

Abstract
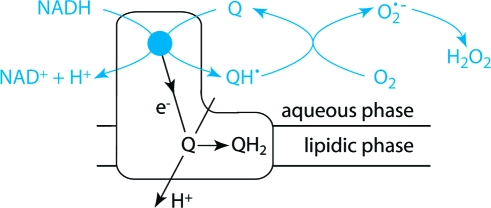
NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria is a complicated, energy-transducing, membrane-bound enzyme that contains 45 different subunits, a non-covalently bound flavin mononucleotide, and eight iron−sulfur clusters. The mechanisms of NADH oxidation and intramolecular electron transfer by complex I are gradually being defined, but the mechanism linking ubiquinone reduction to proton translocation remains unknown. Studies of ubiquinone reduction by isolated complex I are problematic because the extremely hydrophobic natural substrate, ubiquinone-10, must be substituted with a relatively hydrophilic analogue (such as ubiquinone-1). Hydrophilic ubiquinones are reduced by an additional, non-energy-transducing pathway (which is insensitive to inhibitors such as rotenone and piericidin A). Here, we show that inhibitor-insensitive ubiquinone reduction occurs by a ping-pong type mechanism, catalyzed by the flavin mononucleotide cofactor in the active site for NADH oxidation. Moreover, semiquinones produced at the flavin site initiate redox cycling reactions with molecular oxygen, producing superoxide radicals and hydrogen peroxide. The ubiquinone reactant is regenerated, so the NADH:Q reaction becomes superstoichiometric. Idebenone, an artificial ubiquinone showing promise in the treatment of Friedreich’s Ataxia, reacts at the flavin site. The factors which determine the balance of reactivity between the two sites of ubiquinone reduction (the energy-transducing site and the flavin site) and the implications for mechanistic studies of ubiquinone reduction by complex I are discussed. Finally, the possibility that the flavin site in complex I catalyzes redox cycling reactions with a wide range of compounds, some of which are important in pharmacology and toxicology, is discussed.
Complex I (NADH:quinone oxidoreductase) is the first enzyme of the electron transport chain in many aerobically respiring organisms (1,2). In mitochondria, it couples NADH oxidation and ubiquinone reduction to the translocation of four protons across the mitochondrial inner membrane, contributing to the proton motive force that supports ATP synthesis and transport processes. Complex I from bovine mitochondria, a model for the human enzyme, comprises 45 different subunits with a combined mass of almost 1 MDa (3) and nine redox cofactors: a flavin mononucleotide at the active site for NADH oxidation and eight iron−sulfur clusters (4,5). The cofactors are all bound in the hydrophilic domain of the L-shaped enzyme, and the structure of the hydrophilic domain from Thermus thermophilus complex I has been described previously (6). In general, the mechanism of the redox reaction comprises NADH oxidation by hydride transfer to the flavin, followed by reoxidation of the flavin and transfer of the two electrons, along the chain of iron−sulfur clusters, to bound quinone. The mechanisms of quinone reduction and coupled proton translocation remain unknown.
In most mammalian mitochondria, complex I reduces ubiquinone-10 (coenzyme Q10 or Q10), comprising the hydrophilic ubiquinone headgroup and 10 isoprenoid units. The isoprenoid chain renders Q10 extremely hydrophobic, confining it to the membrane and excluding any possibility of it dissociating into the mitochondrial matrix. The extreme hydrophobicity of Q10 also precludes its use in studies of the isolated enzyme, since they require a significant concentration of quinone to be present in predominantly aqueous solutions. Consequently, relatively hydrophilic quinones are used in functional studies of complex I, commonly decylubiquinone (DQ),1 ubiquinone-1 (coenzyme Q1, Q1), and also ubiquinone-0 (coenzyme Q0, Q0) (see Figure 1) (7−12).
Figure 1.

Dependence of the NADH:quinone oxidoreductase activity of isolated complex I on the presence of phospholipids and inhibitors for four different ubiquinones. Rates were determined in the presence (gray bars) and absence (white bars) of 0.4 mg/mL asolectin, without an inhibitor (−) or with 2.3 μM rotenone (R) or 1 μM piericidin (P). Asterisks indicate 23 μM rotenone was used, as 2.3 μM did not fully inhibit the reaction of IDE at the hydrophobic site. Conditions: 100 μM Q, 100 μM NADH, 20 mM Tris-HCl (pH 7.55), 32 °C. Error bars represent the standard deviation of five independent measurements. In the presence of asolectin, the inhibitor sensitivities were approximately 95% (DQ), 90% (Q1), 50% (Q0), and 60% (IDE).
