulting from genomic amplification, may contribute to the BRAF inhibitor resistance of BRAF V600–mutated melanomas, particularly when found in the context of a CDK4 mutation/overexpression.
Introduction
Following the discovery that the overwhelming proportion of melanomas have constitutive activity in the mitogen-activated protein kinase (MAPK) pathway, there has been considerable interest in pharmacologically targeting this pathway using small molecule inhibitors (1, 2). Although there is evidence to suggest that the presence of the BRAF V600E mutation is predictive of response to BRAF/MEK inhibitors (3), recent clinical studies on MEK and BRAF inhibitors have not led to the expected favorable results (4, 5). BRAF/MAPK signaling may be more heterogeneous than first thought and locally regulated by the microenvironment (6, 7). It also is possible that other factors, such as enhanced phosphoinositide-3-kinase/AKT signaling activity, may further influence response to BRAF/MEK inhibition (8). As yet, very little is known about the factors underlying resistance to BRAF inhibition in the BRAF V600E–mutated melanoma population. A greater understanding of the genetic basis of response to BRAF inhibitors is critical in selecting the most appropriate patient population for future clinical studies and developing strategies to overcome inherent resistance.
In the current study, we have turned our attention to alterations in key components of the cell cycle machinery that also may regulate response to BRAF inhibitors. Mutations in BRAF are thought to drive uncontrolled proliferation through the MAPK-induced expression of cyclin D1, which in turn regulates the activity of cyclin-dependent kinase 4 (CDK4)—facilitating cell cycle entry. Thus, increased expression of cyclin D1 and CDK4 or activating mutations in CDK4, are possible mechanisms by which tumor cells may acquire resistance to inhibitors of BRAF.
CDK4 is often deregulated in melanoma through multiple mechanisms. Under physiologic conditions, CDK4 function is negatively regulated through the binding of the CDK inhibitor p16INK4A. Many melanomas are characterized by loss of p16 function, resulting from the acquisition of mutations at the CDKN2A locus, leading to unrestricted CDK4 activity and increased levels of cell proliferation (9). Germ line mutations at codon 24 in CDK4 have been identified in a few melanoma-prone kindred groups, which render the protein resistant to the inhibitory effects of INK4A function (10). Recent studies have identified a group of BRAF/NRAS wild-type melanomas with somatically increased CDK4 copy number (11). Thus, it is likely that CDK4 functions as an oncogene in subgroups of melanomas.
Cyclin D1 regulates proliferation through its ability to bind to and stimulate both CDK4 and CDK6, leading to phosphorylation of the retinoblastoma protein and entry into the cell cycle. Because of this activity, cyclin D1 is thought to be an important oncogene and is amplified in many tumor types, including certain histologic subtypes of melanoma. CCND1 is frequently amplified in acra1 melanoma (44%), as well as in lentigo maligna melanoma (10%) and superficial spreading melanomas (6%; ref. 12). More recent work has shown that increased CCND1 copy number is found in melanomas arising on chronically sun-damaged skin, which lack mutations in both BRAF and NRAS (11). These data support the importance of the overexpression of CCND1 in promoting cell proliferation in subgroups of melanomas.
Several studies have suggested that dysregulation of BRAF, CCND1, and CDK4, either through mutation or amplification, are independent events showing exclusivity in driving the MAPK signaling pathway (11, 13). The current study identifies CDK4 mutations and amplifications of cyclin D1 in concert with the BRAF V600E mutation in metastatic melanoma. It is suggested that increased cyclin D1 expression in particular may contribute to BRAF inhibitor resistance in a subset of BRAF V600E–mutated melanomas.
Materials and Methods
Cell Culture
Human melanoma cells and melanocytes were isolated and cultured as described in ref. 14. The adenoviral vector for cyclin D1 was kindly provided by Dr. Rick Assoian, University of Pennsylvania, Philadelphia, PA. A lentiviral vector encoding CDK4 was generated in the Gene Expression Core of the Wistar Institute. Viral infections were done as previously described in ref. 14.
Adherent Cell Proliferation Analysis
Cells were plated into a 96-well plate at a density of 2.5 × 104 cells per milliliter and left to grow overnight. Cells were treated with increasing concentrations of SB590885 (GlaxoSmithKline) in triplicate, after 72 h, the levels of growth inhibition were examined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay (15). Data show the mean of at least three independent experiments ± SE. To determine the role of cyclin D1 and CDK4 overexpression in resistance to SB590885, 451Lu cells were infected with virus for either cyclin D1 or CDK4. CDK4-overexpressing cells were selected for following puromycin treatment for 48 h. After infection was confirmed by Western blotting, cells were plated out for the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay as described above.
Western Blot Analysis
Proteins were extracted and blotted for as described in ref. 14. After analysis, Western blots were stripped once and re-probed for β-actin to show even protein loading. The antibodies to phosphorylated extracellular signal-regulated kinase (ERK) and total ERK were from Cell Signaling Technology, the antibody to cyclin D1 was from Santa Cruz Biotechnology and the antibody to CDK4 was from Fisher Scientific.
