

Abstract
ATase brings about the short-term regulation of glutamine synthetase (GS) by catalyzing the adenylylation and deadenylylation of GS in response to signals of cellular nitrogen status and energy. The adenylyltransferase (AT) activity of ATase is activated by glutamine and by the unmodified form of the PII signal transduction protein and is inhibited by PII-UMP. Conversely, the adenylyl-removing (AR) activity of ATase is activated by PII-UMP and inhibited by unmodified PII and by glutamine. Here, we show that the enzyme can be reconstituted from two purified polypeptides that comprise the N-terminal two-thirds of the protein and the C-terminal one-third of the protein. Properties of the reconstituted enzyme support recent hypotheses for the sites of regulatory interactions and mechanisms for intramolecular signal transduction. Specifically, our results are consistent with the protein activators (PII and PII-UMP) binding to the enzyme domain with the opposing activity, with intramolecular signal transduction by direct interactions between the N-terminal AR catalytic domain and the C-terminal AT catalytic domain. Similarly, glutamine inhibition of the AR activity involved intramolecular signaling between the AT and AR domains. Finally, our results are consistent with the hypothesis that the AR activity of the N-terminal domain required activation by the opposing C-terminal (AT) domain.
Adenylyltransferase (ATase,1 EC 2.7.7.49) mediates short-term regulation of glutamine synthetase (GS) activity in response to signals of nitrogen, carbon, and energy status in Escherichia coli, by catalyzing the adenylylation and deadenylylation of GS (reviewed in refs (1) and (2)). The ATase was one of the first signal transduction enzymes identified (3,4) and provided the first example of signal transduction by reversible covalent adenylylation (5), yet despite its historical significance and intense early efforts to study the enzyme and signal transduction system (6−8), the complexity of the enzyme and its regulation has provided major challenges to elucidation of its structure and regulation. Its mechanisms of regulation are only beginning to emerge (9−12). The ATase and its substrate, GS, are part of a signal transduction system that includes two additional proteins, uridylyltransferase/uridylyl-removing enzyme (UTase/UR, EC 2.7.7.59) and the PII protein, in which there are two linked cycles of covalent modification, forming a bicyclic system (Figure 1A; reviewed in refs (1), (2), and (13)). The bifunctional UTase/UR protein catalyzes the uridylylation and deuridylylation of the PII protein in one cycle of covalent modification, and PII and PII-UMP regulate the opposing AT and AR activities of the ATase in modification of GS in the other cycle (Figure 1A). The PII and UTase/UR proteins also participate in another signal transduction bicycle with the NRI and NRII proteins that regulate transcription of nitrogen-regulated genes, including the gene encoding GS (reviewed in ref (1)). Thus, GS is subjected to both short-term control by covalent modification and long-term control by regulation of its gene transcription and the synthesis of the enzyme.
Figure 1.

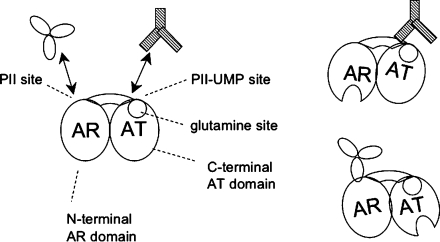
ATase and its role in the signal transduction system controlling glutamine synthetase activity. (A) Circuitry of the UTase/UR-PII-ATase-GS bicyclic signal transduction system controlling GS activity in E. coli. For details, see the text. For the sake of simplicity, only the effects of α-ketoglutarate at high concentrations are depicted and the effects of adenylylate energy charge on PII and PII-UMP activities are not depicted. Activators of catalytic activities are depicted with small arrowheads, while inhibitors of catalytic activities are depicted with small blunt-ended lines (adapted from ref (12)). (B) Model for the effects of activators and inhibitors in controlling the AT and AR activities of ATase. As depicted, the activators and inhibitors function by mediating the transitions between different enzyme forms with different activities. Boxed enzyme species display catalytic activities as follows: E-Gln, E-PII, and E-Gln-PII display AT activity, while E-PII-UMP displays AR activity (adapted from ref (11)). (C) Schematic representation of the proposed domain relationships and regulatory mechanism of ATase. The enzyme is depicted as consisting of two NT domains (an N-terminal AR domain and a C-terminal AT domain), connected by a central region. PII and PII-UMP are depicted as binding to two distinct sites, at the junctions between the NT domains and the central region. Glutamine is depicted as binding to a site on the C-terminal domain, which is near the PII-UMP binding site. (D) Proposed mechanism for PII-UMP activation of the AR activity of ATase. As depicted, PII-UMP alters the conformation of the C-terminal AT domain of ATase, and this in turn interacts with the AR domain of ATase and activates the AR activity. Activation of the AR domain is depicted as the opening of the active site. (E) Proposed mechanism for PII activation of the AT activity of ATase. As depicted, PII alters the conformation of the N-terminal AR domain of the ATase, and this in turn interacts with and activates the C-terminal AT domain of ATase.