The site(s) at which quinone is bound and reduced by complex I remains poorly defined. A possible binding site for the quinone headgroup has been identified in the structure of the hydrophilic domain of complex I from T. thermophilus, close to cluster N2 and the interface with the membrane domain, formed by the 49 kDa and PSST subunits (6) (we use the nomenclature for the subunits of bovine complex I throughout this work). Site-directed mutations in complex I from Yarrowia lipolytica support the importance of the same region in Q binding and reduction (13). A diverse set of hydrophobic compounds, including rotenone and piericidin A, are commonly termed “Q-site inhibitors”, because they inhibit the NADH:quinone oxidoreductase activity of complex I, but not the reduction of hydrophilic electron acceptors such as ferricyanide (14,15). A mutation in the 49 kDa subunit of Rhodobacter capsulatus conferred resistance to rotenone and piericidin A (16), and radiolabeling experiments have localized various inhibitors to the PSST (17), ND1 (17−19), and ND5 (20) subunits. The latter studies demonstrate that subunits which are integral to the membrane are important for quinone reduction also, perhaps because they are required to accommodate the isoprenoid chain.
Although the Q-site inhibitors are potent inhibitors of Q10 reduction by complex I, many studies have observed that relatively hydrophilic ubiquinones are reduced in an “inhibitor-insensitive” reaction also, at a second site, upstream of the Q10-binding site, which is not linked to proton translocation (see, for example, refs (7), (8), (10), and (21)). Here we refer to the physiological, proton-translocating, inhibitor-sensitive Q-site as the “hydrophobic site” and the non-proton-translocating, inhibitor-insensitive site as the “hydrophilic site”. The rates of reduction at the two sites are influenced by the identity and hydrophobicity of the ubiquinone (8−12) and, in studies of the isolated enzyme, by the presence and concentration of phospholipids in the assay medium (7). The location of the hydrophilic Q-site is still debated. Brandt and co-workers have suggested a “hydrophilic entrance” to the hydrophobic site, allowing hydrophilic quinones to bypass the inhibitor block (22), and Tormo and Estornell suggested a large quinone and inhibitor binding site comprising both hydrophobic and hydrophilic domains (23). Degli Esposti and co-workers proposed that the “hydrophilic reaction” is an incomplete reaction at the hydrophobic site (24) or that hydrophilic quinones are reduced by a protein-bound semiquinone species (10). Alternatively, direct interactions with the iron−sulfur clusters, particularly N2, have been suggested (24,25); they are not excluded by the structure of the hydrophilic domain of complex I from T. thermophilus(6) because some of the iron−sulfur clusters are within 14 Å of the solvent accessible surface. Finally, Čénas and co-workers showed that NADH is a competitive inhibitor of 5,8-dioxy-1,4-naphthoquinone reduction, which is insensitive to 2 μM rotenone. They concluded that the naphthoquinone binding site is distinct from, but close to, the NAD+/NADH and ferricyanide binding sites (26).
Here, we show that the hydrophilic Q-binding site in complex I overlaps with the NADH binding site. We describe how the experimental conditions and the identity and hydrophobicity of the ubiquinone enhance or suppress its reaction with the reduced flavin in the site and define the molecular mechanism and consequences of the reduction reaction. The importance of hydrophilic Q-reduction for our understanding of the mechanisms of ubiquinone reduction and reactive oxygen species production by complex I is discussed. The short-chain ubiquinone, idebenone, is included in our studies because it is in clinical trials for the treatment of Friedreich’s Ataxia, a neurodegenerative disease (27), and because it displays a significant rate of reaction at the hydrophilic site in complex I (24,25). Finally, we describe how the mechanism of hydrophilic ubiquinone reduction by the flavin in complex I forms a basis for investigation of the role of complex I in redox cycling reactions which may be important in pharmacology and toxicology.
Experimental Procedures
Complex I was isolated from bovine mitochondria as described previously (28) and quantified using the Pierce bicinchoninic acid assay. Subcomplex Iλ was prepared from complex I as described previously (29), except that the size-exclusion chromatography step was carried out in 0.03% dodecyl β-d-maltopyranoside. EPR spectra recorded at 12 K did not detect cluster N2 (30). Bovine heart submitochondrial particles (SMPs) were prepared using a method based on that of Kotlyar and Vinogradov (31). Q0, Q1, and DQ were from Sigma, and IDE was from Sequoia Research Products. All quinone stock solutions were prepared in ethanol and quantified using an ε275 of 13.7 mM−1 cm−1 (Q1), an ε263 of 13.6 mM−1 cm−1 (Q0), an ε270 of 19.2 mM−1 cm−1 (DQ), and an ε275 of 13.6 mM−1 cm−1 (IDE) (11,32).