Cell Cycle Analysis
Cells were plated into 10 cm dishes at 60% confluency and left to grow overnight before being treated with SB590885 (1 and 3 μmol/L) for 24 h. Cells were ethanol-fixed, stained with propidium iodide, and analyzed as previously described (15). In some experiments, cells were infected with virus for either CDK4, cyclin D1, or CDK4 + cyclin D1 prior to treatment with SB590885 and cell cycle analysis.
Melanoma Sample Collection
Samples were collected from 67 melanomas from patients from the Karolinska Institute (Stockholm, Sweden), Fox Chase Cancer Center (Philadelphia, PA), and the University of Pennsylvania Health System. All samples were collected in full accordance with the Institutional Review Board and Health Insurance Portability and Accountability Act regulations of the participating institutions. The use of the specimens for this study was in accordance with the regulations of the Institutional Review Board of the University of Pennsylvania. All melanomas were of the superficial spreading or nodular type. Screening for mutations by PCR sequencing was carried out for BRAF exon 15 and NRAS exon 3 as previously described (16). CDK4 exon 2 was amplified as in Holland et al. (17). The PCR products were subjected to direct sequencing using BigDye Terminator v1.1 Cycle on an ABI PRISM 3130xl Genetic Analyzer (Applied Biosystem). Sequences were analyzed using Mutation Surveyor, DNA Variant Analysis version 3.1 (SoftGenetics LLC).
Bacterial Artificial Chromosome Array Platform
Comparative genomic hybridization (CGH) was done on the platform with ∼5,000 human bacterial artificial chromosomes (BAC) spaced at an average of 1 Mb across the genome, which was developed at the University of Pennsylvania (18) and previously used in similar studies (19). In the array, CCND1 lies directly on a BAC (RP11-156B3) from 69106701 to 69297637 Mb on chromosome 11. BACs were mapped by sequence-tagged site, BAC end sequence, and fluorescence in situ hybridization analysis according to the hg18 UCSC genome browser released in March 2006 (20) and NCBI build 36.1 released in July 2006. BAC DNA was amplified using oligonucleotide-primed PCR (DOP) primers and clones were spotted on Coming CMT Ultra-Gap slide with at least two replicates on each slide using a Lucidea Array Spotter by Amersham Biosciences, and a spotting solution of 50% DMSO.
Array CGH
Both test and reference DNA were labeled with the opposite dye in a separate experiment (“dye swap”) to account for differences in dye incorporation and provide additional data points for analysis. For hybridization, 1 μg of test DNA and 1 μg of sex-matched pooled normal human DNA (obtained from a set of 10 healthy female or male volunteers) were labeled with either Cy3-dCTP or Cy5-dCTP (Amersham Biosciences, UK) incorporated by random priming (Bioprime Labeling Kit, Invitrogen). The reaction was cleaned with Qiagen MiniElute PCR kit (Qiagen). Equal amounts of test and reference DNA (1 μg each) were mixed and precipitated with Cot1-DNA, 3 mo1/L of sodium acetate (pH 7.0), and ethanol. Arrays were hybridized with 50% deionized formamide, 2× SSC, 2% SDS, 10% dextran sulfate, and 100 μg/μL of yeast tRNA for 72 h at 37°C in a moist chamber on a slowly rocking table. Slides were washed using previously published protocols. Then, arrays were scanned on a GenePix 4000B dual scanner (Axon Instruments).
Data Analysis
Fluorescent data from hybridization images were processed and analyzed with Gene Pix Pro 5.0 (Axon Instruments) to obtain the log2 ratios (tumor/reference) of each slide. Array CGH data were processed through print-tip loess normalization, using the DNMAD application (21), which also allowed us to merge and filter clones between replicates in a slide and in the dye-swap experiment. We filtered out inconsistent replicates (those ones with a log2 ratio distant to the median log2 ratio of the replicates >0.3) and those clones that did not have available data in >70% of the cases. For visualization and detection of copy number alterations, CGH-Explorer v. 3.1b software was used (22).
Statistics
Data show the mean of at least three independent experiments ± SE, unless stated otherwise. Statistically significant results were considered at P ≤ 0.05.