Glutamine synthetase is the key enzyme of ammonia assimilation in E. coli and many other bacteria, and regulation of its activity by reversible covalent modification is required for efficient growth when cells are subjected to changes in the availability of ammonia (14). The adenylylation of GS on Y397 during conditions of nitrogen excess reduces the activity of the GS enzyme and renders the enzyme more sensitive to feedback inhibition (reviewed in ref (2)). This ability to rapidly inactivate GS is important when cells that are nitrogen starved are subjected to ammonia shock; adenylylation prevents a dramatic reduction in the size of the cellular glutamate pool that otherwise would occur as a consequence of ammonia shock (14). The dramatic reduction in the cellular glutamate pool causes a significant growth defect, presumably due to stress at numerous points in metabolism (14). Conversely, deadenylylation of GS∼AMP during conditions of nitrogen limitation results in reactivation of the enzyme. Since GS is a dodecamer, it may contain from zero to 12 adenylyl groups per dodecamer. Early studies suggested that the adenylylation of GS subunits was not processive and that the effects of adenylylation affected only the modified subunit (mentioned in ref (2)), but these issues merit further attention. Under many conditions allowing rapid growth, E. coli contains partially or completely modified GS. This pool of inactive enzyme may be rapidly activated by deadenylylation if it suddenly becomes necessary for the cell to scavenge for ammonia. Apparently, there is a significant selective advantage to maintaining a pool of reserve GS capacity that can be rapidly activated.
The adenylylation and deadenylylation reactions catalyzed by ATase are not the reversal of one another and occur at two distinct active sites (10,15; reviewed in ref (1)). Chemically, the reactions are distinct, as the adenylylation reaction produces PPi and the deadenylylation reaction consumes Pi and produces ADP [it is a phosphorylosis (16)]. The adenylylation and deadenylylation reactions are regulated coordinately, avoiding futile cycling (10,11; Figure 1A,B). The AT reaction is activated by glutamine and by the unmodified form of the PII signal transduction protein. PII and glutamine also inhibit the AR reaction. Conversely, the AR reaction is activated by the uridylylated form of the PII protein, PII-UMP, and this also inhibits the AT activity. Kinetic analysis of the wild-type enzyme and its regulation suggested that the activators and inhibitors had little effect on the binding of substrates and products but instead shifted the enzyme between conformations with different activities (10,11) (Figure 1B). Enzymological and kinetic studies with various mutant forms of the enzyme allowed a detailed hypothesis for the location of binding sites and regulatory mechanisms (10,11) (Figure 1C−E), as described below.
The ATase protein appears to be a monomer and consists of N-terminal and C-terminal NT domains (17) separated by a central region that plays a role in mediating the interactions of the N- and C-terminal domains (10,11,15,18). Point mutations of the NT domain active sites followed by purification and characterization of the altered enzymes showed that the AT activity was eliminated by alteration of the C-terminal active site, while the AR activity was eliminated by alteration of the N-terminal domain active site (10). Appealingly, the isolated C-terminal domain displayed AT activity that was regulated by glutamine (10). These and other data suggest that the glutamine binding site is located within the C-terminal AT domain (10). Conversely, no truncated form of the N-terminal domain had significant AR activity in our hands (10). Others have reported a very low level of AR activity from an N-terminal polypeptide (15,19). Various lines of evidence suggested that the enzyme contained multiple sites for PII and PII-UMP; specifically, a PII site appeared to be associated with the N-terminal domain, while a PII-UMP site appeared to be associated with the C-terminal domain (10,11). In both cases, binding of PII and PII-UMP was greatly improved by the presence of the central part of the ATase, suggesting that either the binding sites were partially within the central region or the central region played a role in stabilizing a conformation of the N- and C-terminal domains (10). Kinetic analysis suggested that the binding of glutamine and PII-UMP to the enzyme was competitive (11). The synergy between PII and glutamine binding required proper interaction of the two domains mediated by the central region of ATase (10). Indeed, mutations within the central region of the protein or simple truncation of the N-terminal two-thirds of the ATase resulted in a dramatic reduction in the glutamine activation constant (Kact) for activation of the AT activity of the C-terminal domain. These observations and others led to the regulatory hypothesis shown in Figure 1C−E. In this hypothesis, activation of the AR activity of the N-terminal domain by PII-UMP is indirect; PII-UMP binding to the C-terminal domain causes the domain to activate the N-terminal domain AR activity (Figure 1D). Furthermore, according to this hypothesis, PII activation of the AT activity is also indirect; PII binds to the N-terminal AR domain, and this domain interacts with the AT domain and transmits the PII activation signal (Figure 1E).
As a prelude to investigating this hypothesis, we tested whether different polypeptides derived from ATase could interact to produce the AR activity. That is, we examined whether the AR activity could be reconstituted from fragments of the ATase. Our earlier studies resulted in several polypeptides that contained various portions of the ATase (10); these were combined in all combinations (10). It was observed that in one case, a pair of polypeptides clearly associated with one another as observed by nondenaturing gel electrophoresis (10). This pair of polypeptides, called ATN6 and ATC3 (Figure 2A), when complexed together, results in the wild-type ATase missing one peptide bond, but in no case was AR activity obtained from combinations of polypeptides, including the case of the ATC3 and ATN6 pair (10). We will show here that our earlier failure to observe AR activity with this pair of polypeptides was due to the presence of ADP in the assay mixtures, and that in fact ATC3 and ATN6 polypeptides reconstitute the ATase, and their complex displays AR activity that is regulated by PII-UMP, PII, and glutamine (see ).
Figure 2.