Kinetic Measurements
Kinetic measurements were carried out at 32 °C in 1 mL cuvettes in 20 mM Tris-HCl (pH 7.55) using an Ocean Optics diode array spectrometer. Complex I was added to a final concentration of ∼15 μg/mL from a 15−20 mg/mL stock solution. When required, asolectin (Fluka) was added to a final concentration of 0.4 mg/mL from a 10 mg/mL stock solution containing 2% (w/v) CHAPS {3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (Anatrace)}. Reactions were initiated by the addition of quinone, from ethanolic stock solutions. SMPs were added to a final concentration of ∼0.05 mg/mL, and 0.026 mg/mL gramicidin [from Bacillus aneurinolyticus (Sigma)] was added as an uncoupler. Rotenone or piericidin A (Sigma) was added to a final concentration of 2.3 or 1 μM, respectively, from ethanolic stock solutions. NADH oxidation was monitored at 340−380 nm (ε = 4.81 mM−1 cm−1), and the initial rate obtained using linear regression over 30 s. Anaerobic assays were performed in a glovebox (Belle Technology, Portesham, U.K.) with <5 ppm O2.
Data were modeled mechanistically using the method described by Yakovlev and Hirst (33). In brief, the qualities of the fits from different parameter combinations was assessed by calculation of least-squares error (LSQE) values (the sum of the squares of the differences between calculated and experimental data points). Wide ranges of possible parameter combinations were assessed using programs coded in C, and the LSQE values were minimized to give the best fit.
NADH:Q stoichiometry measurements were carried out at 32 °C in 200 μL wells in a Molecular Devices microtiter plate reader. Assays were initiated by the addition of NADH (final concentrations of 0, 20, 40, 60, 80, 100, 150, and 200 μM) to solutions containing 50 μM Q, complex I, rotenone, and asolectin as stated. The amount of NADH remaining in each well was used to calculate the amount which had been oxidized at each time point; the NADH oxidized did not depend significantly on its initial concentration.
Amplex Red and cyt c Used To Measure H2O2 and O2− Production by Complex I
H2O2 production was assessed stoichiometrically at 32 °C using the horseradish peroxidase (HRP)-dependent oxidation of Amplex Red to resorufin [ε557−620 = 51.6 ± 2.5 mM−1 cm−1 at pH 7.5 (Sigma)] (34,35). Typically, assays comprised 20 mM Tris-HCl (pH 7.55), 30 μM NADH (Sigma), 2 units/mL HRP (MP Biomedicals, Aurora, OH), 10 μM Amplex Red (Invitrogen), and ∼15 μg/mL complex I. The NADH concentration was 30 μM to minimize artifacts from the Amplex Red detection system (35,36). When required, catalase [from bovine liver (Sigma)] was added to a final concentration of 1000 or 10000 units/mL. Superoxide production was assessed stoichiometrically by monitoring the reduction of 50 μM cyt c [partially acetylated equine heart cytochrome c; ε550−541 = 18.0 ± 0.6 mM−1 cm−1 at pH 7.5 (Sigma)] (35,37). Typically, assays comprised 20 mM Tris-HCl (pH 7.55), 30 μM NADH (Sigma), and ∼15 μg/mL complex I. When required, superoxide dismutase (SOD) [Cu-Zn-SOD from bovine erythrocytes (Sigma)] was added to a final concentration of 50 units/mL.
Results
Quinones Are Reduced at Two Distinct Sites in Bovine Complex I
To investigate the inhibitor-insensitive, nonphysiological pathway of quinone reduction by isolated bovine complex I, four relatively hydrophilic quinones were used: decylubiquinone (DQ), coenzyme Q1 (Q1), coenzyme Q0 (Q0), and idebenone (IDE). Figure 1 shows how the rate of NADH:ubiquinone oxidoreduction by complex I depends on the identity of the quinone, on the presence of exogenous phospholipids, and on the presence of the Q-site inhibitors rotenone and piericidin A, which inhibit only the reaction at the physiological, energy-transducing site. When phospholipids are present, DQ and Q1 both react much more rapidly than Q0 and IDE. In the presence of rotenone or piericidin A, the rates of all four quinones are decreased, but the relative degree of inhibition of DQ (∼95%) and Q1 (∼90%) is much greater than that of Q0 (∼50%) and IDE (∼60%). Previous studies of complex I from bovine heart mitochondria have determined similar values for the rotenone sensitivity of Q1 reduction: ≤95% with Q1 (depending on the phospholipid concentration) (7) and ∼85% with Q1 at concentrations of <50 μM (38). When phospholipids are not present, the rates of all four quinones are decreased, and again, the effect of the phospholipids is much greater for DQ and Q1 than for Q0 and IDE. In the absence of phospholipids, the rates are less susceptible to inhibition by rotenone and piericidin A. Figure 1 supports the existence of two sites for the reduction of quinones by complex I: a hydrophobic site that is activated by phospholipids and inhibited by rotenone and piericidin A and a hydrophilic site that is not inhibited by rotenone or piericidin A. The hydrophobic site is the physiological site of quinone reduction, at which the redox reaction is coupled to proton translocation. Physical models which may help in understanding the effects of phospholipids on the two reactions are discussed below.
