

Abstract
Accelerated molecular dynamics (AMD) is an efficient strategy for accelerating the sampling of molecular dynamics simulations, and observable quantities such as free energies derived on the biased AMD potential can be reweighted to yield results consistent with the original, unmodified potential. In conventional AMD the reweighting procedure has an inherent statistical problem in systems with large acceleration, where the points with the largest biases will dominate the reweighted result and reduce the effective number of data points. We propose a replica exchange of various degrees of acceleration (REXAMD) to retain good statistics while achieving enhanced sampling. The REXAMD method is validated and benchmarked on two simple gas-phase model systems, and two different strategies for computing reweighted averages over a simulation are compared.
Introduction
Free energy is one of the most important quantities in biophysics. The calculation of free energy using molecular dynamics simulations is complicated by the dependence on the amount of the relevant phase space sampled. The complication is more pronounced when two alchemical free energy end points differ by more than a few trivial moieties. The use of restraints to restrict the phase space has proven useful in the convergence of thermodynamic integration, umbrella sampling, and the Bennett acceptance ratio techniques.1−3 Another approach is to enhance phase space sampling instead of restricting the phase space and often relies on the modification of the original Hamiltonian during molecular dynamics simulations.4,5 Accelerated molecular dynamics (AMD), which conventionally modifies the energy landscape by adding a bias to states below an energy threshold, Ecut (eq 1), is an example of the Hamiltonian modification approach and has proven capable of efficiently generating canonical ensembles consistent with experiments on the millisecond time scale.6,7
A potential problem with modifying the Hamiltonian occurs when reweighting an observable O* from the accelerated simulation to find O on the original potential (eq 2 for AMD). If the simulation is highly accelerated and involves a large range of boost factors ΔV, the reweighted average will be dominated by the relatively few points/structures with large values of ΔV in the limit of finite sampling. This statistical problem has recently been quantified as a reduction in the effective number of data points in the simulation.(8) Thus there is a tradeoff between the degree of acceleration and the statistical precision in AMD simulations. The calculation of free energies using thermodynamic integration computes ⟨dV/dλ⟩λ over the course of a simulation, and the calculation of free energy is very sensitive to the statistical accuracy of the computed averages.
In order to take advantage of the sampling efficiency of the AMD method as well as maintain the statistical relevance of every data point, we propose using a replica-exchange framework to couple varying degrees of acceleration. The low degrees of acceleration will not be prone to the reweighting problem and can still take advantage of the high acceleration through replica exchanges. This replica-exchange accelerated molecular dynamics (REXAMD) is a member of the Hamiltonian replica-exchange (HREM) class of simulations, varying from other HREM techniques in the specific Hamiltonian modification scheme. A similar REXAMD approach has recently been applied to studying the effects of neighboring side chains on peptide backbone conformations in short peptides.(9) We demonstrate the REXAMD approach by increasing the convergence rate of thermodynamic integration (TI) for two simple gas-phase model systems, although the method could utilize other free energy calculation methods instead of TI.
Computational Detail
First some terms should be defined. “State” is used to denote a specific level in the replica-exchange scheme. For example, in temperature replica-exchange each state corresponds to a specific temperature, and in REXAMD each state is a modified Hamiltonian described by a set of boost parameters. The term “replica” is used to denote the individual structures that are exchanged between the various REXAMD states. The term “simulation” refers to a specific setup of REXAMD, and the term “run” refers to an instance of a simulation. Simulation is also used to identify the average and standard error computed from multiple runs.
The current replica-exchange framework is a Python program that launches a modified AMBER8 accelerated molecular dynamics simulation(6) for each replica in between Metropolis Monte Carlo exchanges (eq ). The Monte Carlo (MC) exchanges occur every 1000 molecular dynamics (MD) steps, and the pairs that attempt exchanges alternate every other MC period. For example, in a simulation with four states (labeled s0-s3) the simulation would execute 1000 MD steps, attempt MC exchanges between the s0-s1 states and the s2-s3 states, execute 1000 MD steps, attempt a MC exchange between the s1-s2 states, and repeat. The molecular dynamics simulations used a 1 fs−1 time step and were coupled to a 300 K Langevin thermostat with a collision frequency of 10.0 ps−1. The Python program reset the seed number for the AMBER random number generator after every MC exchange.
The boosting scheme is identified as a suffix added to the REXAMD acronym as follows: REXAMDt denotes a boost only to the torsional potential, and REXAMDtT denotes a dual boost scheme applied to the torsional and total potentials.(10) The “-rw” suffix indicates the reported results are from the reweighting of the most accelerated state in a specific simulation. When the “-rw” suffix is not present, the result is coming from the least accelerated state, which in this paper is always no acceleration.
