

Abstract
Plasminogen activator inhibitor type 1 (PAI-1) regulates fibrinolytic activity and mediates vascular atherothrombotic disease. Endothelial cells (ECs) synthesize and secrete PAI-1, but the intracellular signaling pathways that regulate PAI-1 expression are not entirely known. We hypothesize that the phosphatidylinositol 3-kinase (PI3K)/protein kinase Akt pathway, which regulates endothelial function, could modulate PAI-1 expression in ECs. Cultured bovine aortic and human saphenous vein ECs were stimulated with TNF-α, ANG II, insulin, or serum, and PAI-1 expression was determined by Northern and Western analyses. Inhibition of PI3K with wortmannin or LY-294002 enhanced PAI-1 expression induced by these extracellular stimuli. Similarly, overexpression of a dominant-negative mutant of PI3K or Akt increased TNF-α- and insulin-induced PAI-1 expression. The increase in PAI-1 was due to transcriptional and posttranscriptional mechanisms as PI3K inhibitors increased PAI-1 promoter activity and mRNA stability. The induction of PAI-1 by TNF-α and insulin is mediated, in part, by ERK and p38 MAPK. PI3K inhibitors augmented TNF-α- and insulin-induced phosphorylation of these MAPKs. Simvastatin, a 3-hydroxy-3-methylglutaryl-CoA reductase inhibitor, which is known to activate PI3K/Akt, blocks TNF-α- and insulin-induced PAI-1 expression. Treatment with PI3K inhibitors reversed the inhibitor effects of simvastatin on TNF-α- and insulin-induced PAI-1 expression. These findings indicate that the PI3K/Akt pathway acts as a negative regulator of PAI-1 expression in ECs, in part, through the downregulation of MAPK pathways. These results suggest that factors that activate the PI3K/Akt pathway in ECs may have therapeutic benefits for atherothrombotic vascular disease.
Keywords: mitogen-activated protein kinase, cardiovascular disease, statins, cholesterol
PLASMINOGEN ACTIVATOR INHIBITOR type 1 (PAI-1), an inhibitor of tissue-type and urokinase-type plasminogen activators, is a negative regulator of the fibrinolytic system and, thus, promotes thrombosis formation (9, 21). PAI-1 also plays an important role in the atherogenic process and vascular lesion formation (12, 28). PAI-1 expression is upregulated in patients with cardiovascular risk factors, including hypertension, diabetes, and dyslipidemia, and the extent of vascular PAI-1 expression correlates with the severity of atherosclerotic disease (9, 21, 26, 28). Indeed, increased PAI-1 activity has been identified as an independent risk factor for cardiovascular diseases (12, 21). These findings suggest that PAI-1 may be an important mediator of atherothrombotic vascular diseases.
Vascular endothelial cells (ECs) produce and secrete a variety of bioactive molecules, which play important roles in cardiovascular homeostasis and diseases. Although PAI-1 is secreted from various cell types, ECs synthesize and secrete substantial amounts of PAI-1 in response to a number of agonist stimuli, such as TNF-α and ANG II (5, 24). The induction of PAI-1 expression involves intracellular signaling pathways mediated by ERK (8, 29), p38 MAPK (20), Rho/Rho-kinase (14), and protein kinase C (23). However, it is not known whether phosphatidylinositol 3-kinase (PI3K) and protein kinase Akt are involved in the regulation of PAI-1 expression.
The intracellular signaling pathway mediated by PI3K and Akt is involved in the regulation of numerous important cellular functions such as cell survival, proliferation, migration, and gene expressions (2, 10). In ECs, the PI3K/Akt pathway is activated by growth factors such as vascular endothelial growth factor, insulin-like growth factor, and fibroblast growth factor, and by 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors (statins) or fluid shear stress (15, 27). Indeed, the PI3K/Akt pathway mediates various signaling pathways to the activation of endothelial nitric oxide synthase (3, 6). Thus the PI3K/Akt pathway is critical to cardiovascular homeostasis and angiogenesis (15, 27).
Despite the importance of PAI-1 and the PI3K/Akt pathway in ECs, the relation between them has not been shown. This may be clinically important, since statins may exert cholesterol-independent or pleiotropic effects via activation of the PI3K/Akt pathway (31). Indeed, statins downregulate PAI-1 expression in ECs (1, 14), but the mechanism underlying this effect is not fully known. The aim of this study, therefore, was to investigate whether the PI3K/Akt pathway is involved in the regulation of PAI-1 gene expression in ECs and, if so, to determine the mechanisms involved.
MATERIALS AND METHODS
Drugs and reagents
Recombinant human TNF-α, ANG II, and insulin were purchased from Sigma Chemical; LY-294002 and wortmannin from Upstate (Charlottesville, VA); PD-98059 from Cell Signaling Technology (Beverly, MA); and simvastatin from Merck (Whitehouse Station, NJ). Other chemical reagents were purchased from Sigma Chemical.