To compare the rates of reaction of the four ubiquinones at each site, the rates in the presence of phospholipids (without inhibitor) were compared to the rates in the presence of inhibitor (without phospholipids). The “double-knockout” condition was used because it is apparent (see Figure 1, DQ) that neither the omission of phospholipids nor the addition of inhibitor is completely effective at abolishing the reaction at the hydrophobic site. Assuming that the rate observed in the presence of phospholipids is the sum of the rates at the hydrophilic and hydrophobic sites then, for DQ, Q1, Q0, and IDE, the rates at the hydrophobic site are 3.7, 3.3, 0.19, and 0.15 μmol of NADH min−1 mg−1, respectively, and at the hydrophilic site they are 0.03, 0.49, 0.18, and 0.49 μmol of NADH min−1 mg−1, respectively. Importantly, neither the rates at the hydrophobic site (DQ > Q1 ≫ Q0 > IDE) nor the rates at the hydrophilic site (DQ < Q0 ≈ IDE < Q1) correlate in a simple fashion with the hydrophobicity of the ubiquinone [DQ > IDE > Q1 > Q0, based on their cyclohexane/water partition coefficients (11,39)]. Therefore, they are affected by the solubility of the quinone in the external phase and perhaps by molecular interactions between the isoprenoid chains and the two binding sites; these effects are discussed below.
NADH:Q1 oxidoreduction by bovine heart submitochondrial particles (SMPs) confirmed that complex I in the mitochondrial membrane catalyzes the hydrophilic ubiquinone reduction reaction also. The rate of NADH:O2 oxidoreduction (using endogenous UQ10 and measuring the rate of NADH oxidation) was 1.26 μmol min−1 mg−1, and it was 97.8 ± 0.5% sensitive to both rotenone and piericidin A. In the presence of antimycin A (to prevent cycling of the endogenous Q10), the rate of Q1 reduction by the SMPs was 0.54 μmol min−1 mg−1, and in the presence of rotenone or piericidin A, it was ∼0.07 μmol min−1 mg−1, giving an inhibitor sensitivity of ∼87%. Previous studies have investigated the inhibitor sensitivity of a range of ubiquinones using SMPs or broken mitochondria from bovine hearts also. Lenaz and co-workers reported rotenone sensitivity values of 72−77% for Q0, 95−96% for Q1, and 99% for DQ (9), and Degli Esposti and co-workers reported values of 89% (Q1), 94% (DQ), and 63% (IDE) (24). For Q0 and Q1, rotenone sensitivities which increased with Q concentration (from 40 to 63% and from 86 to 90%, respectively) were reported by Di Virgilio and Azzone, reflecting the different apparent KM values of the two quinone binding sites (8). Finally, the rate of NADH:O2 oxidoreduction by our SMPs decreased to 0.28 μmol min−1 mg−1 in 100 μM IDE, consistent with previous work which classified IDE as a complex I inhibitor (24). In fact, IDE is better classified as a competitive substrate; it appears to inhibit because it competes for the active site but then reacts only slowly.
Only the Hydrophilic Reaction Is Exhibited by Subcomplex Iλ
Figure 2 compares NADH:Q1 oxidoreduction by intact complex I and subcomplex Iλ. The preparation of subcomplex Iλ (4) used here contains the flavin, the NADH binding site, and all of the iron−sulfur clusters except the terminal cluster, N2, which is lost when the hydrophilic Iλ domain is resolved. The loss of cluster N2 and the dissociation of the hydrophilic arm together preclude the occurrence of the hydrophobic, physiological quinone reduction reaction in subcomplex Iλ. Figure 2 compares Q1 reduction by complex I and subcomplex Iλ in the presence of exogenous phospholipids and in the presence and absence of rotenone. As expected, Q1 reduction by subcomplex Iλ is independent of the presence of rotenone. The turnover numbers for Q1 reduction by subcomplex Iλ and complex I inhibited by rotenone are the same (at all Q1 concentrations). Therefore, the hydrophilic site of quinone reduction is located “upstream” of cluster N2. It is fully conserved in subcomplex Iλ and acts in a manner independent of the hydrophobic site.