In order to separate the effect of acceleration from the effect of using M replicas, the REXREG control simulations are a replica-exchange between identical regular dynamics potentials. Note that this makes the acceptance probability of MC exchange in eq identically equal to one. The REXREG simulations are analogous to M independent runs from the same starting point with different initial velocities and taking an average result from the M runs.
The replica-exchange efficiency will be monitored based on two criteria. The first criterion is the average acceptance ratio of the replica-exchanges over the course of a run and gives a rough idea of how capable the given replica-exchange scheme is at mixing replicas. The second criterion is the observed relative frequency rmsd metric.(11) This metric compares the observed population frequency of the replicas against the idealized case where each of M replicas spends 1/M of the total time in any given state of the system. The rmsd metric varies from zero for the ideal mixing to 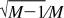 for no mixing. The observed relative frequency metric is more detailed than the average acceptance ratio in monitoring the mixing efficiency of the replica-exchange simulation.
for no mixing. The observed relative frequency metric is more detailed than the average acceptance ratio in monitoring the mixing efficiency of the replica-exchange simulation.
The thermodynamic integration of the model systems was computed using a linear scaling of an all-atom potential (eq 4). Gaussian quadrature integration was used to evaluate the thermodynamic integral from a finite number of ⟨dV/dλ⟩λ calculated at specific λ values (eq 5). The Gaussian quadrature points and weights were taken from the AMBER8 manual.(12) Two strategies were used to calculate ⟨dV/dλ⟩λ at each λ. The first strategy, reweighted periods, calculated the reweighted average of each block of 1000 MD steps in between MC exchanges. These reweighted averages were then averaged together over a complete run to yield ⟨dV/dλ⟩λ for a specific λ. The assumption behind this approach is that the dV/dλ values sampled during 1 ps give rise to ⟨dV/dλ⟩λ for a local region of the conformational space. This strategy becomes exact when the period is longer than the potential energy correlation time of the system. The replica exchange will then balance the occurrence of the local regions. The second strategy, reweighted run, takes an instantaneous dV/dλ and its corresponding ΔV from the MD step immediately prior to a MC exchange. These values are then used to compute a reweighted ⟨dV/dλ⟩λ for the entire simulation. This approach virtually guarantees uncorrelated dV/dλ values at the expense of the number of points being considered in the average. In both strategies each λ was simulated ten times with different random seeds and velocities. An average and standard error for each ⟨dV/dλ⟩λ is then determined and combined into the overall ΔG. The average ΔG is only reported to the first significant digit of the standard error.
Two model systems were studied to validate and benchmark the REXAMD method. Both model systems are symmetric alchemical mutations where the product has an identical structure to the reactant, and thus the ΔG is zero and independent of the force field. Model system A (MSA) is a gas-phase alchemical mutation from ethane-to-ethane (Figure 1A). This system will serve as a positive control to show that REXAMD can reproduce the results of an ergodic regular molecular dynamics simulation. The relative simplicity of the system and the low transition barriers guarantees that the regular molecular dynamics (REXREG) is able to sample the entire conformational space in a short time scale. The thermodynamic integration for MSA uses a 9-point Gaussian quadrature. The MSA REXAMDt simulations used only two replicas: an unmodified potential and an accelerated potential with a torsional boost (Ecut of 5.0 kcal mol−1, α of 2.0 kcal mol−1). Each run was simulated for 8 million MD steps or the equivalent of 8 ns for an unmodified potential.
Figure 1.

Structure of the model systems (A) and (B). The Dm atoms indicate a dummy atom with no nonbonded interactions.
Model System B (MSB) is a highly halogenated butane (Figure 1B). The initial conformation of the system is in a different rotameric state for the two λ end points, as seen in the Newman projections in Figure 1B, and thus requires proper conformational sampling to yield the correct ΔG. The chlorine atoms attached to C2 and C3 were added to make the rotameric sampling more difficult, requiring acceleration to achieve the correct answer within the current time scale of 20 ns. The dual boosting scheme was used for this model system in order to accelerate the large van der Waals interactions experienced in this system. In order to increase the difficulty of converging to the correct result, we are only using a 3-point Gaussian quadrature. The boost parameters for the eight replicas in the MSB REXAMDtT simulations are shown in Table S-I (Supporting Information) and are labeled from s0 to s7 in terms of increasing boost.
Results and Discussion
Model System A
In MSA both the REXREG and REXAMDt simulations were able to efficiently and exhaustively explore the conformational space (data not shown), and the replica mixing was quite efficient (Table 1) within the 8 ns runs. The exhaustive sampling resulted in converged ΔG values within the first ns of the REXREG and REXAMDt simulations (Figure 2). The ΔG results from the entire 8 ns are summarized in Table 2. Recall that “MSA REXAMDt” refers to results taken from the nonaccelerated state and “MSA REXAMDt-rw” refers to the reweighted results of the accelerated state.