Reconstitution of the AR activity of ATase. (A) Schematic depiction of the domains comprising the ATC3 and ATN6 polypeptides. The numbers refer to the first and last amino acid residues in each species using the codon numbers for the wild-type full-length protein. (B) Nondenaturing 14% polyacrylamide gel electrophoresis of polypeptides in isolation and combination. Conditions for gel electrophoresis lacked any known regulators of ATase but included 1 mM MgCl2. Arrows indicate the position of the major ATN6 and ATC3 bands, as well as the position of the complex between these polypeptides. Each sample contained the indicated polypeptides at 6 μM each. Samples were incubated for 10 min at room temperature to allow the formation of complexes, prior to electrophoresis at 4 °C. (C) Reconstitution of the AR activity from the ATC3 and ATN6 polypeptides using an ATP-regenerating system to remove ADP. The initial rate of GS-AMP deadenylylation was measured as described in , with GS-AMP at 8 μM, PII-UMP at 5 μM, ATP at 1 mM, α-ketoglutarate at 1 mM, KPi at 1 mM, pyruvate kinase at 0.022 unit/μL, and PEP at 5 mM. (D) Reconstitution of the AR activity from the ATC3 and ATN6 polypeptides using the noncleavable analogue AMP-PNP in place of ATP. The initial rate of GS-AMP deadenylylation was measured as described in , with GS-AMP at 16 μM, PII-UMP at 5 μM, α-ketoglutarate at 1 mM, KPi at 1 mM, and AMP-PNP at 1 mM.
Since the time of our initial studies of ATase polypeptides (10), it has become apparent that PII not only is a sensor of cellular carbon and nitrogen status, due to its binding of α-ketoglutarate, but also is controlled by the adenylylate energy charge, due to the competitive binding of ADP and ATP to the three nucleotide-binding sites of the PII trimer (12). In reconstituted signal transduction systems and the individual reactions that comprise these larger systems, ADP acted to antagonize the effects of α-ketoglutarate in controlling PII and PII-UMP functions. ADP was a particularly potent inhibitor of the AR reaction, by acting at several levels, including direct inhibition of the activity (it is a product inhibitor) and by powerful inhibition of the ability of PII-UMP to activate the AR activity (12). Furthermore, ADP binding to any unmodified PII that happens to be present in the reconstituted systems results in strong activation of PII binding to ATase and shifting of the enzyme to the AT form, activating the AT and inhibiting the AR activity (12). As these powerful effects of ADP became apparent, we revisited the issue of whether any of our purified polypeptides and combinations of polypeptides displayed AR activity. As we had reported, these polypeptides were of various purity (10). Not unexpectedly, our polypeptides displayed contaminating ATPase activity. When this ATPase activity was counteracted by the presence of an ATP-regenerating system, or when the noncleavable analogue AMP-PNP was used in place of ATP, we observed that one pair of polypeptides, the ATN6 and ATC3 pair, formed a complex that displayed AR activity and other properties of the intact ATase. This reconstituted form of the enzyme allowed us to investigate various aspects of the hypothesis for ATase regulation by glutamine, PII, and PII-UMP.
Materials and Methods
Preparations of ATase, polypeptides derived from ATase, PII, GS, and UTase/UR that were described previously (10−12) were used. ATase and AR reactions were as described previously (10), with detailed conditions for each experiment as described in the figure legends. Briefly, the AT reaction measures the incorporation of the α label from [α-32P]ATP into GS, using TCA precipitation of the incorporated label and trapping of the precipitates onto nitrocellulose filters. The AR reaction measures the release of label from GS-[32P]AMP. This release is quantified by measurement of the release of label from TCA-precipitatable material. For AT and AR activity measurements where an ATP-regenrating system was used, pyruvate kinase (Sigma) was present at a concentration of 0.022 unit/μL and phosphoenolpyruvate (PEP) was present at a concentration of 3−5 mM, as indicated. ATPase activity of the polypeptides derived from ATase was measured using a coupled assay system based on pyruvate kinase and lactate dehydrogenase (20). Nondenaturing gel electrophoresis was performed as described previously (10).
Results
The ATN6 and ATC3 Polypeptides Form a Complex with AR Activity
Previous results showed that the ATC3 and ATN6 polypeptides (Figure 2A) form a 1:1 complex that could be readily detected by nondenaturing polyacrylamide gel electrophoresis (10). Since our previous experiments all contained various regulators of the enzyme (glutamine, PII, α-ketoglutarate, and ATP), we extended our nondenaturing gel electrophoresis analysis to examine the formation of a complex between these polypeptides in the absence of any regulatory species; the complex was readily obtained under these conditions (Figure 2B). Our prior studies did not detect AR activity from the complex of ATC3 and ATN6 (10). Since the AR reaction is extraordinarily sensitive to ADP, we measured the ATPase activity of our purified polypeptides using a coupled enzyme method (20) (Figure S1 of the Supporting Information). The different polypeptides derived from ATase had variable levels of ATPase contamination, and the level of ATPase did not directly correspond to the degree of purification as estimated by visual inspection of gels (Figure S1). To compensate for this ATPase activity, we repeated our measurement of AR activity of polypeptides and combinations of polypeptides, using two different approaches. In one approach, we conducted measurements in the presence of an ATP-regenerating system (Figure 2C). In the other approach, the noncleavable analogue AMP-PNP was used in place of ATP (Figure 2D). Of the polypeptides examined (Figure S1), only the ATN6 and ATC3 pair of polypeptides displayed AR activity when combined (Figure 2C,D). The level of reconstituted AR activity, while easily measurable and ∼15−25-fold above the background, was only ∼1.7% of the wild-type AR activity under similar conditions. Note that the “background” rate in panels C and D of Figure 2, due to the nonspecific loss of the label from the filters, was similar to the rates obtained when ATC3 or ATN6 was present in the absence of the other, indicating that the individual polypeptides lacked significant AR activity. Since the level of reconstituted activity was low relative to the activity of the wild-type enzyme, the experiment shown in Figure 2C was repeated two additional times, and in both cases, the combination of the ATC3 and ATN6 polypeptides provided an AR rate that was more than 15-fold above the background rates, as in Figure 2C (not shown). Furthermore, as shown in panels C and D of Figure 2, the errors for duplicate samples within the same experiment were acceptably low, and generally less than 10%. Thus, AR activity was unambiguously observed. In additional experiments, we examined the dependence of the AR activity on PII-UMP, using the ATP-regenerating system to remove ADP, analogous to the experiments shown in Figure 2C. We observed that omission of PII-UMP resulted in rates similar to the background rate (not shown). That is, as with the wild-type enzyme (10), the AR activity required PII-UMP.