Figure 2.

Complex I inhibited by rotenone catalyzes the reduction of Q1 at the same rate as subcomplex Iλ. Catalysis by complex I (○) is significantly retarded in the presence of rotenone (●). The rates of catalysis by subcomplex Iλ in both the presence (▲) and absence (▽) of rotenone are essentially identical to those from rotenone-inhibited complex I. Conditions: 0.4 mg/mL asolectin, 2.3 μM rotenone, 100 μM NADH, 20 mM Tris-HCl (pH 7.55), 32 °C.
The Hydrophilic Reaction Follows a Ping-Pong Reaction Mechanism at the Flavin Site
Figure 3 shows that the reaction rate for the hydrophilic reaction of Q1, Q0, and IDE decreases as the NADH concentration is increased. The behavior is qualitatively similar to that exhibited by the transhydrogenase reaction catalyzed by complex I, which follows ping-pong kinetics (the two reactants bind, react, and dissociate, one after the other, and both at the flavin site) (33). Inhibition by NADH has been observed also for the reactions of 5,8-dioxy-1,4-naphthoquinone (26) and ferricyanide (40). Therefore, to investigate the proposal that hydrophilic quinone reduction occurs at the flavin site, Figure 3 shows how the rate of NADH:Q oxidoreduction depends on both the NADH and Q concentrations for Q1, Q0, and IDE. In all cases, the rate increases as the Q concentration is increased, indicating that Q does not compete effectively with NADH for the oxidized flavin (KOxQ → ∞). However, the rate increases, then decreases, as the NADH concentration is increased, indicating that NADH does compete effectively with Q for the reduced flavin. The data in Figure 3 suggest two possible reaction mechanisms, which are not mutually exclusive (Scheme 1). In the ping-pong mechanism, the reduced flavin is oxidized by Q in a single two-electron step to form QH2. In the ping-pong-pong mechanism, the reduced flavin is oxidized by Q in two one-electron steps to form two semiquinones (Q•− or QH•). Equations 1 and 2 were derived from Scheme 1, for the ping-pong and ping-pong-pong pathways, respectively, by using the steady-state approximation.
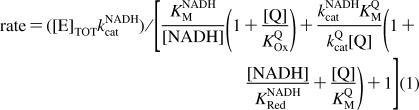 |
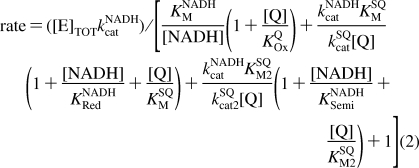 |
First, data sets for the dependence of rate on NADH and Q concentration were fitted for Q1, Q0, and IDE with eq 1, by varying each parameter to minimize the sum of the squares of the errors (the LSQE) between the data points and their equivalent calculated values. The NADH-dependent parameters (KNADHM, kNADHcat, and KNADHRed) were set to the same values for each Q, starting from the values suggested by the transhydrogenase reaction (94 μM, 2700 s−1, and 200 μM, respectively) (33), and the Q-dependent parameters were allowed to vary freely. Satisfactory fits were obtained in all cases, but, for the best fits, Q1 required a lower value of KNADHRed (70 μM) than Q0 and IDE (300 μM) (see Table 1). Allowing KNADHM and kNADHcat to vary did not provide a common best-fit value for KNADHRed. Second, an equivalent analysis was carried out with eq 2. If KNADHSemi = KNADHRed, KSQM = KSQM2, and kSQcat = kSQcat2, the fits using the transhydrogenase parameters were identical to those described above (see Table 1). However, if KNADHRed = 300 μM and KNADHSemi = 70 μM (consistent with tighter NADH binding when the flavin is more oxidized), and if kSQcat1 and kSQcat2 are allowed to differ, good fits could be derived using the same NADH parameters for each Q species. Indeed, differences in kSQcat1 and kSQcat2 are expected to result from the differing reactivity of the one- and two-electron reduced states of the flavin (or of the enzyme, if the electron from the semiflavin is redistributed to the clusters). In summary, the data shown in Figure 3 provide strong evidence that both substrates (NADH and Q) are reacting at the same hydrophilic site in complex I. The data can be explained by mechanisms in which Q is reduced by either one or two electrons; in fact, it is likely that the reactions proceed by both pathways, with the relative contributions of the two varying between the Q species.
Figure 3.