Cell culture
Bovine aortic ECs were cultured in DMEM (Life Technologies GIBCO BRL) containing 10% fetal calf serum (FCS) and an antibiotic mixture of penicillin and streptomycin until optimal confluence was reached. Passages 5-9 were used for all experiments. Human umbilical vein ECs were similarly cultured in M199. Cells were starved for 12 h in medium free of phenol red and serum before stimulation. Then cells were stimulated with TNF-α, ANG II, insulin, or 10% FCS to induce PAI-1 gene expression. In some studies, cells were pretreated with pharmacological inhibitors (LY-294002, wortmannin, PD-98059, or SB-203580) 30 min before agonist stimulation. Preactivated simvastatin was administered 3 h before stimulation. In experiments with simvastatin, LY-294002 was concomitantly added 3 h before agonist stimulation.
Northern blot analysis
Total RNA was prepared from ECs by an acid guanidinium-phenol-chloroform extraction method (TRIzol Reagent, GIBCO BRL). Northern blot analysis was performed with equal amounts of total RNA (10 μg each) as described previously (16). PCR products (cDNA probes) were synthesized using specific primers for human PAI-1 [5′-ATG GCC ATT ACT ACG ACA TCC TGG-3′ (forward) and 5′-CAC AAA GAG GAA GGG TCT GTC CAT-3′ (reverse)] and labeled with [α-32P]dCTP (New England Nuclear Life Science Products). The radioactivity of hybridized bands of PAI-1 mRNA and GAPDH mRNA was quantified using image analyzer software (NIH Image).
Western blot analysis
After each treatment, ECs were lysed in cell lysis buffer containing proteinase inhibitors (20 mM Tris·HCl, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and 1 μg/ml leupeptin). An equivalent amount of extracted protein (10-30 μg) was loaded for SDS-PAGE. Western blot analysis for PAI-1 was performed using a monoclonal antibody for PAI-1 (Santa Cruz Biotechnology). In separate experiments, antibodies for total and phosphorylated Akt (Ser473), antibodies for total and phosphorylated ERK (Thr202/Tyr204), and antibodies for total and phosphorylated p38 MAPK (Thr180/Tyr182; Cell Signaling Technology) were used for detection of each protein. Proteins were visualized using an enhanced chemiluminescence Western blotting luminol reagent (Amersham).
Adenovirus-mediated gene transfer
Adenoviruses (Ad) encoding a dominant-negative (DN) mutant of PI3K (Ad-DN-PI3K, a mutated subunit, p85α) and Akt (Ad-DN-Akt) were provided by Dr. Masato Kasuga (Kobe University Graduate School of Medicine, Kobe, Japan) and Kenneth Walsh (Boston University, Boston, MA), respectively (11, 19). For transfection studies, ECs were incubated with adenovirus vector for 1 h in 400 μl of DMEM containing 5% FCS in a 6-cm culture dish. The cells were then washed and incubated for an additional 24 h and used for experiments. Multiplicity of infection of 100 was used for each experiment.
PAI-1 activity assay
Human aortic ECs (passage 7) were treated with TNF-α for 12 h in the presence or absence of the PI3K inhibitors. The conditioned media were then collected, and PAI-1 activity in the media was measured using a tissue plasminogen activator-coated ELISA kit ZYMUTEST PAI-1 activity assay (DiaPharma, West Chester, OH) according to the manufacturer’s instructions. The concentrations of active PAI-1 was normalized to the amount of intracellular protein in each dish and expressed as nanograms per milligram.
PAI-1 promoter activity
The PAI-1 promoter (-800 bp) linked to luciferase reporter gene (p800LUC) was obtained from David Loskutoff (Scripps Research Institute, La Jolla, CA) (30). ECs were prepared in a 6-cm tissue culture dish. Approximately 500 ng of p800LUC and 4 ng of sea pansy luciferase gene driven by the cytomegalovirus promoter were introduced into ECs by a chemical transfection (FuGene 6 transfection reagent, Roche, Indianapolis, IN). The transfected cells were cultured in DMEM supplemented with 10% FCS for 24 h and then stimulated with TNF-α (10 ng/ml) in serum-free DMEM for 12 h. Luciferase activities were determined by a dual-luciferase reporter assay system (Promega) with a luminometer (model L9501, Berthold).
Statistical analysis
Values are means ± SE of the number of groups represented by n. The data among groups were compared using a one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons. Stability assays for mRNA were analyzed by two-way ANOVA. P < 0.05 was considered statistically significant.
RESULTS
Role of PI3K in TNF-α-induced PAI-1 expression
In a concentration-dependent manner, stimulation with TNF-α for 6 h increased steady-state PAI-1 mRNA expression, with maximal increase at a TNF-α concentration of 10 ng/ml (Fig. 1A). Similarly, in a time-dependent manner, stimulation with TNF-α (10 ng/ml) increased PAI-1 mRNA and protein levels relative to GAPDH. The maximal increase of PAI-1 mRNA expression occurred after 6 h of TNF-α stimulation (Fig. 1B). PAI-1 protein expression was accordingly increased in response to TNF-α (Fig. 1C).