Results
Identification of Melanoma Cell Lines with BRAF Mutations and CDK4 Mutations
To investigate whether CDK4 mutational status influenced BRAF inhibitor response, we screened our cell line panel for CDK4 mutations and identified three cell lines (WM39, WM46, and SK-Mel-28) with an R24C mutation, and one cell line (WM902b) with an R24L mutation (Fig. 1; Table 1). The R24C mutation has been described in familial melanoma kindreds and abrogates the binding of p16 with CDK4. Although R24L has not been previously described, as it is located at the same codon, it presumably is deleterious. Previous studies have shown that the 1205Lu cell line has a K22Q mutation in CDK4 (23). To determine whether the presence of a CDK4 mutation led to BRAF inhibitor resistance, a panel of melanoma cell lines were treated with increasing concentrations of the BRAF inhibitor SB590885 (ref. 24; Fig. 2A). Most of the melanoma cell lines that harbored the BRAF V600E mutation and lacked CDK4 mutations (451Lu, WM35, and WM983) were highly sensitive to SB590885 and had IC50 values of <1 μmol/L. Interestingly, nearly all of the melanoma cell lines with a CDK4 mutation (1205Lu, SK-Mel-28, WM902b, and WM46) showed equivalent SB590885 sensitivity to the control melanoma lines (WM35, 451Lu, and WM983). Of all the cell lines tested, WM39 was the most resistant to the growth-inhibitory effects of SB590885, indicating that there may be other factors responsible for BRAF inhibitor sensitivity on a BRAF V600E/CDK4–mutated background. We next addressed the mechanism responsible for the resistance of the WM39 cell line to BRAF inhibition. Western blotting studies revealed that SB590885 was similarly effective at inhibiting MAPK activity in the resistant melanoma line (WM39) and a sensitive melanoma cell line (451Lu) as shown by the concentration-dependent reduction in phosphorylated ERK levels (Fig. 2C). This finding suggested that the WM39 cell line was able to maintain cell cycling in the absence of MAPK signaling.
Figure 1.
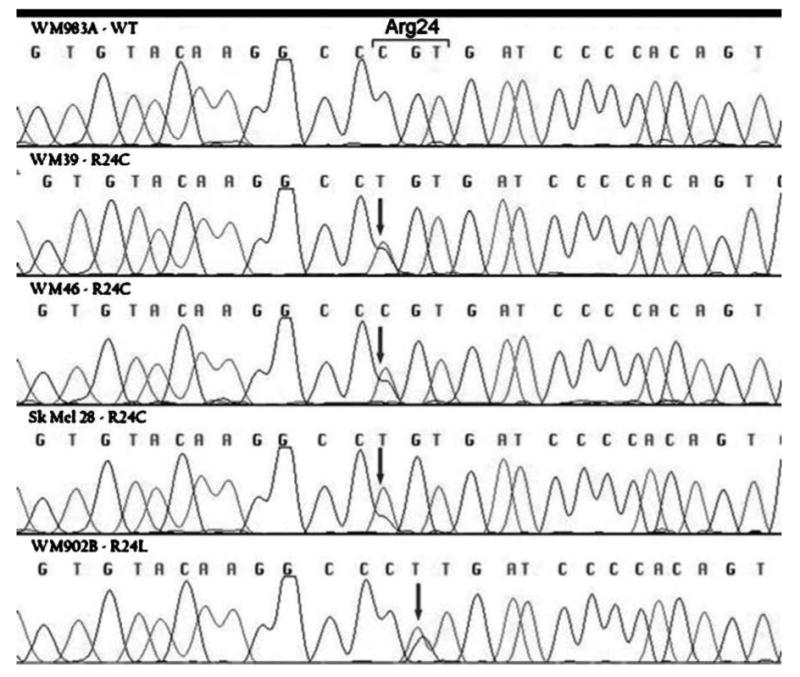
Identification of CDK4 mutations in BRAF V600E – mutated human melanoma cell lines. DNA sequences of CDK4 in a wild-type melanoma cell line (WM983) and R24C CDK4-mutated melanoma cell lines (WM39, WM46, and SK-Mel-28), and an R24L-mutated melanoma cell line (WM902B).
Table 1.
Mutational status of the melanoma cell line panel
| Cell line | BRAF | NRAS | CDK4 |
|---|---|---|---|
| SK-Mel-28 | V600E | WT | R24C |
| WM39 | V600E | WT | R24C |
| WM902b | V600E | WT | R24L |
| WM46 | V600E | WT | R24C |
| 1205Lu | V600E | WT | K22Q |
| WM35 | V600E | WT | WT |
| WM983C | V600E | WT | WT |
| WM164 | V600E | WT | WT |
| 451Lu | V600E | WT | WT |
Abbreviation: WT, wild-type.
Figure 2.