Dependence of the rate of NADH:Q oxidoreduction at the hydrophilic site on NADH and Q concentration. (A) Dependence of NADH:Q1 oxidoreduction on Q1 concentration in 30 (○) and 100 μM NADH (●). (B) Dependence of NADH:Q1 oxidoreduction on NADH concentration at 100 (○) and 250 μM Q1 (●). The data in panels A and B have been fitted using the ping-pong mechanism, using the parameters in row 4 of Table 1. (C) Dependence of NADH:Q0 oxidoreduction on NADH concentration in 100 (○) and 250 μM Q0 (●). (D) Dependence of NADH:IDE oxidoreduction on NADH concentration in 100 (○) and 250 μM IDE (●). The data in panels C and D have been fitted using the ping-pong-pong mechanism, using the parameters in rows 11 and 12 of Table 1. Conditions: 32 °C in 20 mM Tris-HCl (pH 7.55) and 2.3 μM rotenone.
Scheme 1. Mechanism of Quinone Reduction at the Flavin Active Site, Incorporating both the Ping-Pong and Ping-Pong-Pong Reactions, and Double-Substrate Inhibition.

The ping-pong and ping-pong-pong reactions diverge at [FlRed Q]: the ping-pong reaction follows the dotted arrow to produce QH2 and returns directly to [FlOx], and the ping-pong-pong reaction follows the outside pathway to form two semiquinones (QH•). Abbreviations: FlOx/Red, oxidized or reduced state of the flavin; terms in brackets, enzyme-bound species; kcat, first-order rate constant for substrate transformation and product dissociation; KM, Michaelis−Menten constant; KOx, KSemi, and KRed, dissociation constants, referring to the oxidized, semireduced, and reduced enzyme states, respectively.
Table 1. Kinetic Parameters for the Reduction of Q1, Q0, and IDE at the Hydrophilic, Flavin Site in Complex I, Determined by Fitting Experimental Data to the Ping-Pong and Ping-Pong-Pong Mechanisms.
| Ping-Pong Mechanism | ||||||
|---|---|---|---|---|---|---|
| KNADHM (μM) | kNADHcat (s−1) | KNADHRed (μM) | KQM (μM) | kQcat (s−1) | LSQE/LSQEmin | |
| Q1 | 94 | 2700 | 200 | 354 | 56.6 | 2.9 |
| Q0 | 94 | 2700 | 200 | 292 | 15.2 | 1.2 |
| IDE | 94 | 2700 | 200 | 5225 | 660 | 1.7 |
| Q1 | 94 | 2700 | 70 | 200 | 47.4 | 1 |
| Q0 | 94 | 2700 | 300 | 345 | 16.2 | 1 |
| IDE | 94 | 2700 | 3001 | 3770 | 450 | 1 |
| Ping-Pong-Pong Mechanism | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| KNADHM (μM) | kNADHcat (s−1) | KNADHRed (μM) | KNADHSemi (μM) | KSQM (μM) | kSQcat (s−1) | KSQM2 (μM) | kSQcat2 (s−1) | LSQE/LSQEmin | |
| Q1 | 94 | 2700 | 200 | 200 | 354 | 113.2 | 354 | 113.2 | 2.9 |
| Q0 | 94 | 2700 | 200 | 200 | 292 | 30.4 | 292 | 30.4 | 1.2 |
| IDE | 94 | 2700 | 200 | 200 | 5225 | 1320 | 5225 | 1320 | 1.7 |
| Q1 | 94 | 2700 | 300 | 70 | 205 | 500a | 205 | 50.3 | 1 |
| Q0 | 94 | 2700 | 300 | 70 | 250 | 14.2 | 250 | 242.5 | 1 |
| IDE | 94 | 2700 | 300 | 70 | 3880 | 479 | 3880 | 20000a | 1.1 |
Value increases indefinitely without significantly affecting the LSQE value.
NADH Is Oxidized Superstoichiometrically in the Presence of Hydrophilic Quinones
The reaction between NADH and quinone, to form NAD+ and quinol, is a two-electron transfer with a 1:1 stoichiometry, but under conditions which favor quinone reduction at the flavin site, the reaction becomes superstoichiometric (Figure 4). The superstoichiometry of Q1 reduction by complex I has also been noted previously (24,41). Figure 4A shows that 50 μM Q1 is able to oxidize more than 50 μM NADH, whether asolectin and rotenone are present or not. Conditions which favor reaction at the flavin site increase the superstoichiometry, but those that favor the hydrophobic Q-site decrease it. Only quinols are formed at the hydrophobic, physiological Q-site, strongly suggesting that it is semiquinones formed at the flavin site that are responsible for the superstoichiometry, and that the quinols are stable end products. Figure 4B shows that superstoichiometric reactions do not occur unless O2 is present. The rates of NADH oxidation themselves do not depend on the presence of O2, showing that O2 acts indirectly, “recycling” the semiquinone to the quinone, to be reduced again. Figure 4C shows that NADH:DQ oxidoreduction is stoichiometric in both the presence and absence of O2, confirming that the superstoichiometry is associated only with reactions at the flavin site, and Figure 4D compares the reactions of Q1, IDE, and Q0, all of which are superstoichiometric.