Fig. 1.
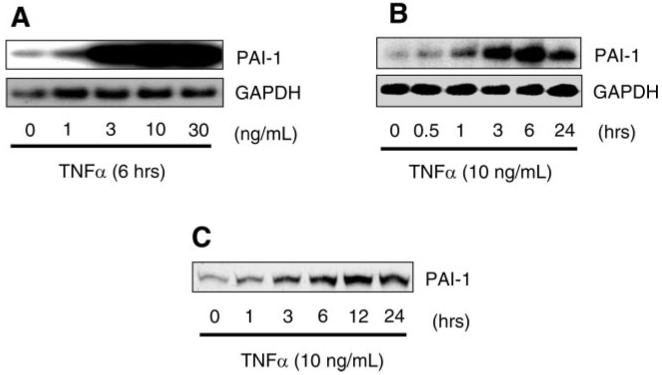
Induction of plasminogen activator inhibitor type 1 (PAI-1) expression by TNF-α in endothelial cells (ECs). A: Northern blots showing dose-dependent induction of PAI-1 mRNA. B: Northern blots showing time-dependent induction of PAI-1 mRNA. C: Western blots showing time-dependent induction of PAI-1 protein expression.
To determine whether PI3K can regulate TNF-α-induced PAI-1 expression, ECs were stimulated with TNF-α in the presence or absence of the PI3K inhibitors wortmannin and LY-294002. Cotreatment with wortmannin or LY-294002 increased TNF-α-stimulated PAI-1 mRNA and protein expression almost twofold (Fig. 2, A and B). Similarly, wortmannin and LY-294002 augmented insulin-induced PAI-1 expression (Fig. 2C). Furthermore, the effects of other stimulators of PAI-1 expression, such as ANG II and FCS, were also enhanced in the presence of PI3K inhibitors (Fig. 2D). This increase in PAI-1 expression correlated with the increase in PAI-1 activity in the presence of PI3K inhibitors (Fig. 2E). These findings indicate that the PI3K pathway inversely regulates PAI-1 expression in ECs.
Fig. 2.
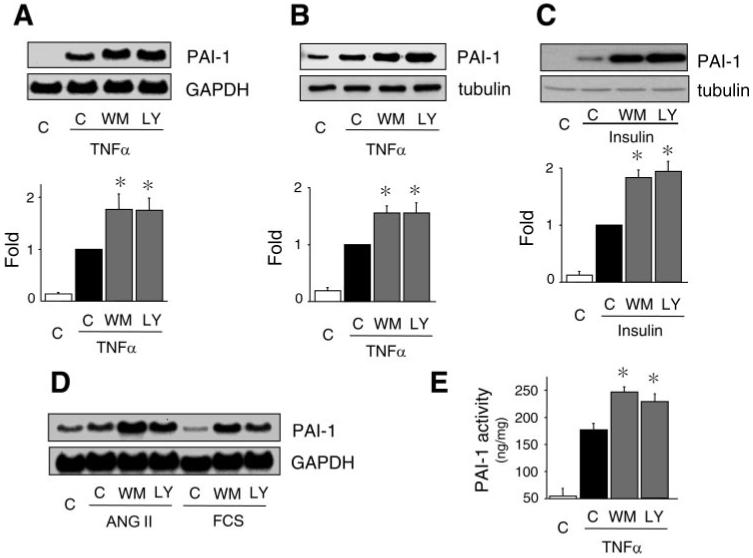
Inhibition of phosphatidylinositol 3-kinase (PI3K) augments TNF-α-induced PAI-1 expression and activity. A: Northern blots showing effects of wortmannin (WM, 50 nmol/l) and LY-294002 (LY, 3 μmol/l) on TNF-α (10 ng/ml, 6 h)-induced PAI-1 mRNA expression. C, control. Values are means ± SE (n = 4). *P < 0.05 vs. TNF-α alone. B: Western blot showing effects of wortmannin or LY-294002 on TNF-α (10 ng/ml, 24 h)-induced PAI-1 protein expression. Values are means ± SE (n = 5). *P < 0.05 vs. TNF-α alone. C: Western blot showing effects of wortmannin or LY-294002 on insulin (1 μmol/l, 6 h)-induced PAI-1 protein expression. Values are means ± SE (n = 3). *P < 0.05 vs. insulin alone. D: Northern blots showing effects of PI3K inhibitors on ANG II- and FCS-induced PAI-1 mRNA expression. Cells were stimulated with ANG II or FCS in the presence or absence of PI3K inhibitors for 6 h. E: effect of wortmannin or LY-294002 on TNF-α-induced PAI-1 activity. At ∼12 h after TNF-α stimulation, PAI-1 activity was measured in conditioned media by ELISA. Values are means ± SE (n = 4). *P < 0.05 vs. TNF-α alone.