As expected on the basis of the wild-type enzyme, the AR activity of the reconstituted enzyme was inhibited by glutamine, by PII, and by the combination of PII and glutamine (Figure 2C,D). We note that the regulation by these effectors was discernibly sharper and more reminiscent of the published results for the wild-type enzyme in the system where AMP-PNP was used in place of ATP (Figure 2D). Under these conditions, combination of PII and glutamine resulted in essentially complete inhibition of the AR activity, as previously observed with the intact enzyme. By contrast, in the experiments where an ATP-regenerating system was used (Figure 2C), regulation by PII, glutamine, and the combination of these two effectors was less dramatic, and the AR activity was not completely inhibited when both PII and glutamine were present. Apparently, a component of the ATP-regenerating system (PEP, pyruvate kinase, or a contaminant of these components) moderately reduces the inhibition of the AR activity. In either case, the inhibition of the AR activity by PII and glutamine provides strong support for the idea that the AR activity measured was due to reconstitution of the ATase.
PII Activation of the AT Activity of ATC3 Was Mediated by the ATN6 Polypeptide
Unlike wild-type ATase, the ATC3 polypeptide had basal AT activity in the absence of any activators (10) (Figure 3). This activity was strongly inhibited by the ATN6 polypeptide, which when present at a saturating concentration almost completely inhibited the basal AT activity (Figure 3). As before, the ATC3 polypeptide was activated by glutamine (10) (Figure 3), and we observed that AT activity in the presence of both glutamine and ATN6 was ∼5-fold higher than the strongly inhibited rate obtained when ATN6 was present in the absence of glutamine (Figure 3A).
Figure 3.

ATN6 mediated the regulation of ATC3 by PII, regulated the basal AT activity of ATC3, and mediated inhibition of the ATC3 AT activity by PII-UMP. (A) ATN6 mediates PII regulation of ATC3 AT activity. The initial rate of GS adenylylation was measured as described in , with GS at 2.5 μM, α-ketoglutarate at 0.05 mM, [α-32P]ATP at 0.5 mM, pyruvate kinase at 0.022 unit/μL, PEP at 3 mM, bovine serum albumin at 0.3 mg/mL, and ATC3 at 0.25 μM. (B) ATN6 inhibits the basal AT activity of ATC3 and mediates PII activation of the ATC3 AT activity. Conditions were as described in and for panel A, except that the concentration of the enzyme ATC3 was increased to 0.8 μM to facilitate examination of the basal AT activity. (C) ATN6 mediates PII-UMP inhibition of the AT activity of ATC3 when glutamine was at the Kact. Conditions were as described in , with GS at 2.5 μM, ATP at 0.5 mM, α-ketoglutarate at 1 mM, ATC3 at 0.025 μM, glutamine at 1.5 mM, pyruvate kinase at 0.022 unit/μL, and PEP at 5 mM. (D) ATN6 mediates PII-UMP inhibition of the AT activity of ATC3 when glutamine was at a saturating level. Conditions were as described in and for panel C, except that glutamine was present at a concentration of 20 mM and ATC3 was present at a concentration of 0.125 μM.
We repeatedly observed that PII failed to activate the AT activity of the ATC3 polypeptide (10); instead, PII reproducibly was a very weak inhibitor of the ATC3 polypeptide AT activity (Figure 3). (In addition to the results shown in Figure 3, this result was obtained in four additional experiments on different days, where under similar conditions the level of inhibition of the ATC3 AT activity by PII ranged from 6 to 12%. Furthermore, as shown in Figure 3, errors for duplicate samples in the same experiment were acceptable and generally less than 10%.) Yet when PII and ATN6 were both present, the AT level was ∼7-fold higher than when ATN6 was present without PII and slightly lower than that in the control experiment lacking both PII and ATN6 (Figure 3). That is, PII appeared to eliminate the inhibition by ATN6. These results could be explained by PII activation of a reconstituted enzyme or by PII causing the dissociation of the ATN6−ATC3 complex, relieving the inhibition of ATC3, but results from experiments where glutamine was present were highly instructive. When ATC3 was present in the absence of ATN6, the AT rate was activated ∼3-fold by glutamine at 1.5 mM, which is the Kact for the ATC3 polypeptide, as opposed to the Kact of 7 mM for the intact enzyme. This activation by glutamine was little affected by PII (Figure 3), but when ATN6, glutamine, and PII were all present, the AT rate was activated 16-fold over the rate obtained with ATC3 alone (Figure 3). This shows that the ATN6 polypeptide mediated the activation of the AT activity of the ATC3 polypeptide by PII, consistent with the hypothesis shown in Figure 1C, and that PII and glutamine worked synergistically to activate the combination of the ATN6 and ATC3 polypeptides, reminiscent of their activation of the native enzyme (9,10).