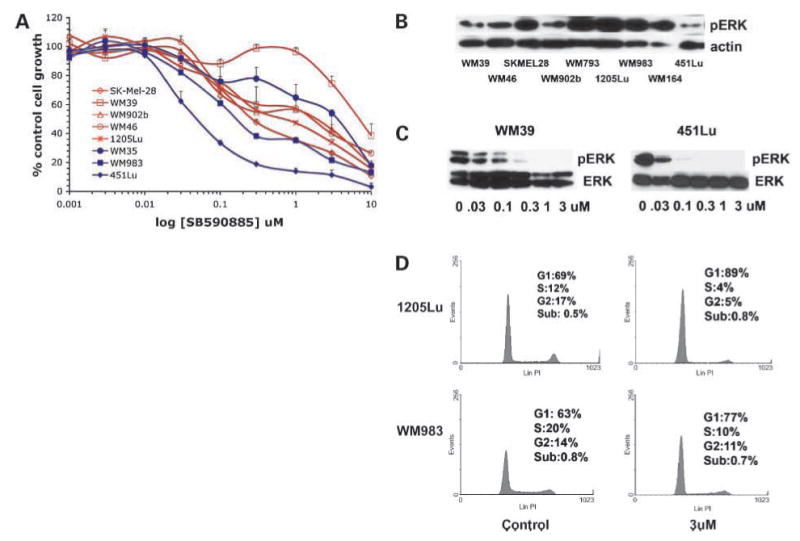
Melanoma cell lines with CDK4 mutations are not resistant to SB590885. A, melanoma cell lines with a CDK4 mutation (WM39, WM46, SK-Mel-28, WM902B, WM793, and 1205Lu: red, open symbols) and melanoma lines without CDK4 mutations (WM983, WM164, and 451Lu; blue, closed symbols) were treated with increasing concentrations of SB590885 (1 nmol/L – 10 μmol/L) for 72 h before being treated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide. Absorbance was read at 540 nm and expressed as a percentage of control absorbance. Points, mean of three independent experiments; bars, SE. B, expression of phosphorylated ERK and actin expression in a panel of melanoma cell lines. C, SB590885 reduces phosphorylated ERK levels in the WM39 and 451Lu cell lines. Cells were treated with increasing concentrations of SB590885 for 1 h, after which, protein was extracted and probed for phosphorylated ERK expression (pERK), equal protein loading was confirmed by levels of total ERK (ERK). D, SB590885 induces a similar level of cell cycle arrest in CDK4-mutated (1205Lu) and CDK4 wild-type (WM983) cell lines. Cells were treated with 1 μmol/L for 24 h prior to fixation, propidium iodide staining, and resolution by flow cytometry.
All six cell lines with the CDK4 mutations (1205Lu, WM793, WM39, WM46, WM902B, and SK-Mel-28) were also BRAF V600E mutation–positive (Table 1) and maintained phosphorylated ERK expression (Fig. 2B). The lack of BRAF inhibitor resistance in melanomas with CDK4 mutations was also seen at the level of the cell cycle arrest, with G1 phase cell cycle arrest following SB590885 treatment in both CDK4-mutated (1205Lu) and CDK4 wild-type (WM983) cell lines (Fig. 2D).
Identification of BRAF V600E – Mutated Melanoma Samples with High Levels of CCND1 Amplification
It is known that cyclin D1 works in concert with CDK4 to drive cell cycle entry. As some subgroups of acral and lentigo meligna melanomas are known to have amplification of cyclin D1, we next looked at whether CCND1 amplification was responsible for the BRAF inhibitor resistance of the WM39 cell line. Using array CGH, we found CCND1 amplification in 17% (11 of 67) of samples; details of amplifications and associations with mutations are found in Table 2. High-level amplification (log2 ratio > 1.5) of CCND1 was identified in 4% (3 of 67) of our melanoma samples; one each with a BRAF mutation, NRAS mutation, and wild-type for both. When the analysis was extended to our cell line panel, we found 25% with an amplification of CCND1 (12 of 47) but only one cell line with a high level (log2 ratio >1.5; Fig. 3A) of CCND1 amplification on a BRAF V600E–mutated background (WM39; Fig. 2B). Sample traces from our array CGH analysis in BRAF-mutated samples and cell line are shown in Fig. 3A. The observed amplification in CCND1 seen in the WM39 cell line was also reflected in increased cyclin D1 protein expression. Levels of cyclin D1 expression were significantly higher than those seen in the 451Lu and WM983 cell lines (Fig. 3B). Interestingly, the levels of cyclin D1 seen in the WM39 cells were comparable to those seen in the 451Lu cell line infected with the cyclin D1 adenovirus (451LU/CCND1; Fig. 3B).
Table 2.
Frequency of CCND1 amplification in melanoma cell lines and melanoma tumor samples
| Total number of samples | 47 Melanoma cell lines (%) | 67 Melanoma tumor samples (%) |
|---|---|---|
| Total CGH CCND1 amplifications | 12/47 (25) | 11/67 (17) |
| CCND1 amplifications, BRAF group | 9/36 (25) | 7/29 (24) |
| CCND1 amplifications, NRAS group | 1/8 (12.5) | 2/18 (11) |
| CCND1 amplifications, BRAF/NRAS wild-type | 2/3 (67) | 2/20 (10) |
Figure 3.