Role of Akt in TNF-α-induced PAI-1 expression
A prominent downstream target of PI3K is protein kinase Akt. Using adenovirus-mediated gene transfer, we found that transfection of ECs with Ad-DN-PI3K or Ad-DN-Akt, but not Ad-LacZ, augmented TNF-α-induced PAI-1 mRNA and protein expression (Fig. 3, A and B). However, basal PAI-1 expression was not appreciably increased with Ad-DN-PI3K or Ad-DN-Akt, probably because of low PI3K and Akt kinase activities under basal conditions. Nevertheless, these findings indicate that inhibition of Akt upregulates TNF-α-induced PAI-1 expression in ECs.
Fig. 3.
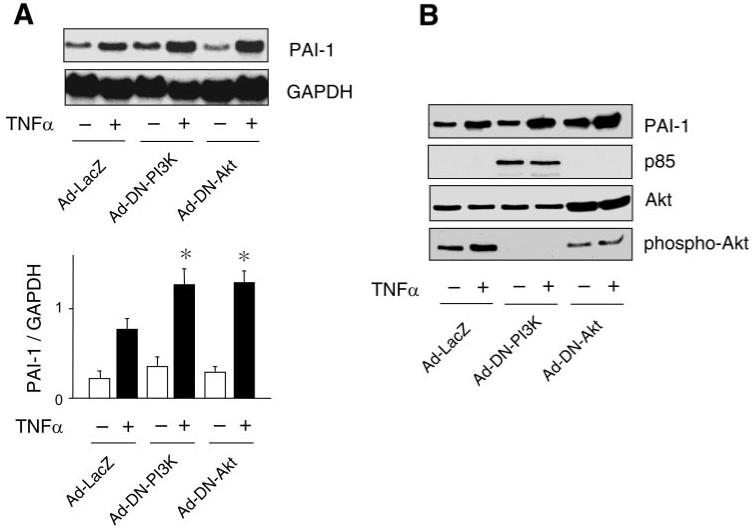
Effects of PI3K and Akt inhibition of PAI-1 expression. A and B: Northern and Western blots, respectively, showing effects of adenoviruses (Ad) encoding LacZ (Ad-LacZ), a dominant-negative (DN) mutant of PI3K (Ad-DN-PI3K), and a dominant-negative mutant of Akt (Ad-DN-Akt) on TNF-α (10 ng/ml)-induced PAI-1 mRNA expression. Blots were evaluated 6 and 24 h after TNF-α stimulation. Values are means ± SE (n = 4). *P < 0.05 vs. Ad-LacZ with TNF-α.
Mechanisms underlying regulation of TNF-α-induced PAI-1 expression by PI3K/Akt
Using a -800-bp PAI-1 promoter luciferase reporter, we found that TNF-α induced PAI-1 promoter activity in ECs (Fig. 4A). Cotreatment with the PI3K inhibitors wortmannin and LY-294002 further increased TNF-α-induced PAI-1 promoter activity, indicating a transcriptional repression of the PAI-1 promoter by PI3K. To determine whether the increase in PAI-1 expression was also due to a posttranscriptional mechanism, we treated ECs with the mRNA synthase inhibitor 5,6-dichlorobenzimidazole riboside (25 μmol/l) (16). Compared with TNF-α alone, addition of the PI3K inhibitors wortmannin and LY-294002 increased the stability of PAI-1 mRNA. These findings indicate that PI3K negatively regulates PAI-1 expression through transcriptional and posttranscriptional mechanisms.
Fig. 4.
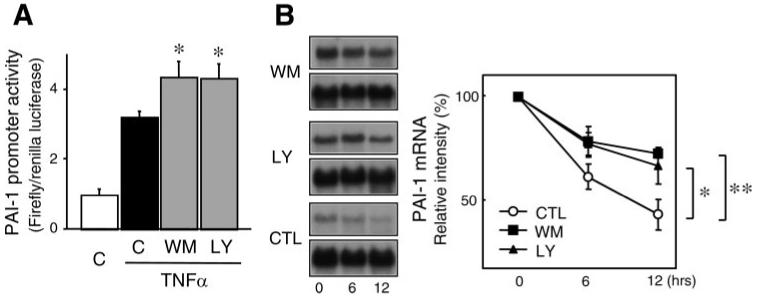
Transcriptional and posttranscriptional regulation of PAI-1 expression by PI3K. A: dual-luciferase reporter assay showing effects of wortmannin (50 nmol/l) and LY-294002 (3 μmol/l) on TNF-α (10 ng/ml)-induced PAI-1 promoter activity in ECs. *P < 0.05 vs. TNF-α alone. B: Northern blots with corresponding analysis showing half-life of PAI-1 mRNA in the presence of mRNA synthase inhibitor 5,6-dichlorobenzimidazole riboside (25 μmol/l), which was added after 6 h of stimulation with TNF-α (10 ng/ml). Ctl, control. Values are means ± SE (n = 4). *P < 0.05. **P < 0.01.