The Basal AT Activity of the ATC3 Polypeptide Was Inhibited by the ATN6 Polypeptide
To study the basal AT activity of the ATC3 polypeptide more carefully, a higher concentration of the ATC3 polypeptide was used (Figure 3B). Under these conditions, the basal activity of the ATC3 polypeptide was again strongly inhibited by the ATN6 polypeptide (∼50-fold). PII and PII-UMP provided very slight inhibition of the ATC3 basal rate. As in Figure 3A, the combination of ATN6 and PII resulted in an AT rate that was nearly that seen in the absence of ATN6 and PII. That is, PII again appeared to eliminate the inhibition afforded by ATN6. Remarkably, when both ATN6 and PII-UMP were present, the basal AT activity of the ATC3 polypeptide was similar to that obtained with ATN6 alone. That is, unlike PII, PII-UMP could not alleviate the inhibition afforded by ATN6. This is reminiscent of the wild-type ATase enzyme, whose AT activity is activated by PII but not by PII-UMP. When ATN6, PII, and PII-UMP were all present simultaneously, an intermediate level of AT activity was obtained that likely reflected the multiple species present.
The ATN6 Peptide Mediated PII-UMP Inhibition of the AT Activity of the ATC3 Peptide
When the basal AT activity of ATC3 was strongly inhibited by ATN6, it was not possible to study further inhibition of the AT activity by PII-UMP. To study the inhibition of the AT activity of the ATC3 polypeptide by PII-UMP and the effects of PII-UMP on the reconstituted enzyme, we examined this inhibition in the presence of glutamine at two concentrations: its Kact for the ATC3 polypeptide [1.5 mM (10) (Figure 3C)] and a high concentration [20 mM (Figure 3D)]. When glutamine was at the ATC3 Kact, ATN6 was a potent inhibitor, reducing the AT activity to ∼14% of the uninhibited rate (Figure 3C). (Since the starting activity was high due to glutamine activation, the inhibited rate was still easily measured, as planned.) Addition of PII-UMP under these conditions resulted in a significant further decrease in the ATase activity, which was easily measured (Figure 3C). By contrast, when glutamine was at a concentration of 20 mM, the ATN6 polypeptide was only a weak inhibitor of the ATC3 AT activity, providing ∼40% inhibition (Figure 3D). Comparison of panels C and D of Figure 3 thus shows that glutamine antagonized the effect of the ATN6 polypeptide. When glutamine was at a concentration of 20 mM, PII-UMP again provided significant inhibition of the AT activity (Figure 3D), but this inhibition was not as great as that obtained when glutamine was at a concentration of 1.5 mM (compare panels C and D of Figure 3). This is as expected, as prior work with the intact ATase showed that PII-UMP and glutamine competed for the enzyme (10). These results show that when glutamine was present, the ATN6 polypeptide mediated inhibition of the AT activity of the ATC3 polypeptide by PII-UMP.
The Reconstituted Enzyme Exhibits Normal Activation by PII
In Figure 3, it was observed that the ATN6 polypeptide mediated the activation of the AT activity by PII and that this activation was synergistic with activation by glutamine. To examine this synergy in more detail, we measured the glutamine Kact for the reconstituted enzyme in the presence and absence of PII. The wild-type enzyme displays strong synergy in activation by PII and glutamine; the presence of either of these activators dramatically reduced the Kact for activation by the other activator (9). To ensure that the ATC3 polypeptide was mostly contained within the reconstituted enzyme, these experiments were conducted under conditions with a large excess of the ATN6 polypeptide (Figure 4). The reconstitited enzyme exhibited a glutamine Kact of ∼ 7 mM (Figure 4A), similar to the that of intact enzyme (9) and higher than the 1.5 mM glutamine Kact of the ATC3 polypeptide (10). When PII was present at a saturating concentration (15 μM), the glutamine Kact of the reconstituted enzyme was ∼0.55 mM (Figure 4B), remarkably reminiscent of values obtained for the wild-type enzyme of 0.45−0.5 mM (9,10). Thus, it appears that the synergy between glutamine and PII exhibited by the intact enzyme was also displayed, essentially in full measure, by the reconstituted enzyme.
Figure 4.

PII activation of the reconstituted enzyme displays synergy with glutamine and biphasic sensitivity to α-ketoglutarate, as observed with the wild-type enzyme. (A) Activation of the AT activity of the reconstituted enzyme by glutamine. Conditions were as described in , with GS at 2.5 μM, ATP at 0.5 mM, α-ketoglutarate at 0.05 μM, ATC3 at 0.25 μM, ATN6 at 10 μM, pyruvate kinase at 0.022 unit/μL, and PEP at 5 mM. As shown, the apparent Kact for glutamine was ∼7 mM. In general, errors for these types of experiments in our laboratory are typically on the order of 10−15%. (B) Activation of the AT activity of the reconstituted enzyme by glutamine in the presence of a saturating concentration of PII. Conditions were as described for panel C, except that PII was present at 15 μM and ATC3 was present at 0.05 μM. As shown, the apparent Kact for glutamine was ∼0.55 mM. In general, errors for these types of experiments in our laboratory are typically on the order of 10−15%. (C) Biphasic effects of α-ketoglutarate on the activation of the AT activity of the reconstituted enzyme. Conditions were as described for panel A, with GS at 2.5 μM, ATP at 0.5 mM, ATC3 at 1.6 μM, ATN6 at 5 μM, PII at 5 μM, PK at 0.022 unit/μL, and PEP at 5 mM. In general, errors for these types of experiments in our laboratory are typically on the order of 10−15%.