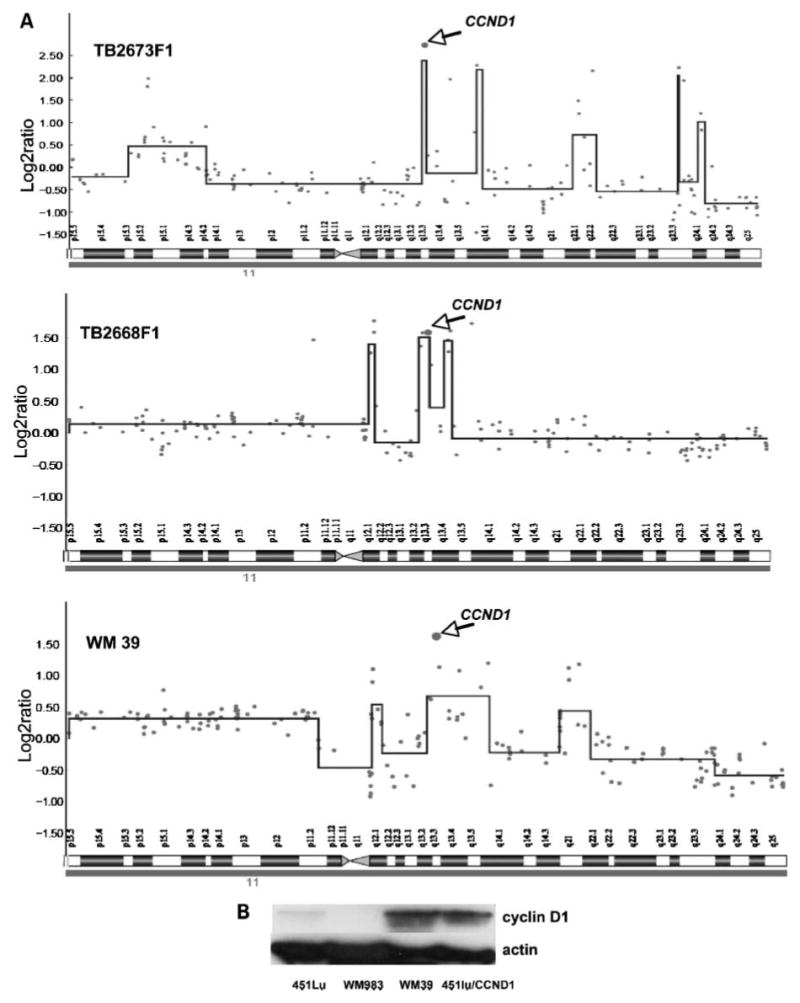
Identification of human melanoma samples and a cell line with high levels of cyclin D1 (CCND1) amplification. A, DNA copy number profiles of chromosome 11 for melanoma tumor samples TB2668F1, TB2673F1, and the melanoma cell line WM39. Arrow, copy number amplification of the BAC on which CCND1 is located. Log2 ratio for each of the genomic clones is plotted according to chromosome position using a “moving average smoother” with a three-clone window. B, the WM39 cell line shows increased expression of cyclin D1 compared with two SB590885-sensitive melanoma cell lines (451Lu and WM983). For comparison, the 451Lu cell line infected with an adenovirus encoding for cyclin D1 (451Lu/CCND1) is also shown. Western blot of cyclin D1 expression, blots were stripped and re-probed for actin to show equal protein loading.
Overexpression of Cyclin D1 Increases Resistance to BRAF Inhibition and Is Enhanced by Increased CDK4 Expression
MAPK activity regulates proliferation through the regulation of cyclin D1 expression. WM39 ceils are highly BRAF inhibitor–resistant and exhibit high levels of CCND1 amplification. As the WM39 cell line also harbors the R24C CDK4 mutation, we next investigated whether cyclin D1 overexpression alone would blunt the growth-inhibitory effects of SB590885, or whether some cooperation was required with CDK4. In these studies, we infected a SB590885-sensitive melanoma cell line (451Lu), with a viral vector encoding for cyclin D1. After 48 hours of incubation with the virus, it was found that cyclin D1 levels were markedly increased (Fig. 4A). Upon treatment of the cells with SB590885 (300 nmol/L), the cyclin D1–overexpressing cells were significantly (P < 0.05) more resistant than the control 451Lu cell line (65 ± 7.0% and 38 ± 2.6% of growth relative to untreated cells, respectively; Fig. 4B). Infection of the 451Lu cells with the cyclin D1 virus also rendered the cells insensitive to the growth-arresting effects of SB590885 treatment (Supplemental Fig. S1). In a final series of experiments, we assessed the effects of coexpressing cyclin D1 and CDK4 in our SB590885-sensirive cell line. Infection of the cells with virus led to the expected increase in CDK4 expression (Fig. 4A), but did not alter the response to SB590885 (cell growth, 40 ± 2.3% relative to untreated controls; Fig. 4B). Furthermore, when the cells were dually infected with viruses for cyclin D1 and CDK4, it was found that the cells became more resistant (P < 0.05) to the growth-inhibitory effects of SB590885 (cell growth, 75.8 ± 8.1% of untreated controls; Fig. 4B). Further analysis revealed that the dual CDK4/cyclin D1–overexpressing cell lines were completely insensitive to SB590885-induced G1 phase cell cycle arrest (Fig. 4C).
Figure 4.