Because ERK and p38 MAPK are activated by TNF-α in ECs (18, 22), we investigated whether these MAPKs could mediate TNF-α-induced PAI-1 expression. Pretreatment with a selective ERK kinase (MEK) inhibitor, PD-98059, or a selective p38 MAPK inhibitor, SB-203580, inhibited TNF-α-induced PAI-1 expression, with the effect of SB-203580 being more prominent (Fig. 5A). Pretreatment with PD-98059 and SB-203580 almost completely blocked TNF-α-induced PAI-1 expression. These effects were relatively specific, since pretreatment with the Rho-kinase inhibitors hydroxyfasudil and Y-27632 had little or no effect on TNF-α-induced PAI-1 expression (data not shown). These results indicate that the MEK/ERK pathway and the p38 MAPK pathway mediate TNF-α-induced PAI-1 expression in ECs. Indeed, inhibition of PI3K with wortmannin or LY-294002 increased TNF-α- and insulin-induced ERK1/2 and p38 MAPK phosphorylation (Fig. 5, B and C), suggesting that the PI3K pathway suppresses the activity of ERK1/2 and p38 MAPK in ECs. Taken together, it is likely that the PI3K/Akt pathway attenuates PAI-1 expression via functional downregulation of TNF-α-induced MEK/ERK- and p38 MAPK-mediated pathways in ECs.
Fig. 5.
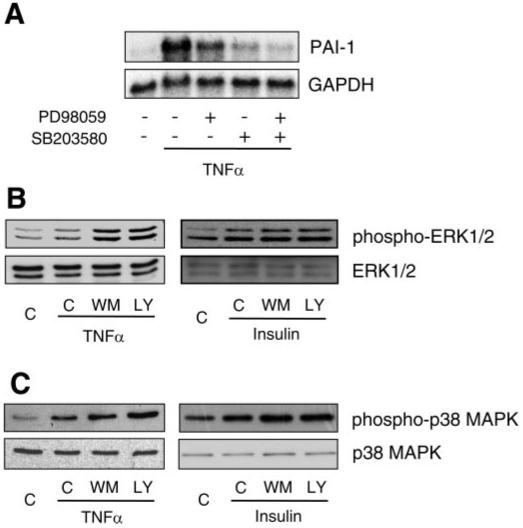
Role of MAPKs in PAI-1 expression. A: Northern blots showing effects of MAPK inhibitors on PAI-1 expression. Pretreatment with an ERK kinase (MEK) inhibitor (PD-98059, 10 μmol/l) or a p38 MAPK inhibitor (SB-203580, 10 μmol/l) suppressed TNF-α (10 ng/ml)-induced PAI-1 expression. Pretreatment with PD-98059 and SB-203580 almost completely suppressed TNF-α (10 ng/ml)-induced PAI-1 expression. B and C: Western blots showing effect of wortmannin (50 nmol/l) and LY-294002 (3 μmol/l) on phosphorylations of ERK1/2 and p38 MAPK. Cells were stimulated with TNF-α (10 ng/ml) or insulin (1 μmol/l) in the presence or absence of wortmannin (50 nmol/l) or LY-294002 (3 μmol/l) for 12 h.
Regulation of PAI-1 expression by statins is mediated by the PI3K pathway
To determine the potential clinical relevance of this pathway, we investigated whether agents such as statins, which are known to activate the PI3K/Akt pathway in ECs, could regulate PAI-1 expression. Indeed, treatment of ECs with a statin, simvastatin, increased Akt phosphorylation and decreased PAI-1 expression (Fig. 6, A and B). Cotreatment with the PI3K inhibitor LY-294002 blocked the downregulation of PAI-1 mRNA and protein expression in response to simvastatin (Fig. 6, B and C). This was associated with reversal of simvastatin-induced downregulation of TNF-α-induced ERK1/2 and p38 MAPK phosphorylation (Fig. 7). These results suggest that the PI3K/Akt pathway plays an important role in the inhibitory effect of simvastatin on TNF-α-induced PAI-1 expression in ECs.
Fig. 6.
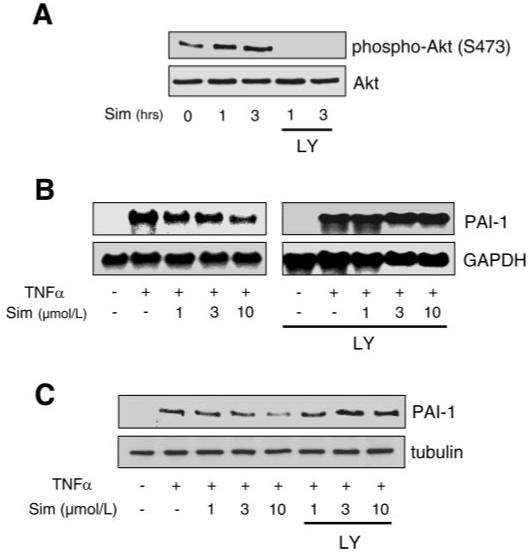
Role of PI3K in the inhibitory effect of simvastatin on TNF-α-induced PAI-1 expression. A: Western blots showing activation (phosphorylation at Ser473) of Akt by simvastatin (Sim). B and C: Northern and Western blots, respectively, showing dose-dependent inhibition of TNF-α-induced PAI-1 expression by simvastatin and its reversal with LY-294002 (3 μmol/l).