Table 1. Summary of the Replica-Exchange Efficiency.
| simulation | acceptanceratioa | observed relativefrequency rmsd |
|---|---|---|
| MSA REXAMDt(8 ns, 2 states) | 39.3 ± 2.2% | 0.00565 ± 0.00420 |
| MSB REXAMDtT(20 ns, 8 states) | 40.2 ± 16.0% | 0.00827 ± 0.00231 |
The average and standard deviation of the acceptance ratios are from the ten runs and the M states. The average and standard deviation of the rmsd of the relative occupancy of the M replicas over the M states, as defined by Abraham et al.,(11) are reported.
Figure 2.

Block average of the MSA thermodynamic integration results when using the reweighted periods strategy. The symbols show the average value of each simulation type, and the shaded region shows the standard error from the ten duplicate runs.
Table 2. ΔG Summary of MSA Thermodynamic Integration Resultsa.
| reweightingstrategy | REXREG | REXAMDt | REXAMDt-rw |
|---|---|---|---|
| periods | +0.002 ± 0.001 | −0.001 ± 0.001 | −0.001 ± 0.001 |
| runs | −0.04 ± 0.01 | +0.02 ± 0.02 | −0.01 ± 0.03 |
The units are in kcal mol−1. The average and standard error from ten simulations are reported for each simulation type.
The statistical precision can be monitored in terms of the number of values that were used in computing ⟨dV/dλ⟩λ. For example, applying the reweighted run strategy to the REXAMDt simulation yields a total of 80,000 data points for each ⟨dV/dλ⟩λ (ten 8 ns trajectories). This strategy resulted in a ΔG of 0.02 ± 0.02 kcal mol−1. In order to produce the same number of points when using the reweighted periods strategy we consider only the first 8 ps of the ten duplicate runs for each λ, which yields a ΔG of 0.02 ± 0.03 kcal mol−1. Note the similarity in both the accuracy and precision of these two results, indicating that exhaustive sampling occurs below the picosecond time scale. The slower ΔG convergence of the reweighted run strategy versus the reweighted periods strategy is due to the slower rate of data collection for the reweighted run strategy.
The REXAMDt-rw simulations also exhibit high accuracy and precision (Figure 2 and Table 2). The average boost applied over the MSA REXAMDt simulations from all of the λ values was 2.0 ± 0.9 kcal mol−1. The small range of boosts (standard deviation of 0.9 kcal mol−1) is predicted to have a relatively small effect on the reweighted precision as predicted by Shen and Hamelberg.(8) The reweighted periods strategy reduces the effective number of instantaneous dV/dλ values from 80 million to 16 million for each ⟨dV/dλ⟩λ, and the REXAMDt-rw simulations exhibit marginally worse precision than the REXAMDt (Table 2). A similar effect is observed in the reweighted runs strategy (a reduction from 80,000 to approximately 15,000).
Model System B
The 20 ns MSB REXAMDtT simulations are well mixed (Table 1, Figures S-I and S-II (Supporting Information)). The regular molecular dynamics (REXREG) was unable to efficiently sample the conformational space (Figure S-III in the Supporting Information) and still shows a substantially nonzero ΔG after the 20 ns for both the reweighted periods and reweighted runs strategies (Table 3). The slow convergence of the REXREG result can also be seen in the block averaging of ΔG in Figure 3. In contrast, the REXAMDtT simulations were able to efficiently sample the conformational space Figure S-IV in the Supporting Information. The ΔG was consistently within 0.1 kcal mol−1 of zero after 2.9 and 5.5 ns for the reweighted periods strategy and the reweighted runs strategy, respectively.
Table 3. ΔG Summary of MSB Thermodynamic Integration Resultsa.
| reweightingstrategy | REXREG | REXAMDtT | REXAMDtT-rw |
|---|---|---|---|
| periods | +0.12 ± 0.08 | +0.04 ± 0.01 | +0.08 ± 0.06 |
| runs | +0.16 ± 0.07 | +0.03 ± 0.04 | −9 ± 7 |
Units are in kcal mol−1. The average and standard error from ten simulations are reported for each simulation type.
Figure 3.

Block average of the MSB thermodynamic integration results from the reweighted periods strategy. The symbols show the average value of each simulation type, and the shaded region shows the standard error for each simulation type.