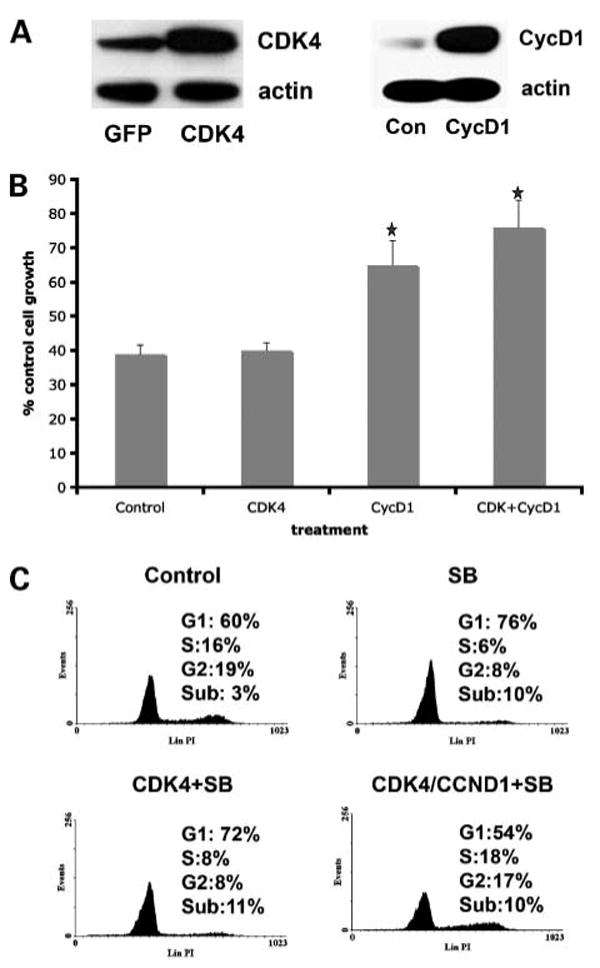
Overexpression of cyclin D1 reduces sensitivity to the BRAF inhibitor SB590885. A, infection of 451Lu melanoma cells with either virus for cyclin D1 or for CDK4. Proteins were extracted from green fluorescent protein (GFP, control), cyclin D1 (CycD1), and CDK4-infected 451Lu melanoma cells before being resolved by Western blotting. Blots were stripped and re-probed for actin to show equal protein loading. B, overexpression of cyclin D1 alone and in combination with CDK4 reduces the growth-inhibitory effect of SB590885. Cells that were infected with the green fluorescent protein virus, virus encoding for cyclin D1, virus for CDK4 or the two viruses encoding for CDK4 and cyclin D1 before being treated with SB590885 (300 nmol/L) for 72 h and analyzed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. Data shows the mean inhibition of cell growth following drug treatment relative to untreated controls (*, P < 0.05 relative to growth inhibition in cells transfected with viral control). C, cell cycle profiles of cells treated with SB590885 that overexpress CDK4 or CDK4/cyclin D1. Cells were infected with virus as described in B before being treated with SB590885 (3 μmol/L) for 24 h. After this time, cells were fixed, harvested, and stained with propidium iodide before being analyzed by flow cytometry.
Discussion
The past 2 years have seen progress in defining new molecular subcategories of melanoma based on patterns of gene amplification and mutation (3, 11, 25). However, how these different mutational profiles and molecular sub-groupings translate into novel strategies for treatment have not been well explored. The initial flurry of excitement following the identification of activating BRAF mutations in melanoma has been tempered by the relatively poor performance of BRAF and MEK inhibitors in early clinical studies (4, 5). Although BRAF is clearly an important oncogene in melanoma, and the MAPK pathway an excellent therapeutic target, there may be other alterations in cell proliferation pathways that convey resistance to BRAF inhibitors. Herein, we identify a distinct group of melanoma cell lines harboring both the BRAF V600E mutation and an activating mutation in CDK4. Additionally, we identified a group of human melanoma samples that have both a BRAF V600E mutation and amplified CCND1, as well as one cell line with a BRAF V600E mutation, amplified CCND1 and a CDK4 mutation.
Typically, BRAF inhibitors block cell cycle entry through the inhibition of MAPK activity leading to decreased cyclin D1 expression and an up-regulation of the CDK inhibitor p27KIP1(8, 26). As the cooperation of cyclin D1 and CDK4 is required for progression through the cell cycle, we hypothesized that activating mutations in CDK4 may increase resistance to BRAF inhibitors. However, we found that the presence of CDK4 mutations did not alter responsiveness to cell cycle arrest following BRAF inhibitor treatment. All of the mutations in CDK4 described in this study (K22Q, R24C, and R24L) had little effect on the responses seen in SB590885 treatment.
A further possible mechanism underlying the resistance to BRAF inhibition could be the increased expression of cyclin D1 resulting from CCND1 amplification. Prior work has shown that groups of non–BRAF/NRAS mutated melanomas arising from chronically sun-damaged skin and mucosal sites have increased copy numbers of CCND1 (11). These studies, based only on primary melanomas, suggest that CDK4 and CCND1 are independent oncogenes in melanomas without BRAF and NRAS mutations (11). In contrast, our data show that there is a subpopulation of metastatic melanomas that carry mutations in either BRAF or NRAS and amplification in CCND1, in some cases at high levels. These data suggest that an increased complexity of genetic changes evolves as the tumor progresses and dysregulation of a pathway may occur at several different proteins.