Another characteristic and unusual aspect of the activation of the AT activity by PII is that the response to α-ketoglutarate is biphasic (9−11). The effector α-ketoglutarate binds to PII and controls its ability to activate its receptors (reviewed in refs (1) and (21)). Remarkably, in the case of ATase, the effects of α-ketoglutarate are not related to changes in the binding of PII to the ATase (11). Apparently, α-ketoglutarate regulates a step in the activation process that occurs after the binding of PII to the ATase. The biphasic nature of the response to α-ketoglutarate is due to the anticooperativity of binding of this effector in the presence of ATP, as studied here. PII trimers that are liganded to a single molecule of α-ketoglutarate are very effective in activating the AT activity, while PII trimers that are saturated with three molecules of α-ketoglutarate are not effective in activating the AT activity. Thus, when PII and ATase are present, as the level of α-ketoglutarate is increased the AT activity rises and then declines as PII trimers become liganded by one molecule of α-ketoglutarate and then become saturated with α-ketoglutarate, respectively, resulting in a biphasic rate versus saturation plot (9−11). Since biphasic rate versus saturation plots are unusual, we examined whether our reconstituted enzyme displayed this phenomenon and observed that it did (Figure 4C). This is consistent with the activation of the reconstituted enzyme by PII representing the same processes occurring in the intact wild-type enzyme.
The ATN6 Polypeptide Was an Indirect Inhibitor and Activator of ATase That Bound to PII or PII-UMP
Since the intact full-length ATase contains the AT domain found in the ATC3 polypeptide, we also investigated whether ATN6 could interact with the full-length ATase. We did not obtain any evidence from nondenaturing gel electrophoresis experiments that ATN6 and ATase could interact (not shown). The ATN6 polypeptide, at high concentration, had little effect on the glutamine-activated AT activity of ATase but resulted in ∼5-fold inhibition of the PII-activated AT activity of ATase (Figure 5A). Similarly, ATN6 resulted in a significant reduction in the AT activity of ATase when both PII and glutamine were present (Figure 5A). This inhibition of PII activation afforded by ATN6 was most likely due to the binding and sequestration of PII by ATN6. Similarly, under certain conditions, ATN6 can serve as an indirect activator of the AT activity of ATase, by its ability to bind and sequester the inhibitor PII-UMP (Figure 5B). For example, when glutamine is the sole activator of the ATase activity, PII-UMP provides significant inhibition of the AT activity of ATase (Figure 5B). Prior work showed that this inhibition by PII-UMP was competitive with glutamine, suggesting that only one of these two effectors can be bound to the ATase at any given time (10,11). In the presence of glutamine and PII-UMP, addition of ATN6 provided modest activation of the AT activity. Presumably, it did so because of its ability to bind and sequester the inhibitor, PII-UMP.
Figure 5.

ATN6 acted as an indirect activator or inhibitor of ATase by sequestering inhibitors and activators. (A) ATN6 acts as an inhibitor of ATase, by sequestering the activator PII. The initial rate of GS adenylylation by full-length, wild-type ATase (0.2 μM) was determined as described in , with GS at 2.5 μM, α-ketoglutarate at 0.05 mM, [α-32P]ATP at 0.5 mM, pyruvate kinase at 0.022 unit/μL, and PEP at 3 mM. In general, errors for these types of experiments in our laboratory are typically on the order of 10−15%. (B) ATN6 acts as an activator of ATase, by sequestering the inhibitor PII-UMP. Procedures and conditions were as described for panel A, except that ATase was present at 1.0 μM. In general, errors for these types of experiments in our laboratory are typically on the order of 10−15%.
Discussion
We describe a reconstituted form of the ATase protein that differs from the normal wild-type enzyme by the lack of the peptide bond normally found between amino acid residues 608 and 609 in the 946-amino acid ATase. This reconstituted form of the enzyme, formed from separately purified polypeptides, allowed us to examine a detailed hypothesis for regulation of the ATase that resulted from prior studies. Specifically, we examine the roles of interactions between the AT and AR catalytic domains of ATase in regulation by glutamine, PII, and PII-UMP.
The isolated ATC3 polypeptide, corresponding approximately to the catalytic C-terminal AT domain of ATase, displayed basal AT activity in the absence of any activators, and this AT activity was further stimulated by glutamine, with a glutamine Kact of ∼1.5 mM. By contrast, the full-length, wild-type ATase displays little basal activity and is activated by glutamine with a Kact of ∼7 mM. Here, we observed that the reconstituted enzyme displays properties similar to those of the intact enzyme low basal activity and glutamine activation with a Kact of ∼7 mM. Thus, the ATN6 polypeptide clearly acted to inhibit the AT activity of the ATC3 polypeptide and to influence the binding of glutamine. Similar effects on the binding of glutamine were previously described for mutations in the central region of ATase that, presumably, altered the interactions of the two ATase catalytic domains (10). Together, these observations are most easily explained by the hypothesis that the catalytic domains have extensive interactions and regulate each other‘s activities (Figure 1C−E). Indeed, the ability of the ATN6 polypeptide to inhibit the basal activity of ATC3 was antagonized by glutamine. Thus, we argue that glutamine not only activated the AT activity of the C-terminal AT domain but also altered the conformation of this domain in such a way that the interactions between domains of ATase were altered. In the case of the reconstituted enzyme, glutamine may regulate the fraction of the polypeptides that are in the complex relative to the fraction that are uncomplexed.