The reweighting procedure was applied to the state with the highest degree of acceleration, s7, because this state is the most independent of the other states in terms of convergence. The most accelerated state is also expected to have the highest range of ΔV boost factors and therefore exhibit the largest reweighting problem.(8) This prediction can be seen in the poor accuracy and precision of the ΔG of reweighted runs for REXAMDtT-rw (Table 3, Figure 4). The effective numbers of data points for the s7 states are shown in Table S-I (Supporting Information) and demonstrate the source of the poor statistics. For example, the λ of 0.5 simulations had a standard deviation of boost values of 13 kcal/mol, and only 30 of the 200,000 data points from the ten duplicate runs contributed to ⟨dV/dλ⟩λ=0.5.
Figure 4.

Block average of the MSB thermodynamic integration results from the reweighted runs strategy shown on two different scales. The symbols show the average value of each simulation type, and the shaded region shows the standard error for each simulation type. The top plot shows the REXAMDtT-rw results on scale and shows how poor the statistics are after reweighting. The bottom plot shows the REXAMDtT results on scale and shows how quickly the REXAMD technique converges to within statistical accuracy.
The reweighted periods strategy for REXAMDtT-rw has at least one effective point in each 1 ps period and therefore at least 200,000 data points for each ⟨dV/dλ⟩λ when the ten duplicate runs are considered. Compared to the reweighted runs strategy, the increase of the effective number of points results in the increase of the accuracy and precision of the computed ΔG by 2 orders of magnitude (Table 3). The effective number of points is still less than that of REXAMDtT, which has 200 million data points, and the accuracy and precision of REXAMDtT are still better than those of REXAMDtT-rw when using the same averaging strategy (Table 3, Figure 3).
Conclusion
The REXAMD method has been shown to efficiently accelerate conformational sampling while avoiding the statistical reweighting problem inherent in AMD. The REXAMD method was validated on the simple model system A. In the more complex model system B the dual boost REXAMD scheme showed marked improvement over the regular molecular dynamics approach as well as better statistical accuracy and precision in comparison to the reweighted results of the accelerated replicas. We are currently researching the application of this method to more complicated systems.
Acknowledgments
We would like to acknowledge Dr. Xiaolin Cheng for insightful discussions and Robert Swift, Arneh Babakhani, and Morgan Lawrenz for manuscript editing. M.F. was supported in part by an NIH Molecular Biophysics Training Grant (GM-08326) and subsequently by an NSF Graduate Research Fellowship. Funding by NIH GM31749, NSF MCB-0506593, and MCA93S013 (to J.A.M.) also supports this work. Additional support from the Howard Hughes Medical Institute, San Diego Supercomputing Center, the National Biomedical Computational Resource, and the Center for Theoretical Biological Physics is gratefully acknowledged.
Supporting Information Available
Additional figures and tables with more detailed information about the replica-exchange efficiency and acceleration parameters. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Fujitani H.; Tanida Y.; Ito M.; Jayachandran G.; Snow C. D.; Shirts M. R.; Sorin E. J.; Pande V. S. J. Chem. Phys. 2005, 123, 084108. [DOI] [PubMed] [Google Scholar]
- Hamelberg D.; McCammon J. A. J. Am. Chem. Soc. 2004, 126, 7683–7689. [DOI] [PubMed] [Google Scholar]
- Wang J.; Deng Y.; Roux B. Biophys. J. 2006, 91, 2798–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Fajer M.; Yang W. J. Chem. Phys. 2007, 126, 024106. [DOI] [PubMed] [Google Scholar]
- Woods C. J.; Essex J. W.; King M. A. J. Phys. Chem. B 2003, 107, 13703–13710. [Google Scholar]
- Hamelberg D.; Mongan J.; McCammon J. A. J. Chem. Phys. 2004, 120, 11919–11929. [DOI] [PubMed] [Google Scholar]
- Markwick P. R. L.; Bouvignies G.; Blackledge M. J. Am. Chem. Soc. 2007, 129, 4724–4730. [DOI] [PubMed] [Google Scholar]
- Shen T.; Hamelberg D. J. Chem. Phys. 2008, 129, 034103. [DOI] [PubMed] [Google Scholar]
- Xu C.; Wang J.; Liu H. J. Chem. Theory Comput. 2008, 4, 1348–1359. [DOI] [PubMed] [Google Scholar]
- Hamelberg D.; de Oliveira C. A. F.; McCammon J. A. J. Chem. Phys. 2007, 127, 155102. [DOI] [PubMed] [Google Scholar]
- Abraham M. J.; Gready J. E. J. Chem. Theory Comput. 2008, 4, 1119–1128. [DOI] [PubMed] [Google Scholar]
- Pearlman D. A.; Case D. A.; Caldwell J. W.; Ross W. S.; Cheatham T. E.; DeBolt S.; Ferguson D.; Seibel G.; Kollman P. Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.