Fig. 7.
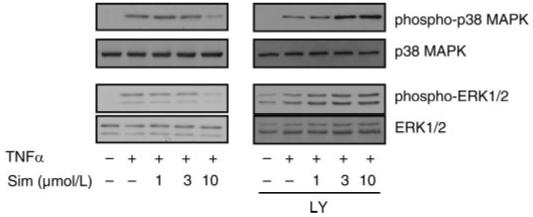
Role of PI3K in the inhibitory effect of simvastatin on TNF-α-induced ERK and p38 MAPK phosphorylation. Western blots show dose-dependent inhibition of TNF-α-induced ERK1/2 and p38 MAPK phosphorylation by simvastatin and its reversal with LY-294002 (3 μmol/l).
DISCUSSION
We have shown that inhibition of the PI3K/Akt pathway with pharmacological inhibitors or dominant-negative mutants augments agonist-induced PAI-1 expression in ECs. The increase in PAI-1 expression via PI3K inhibition occurs at the level of increased PAI-1 gene transcription and prolongation of PAI-1 mRNA half-life. Since PI3K negatively regulates the MEK/ERK and p38 MAPK pathways, these effects may be responsible for the induction of PAI-1 by TNF-α. However, the prolongation of PAI-1 mRNA half-life is not known but may be due to regulation of protein(s) that bind(s) the PAI-1 3′-untranslated region by PI3K. The clinical relevance of this pathway may be reflected in some of the vascular protective effects of statins, which stimulate the PI3K/Akt pathway and downregulate PAI-1 expression. These results indicate that the PI3K/Akt pathway in ECs may not only be important for the activation of endothelial nitric oxide synthase, but it may also be involved in the downregulation of proatherothrombotic genes such as PAI-1.
Although growth factor-mediated receptor tyrosine kinase activation is the major upstream event for PI3K/Akt activation, recent studies indicate that TNF-α and ANG II are also able to activate the PI3K/Akt pathway in ECs (17, 18, 22). Since inhibition of the PI3K/Akt pathway augments PAI-1 expression induced by different agonists, it is possible that the PI3K/Akt pathway acts as an intrinsic suppressor of PAI-1 induction under various stimuli such as TNF-α and growth factors. Indeed, treatment with PI3K inhibitors exacerbates cytokine-induced hypercoagulation in a mouse model of endotoxemia (25). Because the PI3K/Akt pathway also increases endothelial production of nitric oxide (3, 6, 15) and suppresses endothelial expression of tissue factor, another important molecule for thrombosis (4, 11), it is likely that the PI3K/Akt pathway in ECs exerts antithrombotic and vascular protective effects at multiple levels and through various other pathways.
The finding that the PI3K/Akt pathway negatively regulates the MEK/ERK and p38 MAPK pathways indicates an important cross talk between pro- and antithrombotic pathways in ECs (7, 17). Since TNF-α activates both of these pathways, it is possible that the activation of PI3K/Akt could attenuate the induction of PAI-1 expression via MEK/ERK and p38 MAPK. Indeed, inhibition of PI3K augments TNF-α-induced PAI-1 expression. These results indicate that the PI3K/Akt pathway has suppressor effects on MAPKs in ECs, especially by agents that are known to activate both pathways. However, it remains to be determined how the PI3K/Akt pathway inhibits MAPKs and how the balance between the pro- and antithrombotic pathways is regulated.
Statins have been shown to potentiate fibrinolytic activity and, clinically, to reduce the incidence of acute coronary syndromes (13, 31). Statins have many biological actions on vascular wall cells, and the activation of the PI3K/Akt pathway in ECs may contribute substantially to some of their beneficial effects in preventing cardiovascular diseases (15, 32). Indeed, simvastatin increased Akt phosphorylation and inhibited TNF-α-induced PAI-1 expression, effects that were blocked by PI3K inhibitors. These findings suggest that the PI3K/Akt pathway may mediate some inhibitory effects of statins on PAI-1 expression. It is also possible that other cholesterol-independent effects of statins, such as inhibition of the Rho/Rho-kinase pathway, may also be involved in the regulation of PAI-1 expression (14, 32). Indeed, inhibition of Rho/Rho-kinase leads to activation of the PI3K/Akt pathway in ECs (31), which could affect PAI-1 expression.
In summary, the endothelial PI3K/Akt pathway plays an important role in regulating fibrinolytic balance in the vascular wall. Further studies, which can translate these basic mechanisms to the clinical setting, could yield a promising therapeutic target for patients with atherothrombotic vascular diseases.