Figure 4.

NADH is oxidized superstoichiometrically in aerobic quinone-containing solutions. Complex I was incubated in the presence of ∼50 μM Q (dotted lines) in varying concentrations of NADH (from 0 to 200 μM). The NADH concentrations were measured at 2 min intervals, and the amount of NADH oxidized was determined by comparing measured and initial NADH concentrations. (A) NADH oxidation by 50 μM Q1 in an aerobic solution in the presence or absence of rotenone (Rot.) and/or asolectin (Aso.). NADH is oxidized superstoichiometrically. (B) An experiment equivalent to that conducted for panel A but in an anaerobic solution (the symbols are conserved). NADH is not oxidized superstoichiometrically. (C) NADH oxidation by 50 μM DQ in the presence of asolectin, in anaerobic (○) and aerobic (●) solutions. NADH is not oxidized superstoichiometrically. (D) NADH oxidation by 50 μM Q1, IDE, and Q0 in an aerobic solution in the presence of rotenone and asolectin. NADH is oxidized superstoichiometrically. Conditions: 20 mM Tris-HCl, pH 7.55, 32 °C, 2.3 μM rotenone, 0.4 mg/mL asolectin.
Reactive Oxygen Species Are Produced by the Reduction of Quinones at the Flavin Site
Figure 5A shows that quinone reduction at the flavin site in complex I leads to H2O2 production. The addition of NADH to complex I produces low levels of H2O2 when O2 is present (35). When DQ is added, the rates of H2O2 production do not change, but they are enhanced significantly upon addition of Q1. In the absence of O2 there is a background reaction between one or more reduced quinone species and the Amplex Red detection system which is not sensitive to the addition of catalase, and which is observed for Q1, Q0, and IDE (see Table 2). In the presence of O2, the addition of Q1 produces an approximately 10-fold increase in the rate of H2O2 production, to ∼300 nmol min−1 mg−1. This observation is consistent with the formation of semiquinones which react with O2 to form superoxide (and subsequently H2O2) in redox cycling reactions (see Scheme 2) (42). Note that, because the rate of NADH oxidation is independent of the presence of O2, the reaction of O2 is not with the enzyme or with an enzyme-bound quinone or semiquinone species, but with the products of the enzyme-catalyzed reaction.
Figure 5.

Effects of DQ, Q1, and O2 on resorufin formation and cyt c reduction, catalyzed by complex I. (A) Assay traces showing resorufin formation under aerobic and anaerobic conditions. The assays began in a solution containing complex I; NADH was added after 30 s (to reveal a clear distinction between the anaerobic and aerobic experiments), and then either DQ or Q1 was added. The addition of DQ did not affect the rate of resorufin formation, but the addition of Q1 caused it to increase, especially in the presence of O2. (B) Assay traces showing cyt c reduction under aerobic and anaerobic conditions. The assays began in a solution containing complex I; NADH was added after 30 s (again revealing a clear distinction between the anaerobic and aerobic experiments), and then either DQ or Q1 was added. In both cases, the addition of Q1 caused significant increases in the observed rate. Conditions: 20 mM Tris-HCl, pH 7.55, 32 °C, 30 μM NADH, 100 μM Q, +O2 = atmospheric O2, −O2 = anaerobic glovebox, 2.3 μM rotenone, 50 μM cyt c, 2 units/mL HRP, 10 μM Amplex Red.
Table 2. Resorufin Formation (H2O2 formation) and cyt c Reduction (O2•− formation) in the Presence of Hydrophilic Quinones Depend on the Presence of Asolectin, Rotenone, and O2a.