The activation of the wild-type ATase by PII and glutamine displays strong synergy, in that each activator dramatically decreases the Kact for the other (9). Experiments in which the binding of PII to the ATase was directly examined by gel-filtration chromatography or nondenaturing gel electrophoresis indicated that under certain conditions, glutamine was required to detect the complex of PII and ATase (10). Furthermore, the form of the enzyme liganded to both activators displayed the highest Kcat(11). This synergy between glutamine and PII required proper interaction of the two catalytic domains of ATase; mutations altering the central region could eliminate activation by PII while having essentially no effect on the binding of PII. Thus, we hypothesized that PII binding to the N-terminal part of ATase caused an alteration of the AR catalytic domain that in turn was communicated to the AT domain via extensive interactions between the two catalytic domain of ATase (Figure 1E). Here, we observed that the ATN6 polypeptide mediated regulation of the AT activity by PII, and that this activation by PII was synergistic with glutamine. This is entirely consistent with and provides strong support for the hypothesis. Furthermore, as predicted by the hypothesis, PII had a dramatic effect on the glutamine Kact of the reconstituted enzyme, almost exactly as obtained with the wild-type enzyme. Other aspects of PII activation of the reconstituted enzyme, such as the biphasic effects of α-ketoglutarate, were as expected on the basis of the wild-type enzyme, suggesting that PII activates the reconstituted enzyme by the same mechanisms used with the intact enzyme.
We also showed that the AR activity of the N-terminal domain of ATase was displayed by our reconstituted enzyme, although at a level much lower than that displayed by the wild-type enzyme. This AR activity was not detectable with the ATN6 polypeptide, and thus, for this activity, the ATC3 polypeptide may be considered to be an essential activator of the ATN6 polypeptide. This is consistent with the hypothesis (Figure 1D). At this time, we have no explanation for the low level of AR activity displayed by the reconstituted enzyme, relative to the intact wild-type enzyme. Perhaps the absence of a peptide bond between amino acid residues 608 and 609 of the enzyme reduces the ability of the enzyme to obtain the optimal conformation for AR activity. The AR activity of the reconstituted enzyme required PII-UMP, as is the case with the wild-type protein. We expect that the ATC3 polypeptide may be required for the binding of PII-UMP by the complex, although our results are also consistent with the possibility that PII-UMP binds to the N-terminal ATN6 polypeptide, with ATC3 being required to allow the active enzyme conformation. Similarly, inhibition of the AT activity of the ATC3 polypeptide by PII-UMP also required the ATN6 polypeptide. This, again, might indicate that the interactions of the catalytic domains within the reconstituted enzyme were required to obtain the enzyme conformation that binds to PII-UMP.
Finally, it is very clear that the reconstituted enzyme, like the wild-type enzyme, was able to distinguish between PII and PII-UMP. For example, PII was an activator of the AT activity of the reconstituted enzyme, while having only minor effects on the isolated ATC3 polypeptide. PII-UMP was an inhibitor of the AT activity of the reconstituted enzyme, while having only minor effects on the isolated ATC3 polypeptide. Thus, the interaction of ATC3 with ATN6 to reconstitute the enzyme results in sensitivity not only to PII and PII-UMP but also to the opposing effects of these regulators. These results again imply that the same regulatory processes occurring in the intact wild-type enzyme are also occurring in the reconstituted version of the enzyme.
Acknowledgments
We thank Patrick O’Brien for reviewing an earlier version of this work and for providing numerous helpful suggestions.
Supporting Information Available
SDS−PAGE for each polypeptide that was examined and the ATPase activity obtained from each polypeptide preparation (Figure S1) and the map of the peptides compared to the intact ATase (Figure S2). This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: ATase, adenylyltransferase (EC 2.7.7.49), product of glnE; AT, adenylyltransferase activity of ATase; AR, adenylyl-removing (deadenylylation) activity of ATase; UTase/UR, uridylyltransferase/uridylyl-removing enzyme (EC 2.7.7.59), product of glnD; PII, signal transduction protein, product of glnB; PII-UMP, uridylylated form of PII; GS, glutamine synthetase, product of glnA; GS∼AMP, adenylylated form of GS; NRII, nitrogen regulator II, NtrB, product of glnL (ntrB); NRI, nitrogen regulator I, NtrC, product of glnG (ntrC); NT, nucleotidyl transferase domain; AMP-PNP, adenylyl imidodiphosphate.