Acknowledgments
GRANTS
This work was supported by National Institutes of Health Grants HL-52233, HL-70274, HL-080187, and DK-62729 and the American Heart Association Bugher Foundation Award. Y. Mukai is a recipient of the Merck-Banyu Fellowship in Cardiovascular Medicine. C.-Y. Wang is a recipient of Chang Gung Memorial Hospital Research Grant CMRPG34016.
REFERENCES
- 1.Bourcier T, Libby P. HMG CoA reductase inhibitors reduce plasminogen activator inhibitor-1 expression by human vascular smooth muscle and endothelial cells. Arterioscler Thromb Vasc Biol. 2000;20:556–562. doi: 10.1161/01.atv.20.2.556. [DOI] [PubMed] [Google Scholar]
- 2.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 3.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 4.Eto M, Kozai T, Cosentino F, Joch H, Luscher TF. Statin prevents tissue factor expression in human endothelial cells: role of Rho/Rho-kinase and Akt pathways. Circulation. 2002;105:1756–1759. doi: 10.1161/01.cir.0000015465.73933.3b. [DOI] [PubMed] [Google Scholar]
- 5.Feener EP, Northrup JM, Aiello LP, King GL. Angiotensin II induces plasminogen activator inhibitor-1 and -2 expression in vascular endothelial and smooth muscle cells. J Clin Invest. 1995;95:1353–1362. doi: 10.1172/JCI117786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gratton JP, Morales-Ruiz M, Kureishi Y, Fulton D, Walsh K, Sessa WC. Akt down-regulation of p38 signaling provides a novel mechanism of vascular endothelial growth factor-mediated cytoprotection in endothelial cells. J Biol Chem. 2001;276:30359–30365. doi: 10.1074/jbc.M009698200. [DOI] [PubMed] [Google Scholar]
- 8.Hamaguchi E, Takamura T, Shimizu A, Nagai Y. Tumor necrosis factor-α and troglitazone regulate plasminogen activator inhibitor type 1 production through extracellular signal-regulated kinase- and nuclear factor-κB-dependent pathways in cultured human umbilical vein endothelial cells. J Pharmacol Exp Ther. 2003;307:987–994. doi: 10.1124/jpet.103.054346. [DOI] [PubMed] [Google Scholar]
- 9.Huber K, Christ G, Wojta J, Gulba D. Plasminogen activator inhibitor type-1 in cardiovascular disease. Status report 2001. Thromb Res. 2001;103(Suppl 1):S7–S19. doi: 10.1016/s0049-3848(01)00293-6. [DOI] [PubMed] [Google Scholar]
- 10.Kandel ES, Hay N. The regulation and activities of the multifunctional serine/threonine kinase Akt/PKB. Exp Cell Res. 1999;253:210–229. doi: 10.1006/excr.1999.4690. [DOI] [PubMed] [Google Scholar]
- 11.Kim I, Oh JL, Ryu YS, So JN, Sessa WC, Walsh K, Koh GY. Angiopoietin-1 negatively regulates expression and activity of tissue factor in endothelial cells. FASEB J. 2002;16:126–128. doi: 10.1096/fj.01-0556fje. [DOI] [PubMed] [Google Scholar]
- 12.Kohler HP, Grant PJ. Plasminogen-activator inhibitor type 1 and coronary artery disease. N Engl J Med. 2000;342:1792–1801. doi: 10.1056/NEJM200006153422406. [DOI] [PubMed] [Google Scholar]
- 13.Krysiak R, Okopien B, Herman Z. Effects of HMG-CoA reductase inhibitors on coagulation and fibrinolysis processes. Drugs. 2003;63:1821–1854. doi: 10.2165/00003495-200363170-00005. [DOI] [PubMed] [Google Scholar]
- 14.Kunieda Y, Nakagawa K, Nishimura H, Kato H, Ukimura N, Yano S, Kawano H, Kimura S, Nakagawa M, Tsuji H. HMG CoA reductase inhibitor suppresses the expression of tissue factor and plasminogen activator inhibitor-1 induced by angiotensin II in cultured rat aortic endothelial cells. Thromb Res. 2003;110:227–234. doi: 10.1016/s0049-3848(03)00346-3. [DOI] [PubMed] [Google Scholar]
- 15.Kureishi Y, Luo Z, Shiojima I, Bialik A, Fulton D, Lefer DJ, Sessa WC, Walsh K. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–1010. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laufs U, Endres M, Stagliano N, Amin-Hanjani S, Chui DS, Yang SX, Simoncini T, Yamada M, Rabkin E, Allen PG, Huang PL, Bohm M, Schoen FJ, Moskowitz MA, Liao JK. Neuroprotection mediated by changes in the endothelial actin cytoskeleton. J Clin Invest. 2000;106:15–24. doi: 10.1172/JCI9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madge LA, Pober JS. A phosphatidylinositol 3-kinase/Akt pathway, activated by tumor necrosis factor or interleukin-1, inhibits apoptosis but does not activate NF-κB in human endothelial cells. J Biol Chem. 2000;275:15458–15465. doi: 10.1074/jbc.M001237200. [DOI] [PubMed] [Google Scholar]