In general, tumor cell lines show the same genetic and genomic changes as in the “parent” tumor from which they were established, but at an increased frequency in the cell lines (27). In contrast to these findings, we identified a higher frequency of melanoma samples with high-level CCND1 amplification than cell lines. This finding, however, will need to be verified in additional sample sets, as it is possible that it is due to sampling artifact(s). Nonetheless, these data suggest a possible reason for the apparently good preclinical activity of BRAF inhibitors. The only cell line with a high level of CCND1 amplification comparable to that seen in the melanoma samples was WM39. Interestingly, this line contained coexistent CDK4 R24C and BRAF V600E mutations, and was highly resistant to SB590885. To investigate whether increased cyclin D1 expression could regulate SB590885 resistance independently of CDK4 mutational status, we overexpressed cyclin D1 in an otherwise BRAF inhibitor–sensitive cell line. The increased expression of cyclin D1 led to a blunting of the antiproliferative effect of SB590885, suggesting that melanoma cell lines with high basal levels of cyclin D1 are less dependent on BRAF/MEK signaling to drive cell cycle entry. We further showed that this effect was enhanced by the coinfection of cyclin D1 and CDK4, with the cells being rendered highly SB590885-resistant.
It has been previously suggested that the presence of the BRAF V600E mutation conveys sensitivity to BRAF/MEK inhibition (3, 24). Whereas this is certainly true in some cases, the data presented herein seems to show that increased expression in other components of the cell cycle machinery, such as CCND1 amplification, may contribute to a resistant phenotype on a BRAF V600E–mutated background. Seventeen percent of melanoma samples with the BRAF V600E mutation also exhibit some degree of CCND1 amplification, suggesting that this could be an important mechanism of resistance in the clinical population. Our results are consistent with some of the early data coming from clinical trials, in which it has been shown that good levels of intratumoral phosphorylated ERK inhibition can be achieved in the absence of any clinical response (4, 5).
In summary, we have shown that although BRAF V600E–mutated melanomas harbor both mutations in CDK4 and amplifications in cyclin D1, the level of cyclin D1, particularly when seen in concert with increased CDK4, seems to preferentially regulate responsiveness to small molecule BRAF inhibitors. It can therefore be expected that melanoma patients with both BRAF V600E mutations and high levels of CCND1 amplification will show resistance to BRAF inhibition.
Supplementary Material
Acknowledgments
We thank the patients who contributed melanoma samples for this study, without which it would have been possible, and C. Angelica Medina for providing research support on previous iterations of the genetic and genomic data.
Grant support: The NIH (CA76674, CA25874, CA10815, CA93372, CA47159, CA80999, CA098101, CA117881, GM071695) and funds from the Commonwealth Universal Research Enhancement Program, Pennsylvania Department of Health. K.S.M. Smalley is the recipient of a Career Development Award from the National Cancer Institute-Specialized Programs of Research Excellence (CA93372).
Footnotes
Disclosure of Potential Conflicts of Interest: A.J. King is an employee of GlaxoSmithKline. All other authors report no conflicts of interest.
References
- 1.Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007;445:851–7. doi: 10.1038/nature05661. [DOI] [PubMed] [Google Scholar]
- 2.Smalley KSM. A pivotal role for ERK in the oncogenic behaviour of malignant melanoma? Int J Cancer. 2003;104:527–32. doi: 10.1002/ijc.10978. [DOI] [PubMed] [Google Scholar]
- 3.Solit DB, Garraway LA, Pratilas CA, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–62. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rinehart J, Adjei AA, Lorusso PM, et al. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J Clin Oncol. 2004;22:4456–62. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 5.Lorusso PM, Adjei AA, Varterasian M, et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI-1040 in patients with advanced malignancies. J Clin Oncol. 2005;23:5281–93. doi: 10.1200/JCO.2005.14.415. [DOI] [PubMed] [Google Scholar]
- 6.Zhuang L, Lee CS, Scolyer RA, et al. Activation of the extracellular signal regulated kinase (ERK) pathway in human melanoma. J Clin Pathol. 2005;58:1163–9. doi: 10.1136/jcp.2005.025957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haass NK, Sproesser K, Nguyen TK, et al. The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin Cancer Res. 2008;14:230–9. doi: 10.1158/1078-0432.CCR-07-1440. [DOI] [PubMed] [Google Scholar]