Supplementary Material
References
- Ninfa A. J.; Jiang P.; Atkinson M. R.; Peliska J. A. (2000) Integration of antagonistic signals in the regulation of bacterial nitrogen assimilation. Curr. Top. Cell. Regul. 36, 32–76. [DOI] [PubMed] [Google Scholar]
- Rhee S. G., Chock P. B., and Stadtman E. R. (1989) Regulation of Escherichia coli glutamine synthetase. In Advances in Enzymology (Meister A., Ed.) Vol. 62, pp 37−92, John Wiley and Sons, New York. [DOI] [PubMed] [Google Scholar]
- Kingdon H. S.; Shapiro B. M.; Stadtman E. R. (1967) Regulation of glutamine synthetase. VIII. ATP:glutamine synthetase adenylyltransferase, an enzyme that catalyzes alterations in the regulatory properties of glutamine synthetase. Proc. Natl. Acad. Sci. U.S.A. 58, 1703–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff I.; Mecke D.; Holzer H. (1967) Mechanism of the enzymatic inactivation of glutamine synthetase from E. coli. Biochem. Biophys. Res. Commun. 28, 740–745. [DOI] [PubMed] [Google Scholar]
- Shapiro B. M.; Stadtman E. R. (1968) 5′-Adenylyl-O-tyrosine, the novel phosphodiester residue of adenylylated glutamine synthetase from Escherichia coli. J. Biol. Chem. 243, 3769–3771. [PubMed] [Google Scholar]
- Caban C. E.; Ginsberg A. (1976) Glutamine synthetase adenylyltransferase from Escherichia coli: Purification and physical and chemical properties. Biochemistry 15, 1569–1580. [DOI] [PubMed] [Google Scholar]
- Adler S. P.; Purich D.; Stadtman E. R. (1975) Cascade control of Escherichia coli glutamine synthetase. J. Biol. Chem. 250, 6264–6272. [PubMed] [Google Scholar]
- Engleman E. G.; Francis S. H. (1978) Cascade control of E. coli glutamine synthetase. Arch. Biochem. Biophys. 191, 602–612. [DOI] [PubMed] [Google Scholar]
- Jiang P.; Peliska J. A.; Ninfa A. J. (1998) The regulation of glutamine synthetase covalent modification revisited: Role of 2-ketoglutarate in the regulation of glutamine synthetase adenylylation state. Biochemistry 37, 12802–12810. [DOI] [PubMed] [Google Scholar]
- Jiang P.; Pioszak A. A.; Ninfa A. J. (2007) Structure/function analysis of glutamine synthetase adenylyltransferase (ATase, E.C. 2.7.7.49) of Escherichia coli. Biochemistry 46, 4117–4132. [DOI] [PubMed] [Google Scholar]
- Jiang P.; Mayo A. E.; Ninfa A. J. (2007) Escherichia coli glutamine synthetase adenylyltransferase (ATase, E.C. 2.7.7.49): Kinetic characterization of regulation by PII, PII-UMP, glutamine, and α-ketoglutarate. Biochemistry 46, 4133–4146. [DOI] [PubMed] [Google Scholar]
- Jiang P.; Ninfa A. J. (2007) The Escherichia coli PII signal transduction protein controlling nitrogen assimilation acts as a sensor of adenylate energy charge in vitro. Biochemistry 46, 12976–12986. [DOI] [PubMed] [Google Scholar]
- Rhee S. G., Bang W. G., Koo J. H., Min K. H., and Park S. C. (1988) Regulation of glutamine synthetase activity and its biosynthesis in Escherichia coli: Mediation by three cycles of covalent modification. In Enzyme dynamics and regulation (Chock P. B.; Huang C. Y.; Tsou C. L., and Wang J. H., Eds.) pp 136−145, Springer-Verlag, Berlin. [Google Scholar]
- Kustu S.; Hirschman J.; Burton D.; Jelsko J.; Meeks J. C. (1984) Covalent modification of bacterial glutamine synthetase: Physiological significance. Mol. Gen. Genet. 197, 309–317. [DOI] [PubMed] [Google Scholar]
- Jaggi R.; van Heeswijk W. C.; Westerhoff H. V.; Ollis D.; Vasudevan S. G. (1997) The two opposing activities of adenylyltransferase reside in distinct homologous domains, with intramolecular signal transduction. EMBO J. 16, 5562–5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson W. B.; Stadtman E. R. (1970) Glutamine synthetase deadenylylation: A phosphorolytic reaction yielding ADP as nucleotide. Biochem. Biophys. Res. Commun. 41, 704–709. [DOI] [PubMed] [Google Scholar]
- Holm L.; Sander C. (1995) DNA polymerase β belongs to an ancient nucleotidyltransferase superfamily. Trends Biochem. Sci. 20, 345–347. [DOI] [PubMed] [Google Scholar]
- Clancy P.; Xu Y.; van Heeswijk W. C.; Vasudevan S. G.; Ollis D. L. (2007) The domains carrying the opposing activities in adenylyltransferase are separated by a central regulatory domain. FEBS J. 274, 2865–2877. [DOI] [PubMed] [Google Scholar]
- Xu Y.; Zhang R.; Joachimiak A.; Carr P.; Huber T.; Vasudevan S.; Ollis D. (2004) Structure of the N-terminal domain of Escherichia coli glutamine synthetase adenylyltransferase. Structure 12, 861–869. [DOI] [PubMed] [Google Scholar]
- Norby J. G. (1988) Coupled assay of Na+,K+-ATPase. Methods Enzymol. 156, 116–119. [DOI] [PubMed] [Google Scholar]
- Ninfa A. J.; Jiang P. (2005) PII signal transduction proteins: Sensors of α-ketoglutarate that regulate nitrogen metabolism. Curr. Opin. Microbiol. 8, 168–173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.