- 18.Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp Mol Pathol. 2001;70:317–325. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]
- 19.Miyake K, Ogawa W, Matsumoto M, Nakamura T, Sakaue H, Kasuga M. Hyperinsulinemia, glucose intolerance, and dyslipidemia induced by acute inhibition of phosphoinositide 3-kinase signaling in the liver. J Clin Invest. 2002;110:1483–1491. doi: 10.1172/JCI15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Norata GD, Banfi C, Pirillo A, Tremoli E, Hamsten A, Catapano AL, Eriksson P. Oxidised-HDL3 induces the expression of PAI-1 in human endothelial cells. Role of p38 MAPK activation and mRNA stabilization. Br J Haematol. 2004;127:97–104. doi: 10.1111/j.1365-2141.2004.05163.x. [DOI] [PubMed] [Google Scholar]
- 21.Nordt TK, Peter K, Ruef J, Kubler W, Bode C. Plasminogen activator inhibitor type-1 (PAI-1) and its role in cardiovascular disease. Thromb Haemost. 1999;82(Suppl 1):14–18. [PubMed] [Google Scholar]
- 22.Ohashi H, Takagi H, Oh H, Suzuma K, Suzuma I, Miyamoto N, Uemura A, Watanabe D, Murakami T, Sugaya T, Fukamizu A, Honda Y. Phosphatidylinositol 3-kinase/Akt regulates angiotensin II-induced inhibition of apoptosis in microvascular endothelial cells by governing survivin expression and suppression of caspase-3 activity. Circ Res. 2004;94:785–793. doi: 10.1161/01.RES.0000121103.03275.EC. [DOI] [PubMed] [Google Scholar]
- 23.Ren S, Shatadal S, Shen GX. Protein kinase C-β mediates lipoprotein-induced generation of PAI-1 from vascular endothelial cells. Am J Physiol Endocrinol Metab. 2000;278:E656–E662. doi: 10.1152/ajpendo.2000.278.4.E656. [DOI] [PubMed] [Google Scholar]
- 24.Sawdey M, Podor TJ, Loskutoff DJ. Regulation of type 1 plasminogen activator inhibitor gene expression in cultured bovine aortic endothelial cells. Induction by transforming growth factor-β, lipopolysaccharide, and tumor necrosis factor-α. J Biol Chem. 1989;264:10396–10401. [PubMed] [Google Scholar]
- 25.Schabbauer G, Tencati M, Pedersen B, Pawlinski R, Mackman N. PI3K-Akt pathway suppresses coagulation and inflammation in endotoxemic mice. Arterioscler Thromb Vasc Biol. 2004;24:1963–1969. doi: 10.1161/01.ATV.0000143096.15099.ce. [DOI] [PubMed] [Google Scholar]
- 26.Schneiderman J, Sawdey MS, Keeton MR, Bordin GM, Bernstein EF, Dilley RB, Loskutoff DJ. Increased type 1 plasminogen activator inhibitor gene expression in atherosclerotic human arteries. Proc Natl Acad Sci USA. 1992;89:6998–7002. doi: 10.1073/pnas.89.15.6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circ Res. 2002;90:1243–1250. doi: 10.1161/01.res.0000022200.71892.9f. [DOI] [PubMed] [Google Scholar]
- 28.Sobel BE, Taatjes DJ, Schneider DJ. Intramural plasminogen activator inhibitor type-1 and coronary atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:1979–1989. doi: 10.1161/01.ATV.0000091250.53231.4D. [DOI] [PubMed] [Google Scholar]
- 29.Takeda K, Ichiki T, Tokunou T, Iino N, Fujii S, Kitabatake A, Shimokawa H, Takeshita A. Critical role of Rho-kinase and MEK/ERK pathways for angiotensin II-induced plasminogen activator inhibitor type-1 gene expression. Arterioscler Thromb Vasc Biol. 2001;21:868–873. doi: 10.1161/01.atv.21.5.868. [DOI] [PubMed] [Google Scholar]
- 30.Van Zonneveld AJ, Curriden SA, Loskutoff DJ. Type 1 plasminogen activator inhibitor gene: functional analysis and glucocorticoid regulation of its promoter. Proc Natl Acad Sci USA. 1988;85:5525–5529. doi: 10.1073/pnas.85.15.5525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolfrum S, Dendorfer A, Rikitake Y, Stalker TJ, Gong Y, Scalia R, Dominiak P, Liao JK. Inhibition of Rho-kinase leads to rapid activation of phosphatidylinositol 3-kinase/protein kinase Akt and cardiovascular protection. Arterioscler Thromb Vasc Biol. 2004;24:1842–1847. doi: 10.1161/01.ATV.0000142813.33538.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolfrum S, Jensen KS, Liao JK. Endothelium-dependent effects of statins. Arterioscler Thromb Vasc Biol. 2003;23:729–736. doi: 10.1161/01.ATV.0000063385.12476.A7. [DOI] [PubMed] [Google Scholar]