- 8.Smalley KS, Haass NK, Brafford PA, Lioni M, Flaherty KT, Herlyn M. Multiple signaling pathways must be targeted to overcome drug resistance in cell lines derived from melanoma metastases. Mol Cancer Ther. 2006;5:1136–44. doi: 10.1158/1535-7163.MCT-06-0084. [DOI] [PubMed] [Google Scholar]
- 9.Hacker E, Muller HK, Irwin N, et al. Spontaneous and UV radiation-induced multiple metastatic melanomas in Cdk4R24C/R24C/TPras mice. Cancer Res. 2006;66:2946–52. doi: 10.1158/0008-5472.CAN-05-3196. [DOI] [PubMed] [Google Scholar]
- 10.Hayward NK. Genetics of melanoma predisposition. Oncogene. 2003;22:3053–62. doi: 10.1038/sj.onc.1206445. [DOI] [PubMed] [Google Scholar]
- 11.Curtin JA, Fridlyand J, Kageshita T, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47. doi: 10.1056/NEJMoa050092. [DOI] [PubMed] [Google Scholar]
- 12.Sauter ER, Yeo UC, von Stemm A, et al. Cyclin D1 is a candidate oncogene in cutaneous melanoma. Cancer Res. 2002;62:3200–6. [PubMed] [Google Scholar]
- 13.Muthusamy V, Hobbs C, Nogueira C, et al. Amplification of CDK4 and MDM2 in malignant melanoma. Genes Chromosomes Cancer. 2006;45:447–54. doi: 10.1002/gcc.20310. [DOI] [PubMed] [Google Scholar]
- 14.Smalley KS, Brafford P, Haass NK, Brandner JM, Brown E, Herlyn M. Up-regulated expression of zonula occludens protein-1 in human melanoma associates with N-cadherin and contributes to invasion and adhesion. Am J Pathol. 2005;166:1541–54. doi: 10.1016/S0002-9440(10)62370-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smalley KS, Contractor R, Haass NK, et al. Ki67 expression levels are a better marker of reduced melanoma growth following MEK inhibitor treatment than phospho-ERK levels. Br J Cancer. 2007;96:445–9. doi: 10.1038/sj.bjc.6603596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards RH, Ward MR, Wu H, et al. Absence of BRAF mutations in UV-protected mucosal melanomas. J Med Genet. 2004;41:270–2. doi: 10.1136/jmg.2003.016667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holland EA, Schmid H, Kefford RF, Mann GJ. CDKN2A (P16(INK4a)) and CDK4 mutation analysis in 131 Australian melanoma probands: effect of family history and multiple primary melanomas. Genes Chromosomes Cancer. 1999;25:339–48. [PubMed] [Google Scholar]
- 18.Greshock J, Naylor TL, Margolin A, et al. 1-Mb resolution array-based comparative genomic hybridization using a BAC clone set optimized for cancer gene analysis. Genome Res. 2004;14:179–87. doi: 10.1101/gr.1847304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naylor TL, Greshock J, Wang Y, et al. High resolution genomic analysis of sporadic breast cancer using array-based comparative genomic hybridization. Breast Cancer Res. 2005;7:R1186–98. doi: 10.1186/bcr1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karolchik D, Baertsch R, Diekhans M, et al. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–4. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vaquerizas JM, Dopazo J, Diaz-Uriarte R. DNMAD: web-based diagnosis and normalization for microarray data. Bioinformatics. 2004;20:3656–8. doi: 10.1093/bioinformatics/bth401. [DOI] [PubMed] [Google Scholar]
- 22.Lingjaerde OC, Baumbusch LO, Liestol K, Glad IK, Borresen-Dale AL. CGH-Explorer: a program for analysis of array-CGH data. Bioinformatics. 2005;21:821–2. doi: 10.1093/bioinformatics/bti113. [DOI] [PubMed] [Google Scholar]
- 23.Yang G, Rajadurai A, Tsao H. Recurrent patterns of dual RB and p53 pathway inactivation in melanoma. J Invest Dermatol. 2005;125:1242–51. doi: 10.1111/j.0022-202X.2005.23931.x. [DOI] [PubMed] [Google Scholar]
- 24.King AJ, Patrick DR, Batorsky RS, et al. Demonstration of a genetic therapeutic index for tumors expressing oncogenic BRAF by the kinase inhibitor SB-590885. Cancer Res. 2006;66:11100–5. doi: 10.1158/0008-5472.CAN-06-2554. [DOI] [PubMed] [Google Scholar]
- 25.Curtin JA, Busam K, Pinkel D, Bastian BC. Somatic activation of KIT in distinct subtypes of melanoma. J Clin Oncol. 2006;24:4340–6. doi: 10.1200/JCO.2006.06.2984. [DOI] [PubMed] [Google Scholar]
- 26.Bhatt KV, Spofford LS, Aram G, McMullen M, Pumiglia K, Aplin AE. Adhesion control of cyclin D1 and p27Kip1 levels is deregulated in melanoma cells through BRAF-MEK-ERK signaling. Oncogene. 2005;24:3459–71. doi: 10.1038/sj.onc.1208544. [DOI] [PubMed] [Google Scholar]
- 27.Greshock J, Nathanson K, Martin AM, et al. Cancer cell lines as genetic models of their parent histology: analyses based on array comparative genomic hybridization. Cancer Res. 2007;67:3594–600. doi: 10.1158/0008-5472.CAN-06-3674. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.