| Resorufin Formation (nmol min−1 mg−1) | |||||||
|---|---|---|---|---|---|---|---|
| Aso | Rot | O2 | CAT | DQ | Q1 | Q0 | IDE |
| + | − | + | − | 33.9 ± 0.1 | 303.8 ± 21.0 | 29.7 ± 3.2 | 119.7 ± 1.3 |
| + | + | + | − | 29.4 ± 6.6 | 234.6 ± 1.2 | 26.2 ± 0.8 | 123.0 ± 0.3 |
| − | − | + | − | 40.9 ± 4.6 | 307.7 ± 4.4 | 38.0 ± 2.5 | 246.6 ± 11.4 |
| − | + | + | − | 41.5 ± 2.0 | 300.1 ± 32.8 | 38.5 ± 5.7 | 257.8 ± 12.9 |
| − | + | + | + | 3.6 ± 0.3 | 23.0 ± 1.2 | 16.0 ± 0.7 | 23.5 ± 2.3 |
| − | + | − | − | 1.0 ± 0.4 | 24.9 ± 1.6 | 9.6 ± 0.8 | 25.4 ± 3.6 |
| Cyt c Reduction (nmol min−1 mg−1) | |||||||
|---|---|---|---|---|---|---|---|
| Aso | Rot | O2 | SOD | DQ | Q1 | Q0 | IDE |
| − | + | + | − | 48.4 ± 3.3 | 532.0 ± 32.6 | 276.8 ± 5.0 | 432.7 ± 3.0 |
| − | + | + | + | 6.4 ± 2.5 | 203.4 ± 6.8 | 125.7 ± 2.5 | 208.6 ± 18.3 |
| − | + | − | − | 26.2 ± 3.0 | 609.0 ± 14.6 | 315.3 ± 4.7 | 461.6 ± 9.5 |
| − | + | − | + | 24.3 ± 0.9 | 552.4 ± 5.2 | 263.1 ± 1.9 | 419.5 ± 23.2 |
Conditions: 20 mM Tris-HCl, pH 7.55, 32 °C, 30 μM NADH, 100 μM Q, +O2 = atmospheric O2, −O2 = anaerobic glovebox, Aso = 0.4 mg/mL asolectin, Rot = 2.3 μM rotenone, or 10 μM rotenone for IDE, CAT = 10000 units/mL catalase, SOD = 50 units/mL Cu-Zn-SOD, 50 μM cyt c, 2 units/mL HRP, 10 μM Amplex Red.
Scheme 2. Production, Interconversion, and Detection of the Reactive Oxygen and Semiquinone Species Produced by the Reduced Flavin in Complex I.

Figure 5B shows equivalent experiments using acetylated cytochrome c (cyt c), an established system for the detection of superoxide (37). In the absence of a quinone, the rates of cyt c reduction are low and abolished in the absence of O2(35). When DQ is added, they do not change significantly, though a small increase in the absence of O2 is observed when phospholipids are absent. When Q1 is added, a significant increase is observed, both in the presence and in the absence of O2. In addition to being reduced by superoxide, cyt c is reduced directly by semiquinone radicals also, with the interpretation of experimental data complicated further by equilibria being established between O2 and semiquinone, and superoxide and quinone (42). We note that an increase in the level of superoxide production in the presence of hydrophilic quinones has been observed previously (24,25), and that hydrophilic quinones have been suggested to mediate O2 reduction by the flavin in complex I also (43).
Table 2 presents a comprehensive summary of data from the Amplex Red and cyt c detection systems for each of the four Q species. DQ exhibits the slowest rates of reaction, dominated by the direct reaction of the reduced flavin with O2(35), and enhanced slightly by semiquinone formation only in the absence of asolectin. For Q1, both the Amplex Red and cyt c detection systems are consistent with significant semiquinone formation, and with interconversions between semiquinone and O2, and superoxide and quinone. As expected, one-electron cyt c reduction (in the absence of asolectin and in the presence of rotenone) is close to twice as fast as two-electron H2O2 formation, and cyt c reduction is not abolished by SOD. We note that the level of H2O2 formation by Q1 in the presence of rotenone and asolectin together is ∼20% lower than in the presence of either rotenone or asolectin alone. This observation is difficult to explain, but it may be because either asolectin or the steady-state level of reduced flavin affects the ratio of quinol to semiquinone formation at the flavin site, and their stability and reactivity in solution also. The rates of H2O2 production by IDE are comparable to those of Q1, but they are significantly enhanced in the absence of asolectin, consistent with the increase in the rate of the hydrophilic reaction observed under the same conditions (see Figure 1). Q0 (and IDE, to a lesser extent) causes significantly more cyt c reduction than H2O2 formation. It is possible that the hydrophilic Q0 semiquinone is more stable in aqueous solution, so that superoxide formation is less favored or the semiquinone dismutates directly. Scheme 2 summarizes the reaction pathways which originate at the flavin site in complex I.
Discussion
Identification of the Flavin Site as the Hydrophilic Q-Binding Site
Two pieces of evidence strongly suggest that the hydrophilic site of quinone reduction in complex I is the flavin site at which NADH is oxidized: the hydrophilic reaction is conserved in a preparation of subcomplex Iλ which lacks the terminal iron−sulfur cluster N2 and the hydrophobic Q-binding site, and it is inhibited by high NADH concentrations and can be explained mechanistically using ping-pong or ping-pong-pong kinetics (using parameters derived from the study of transhydrogenation reactions at the flavin). Previously, the hydrophilic site of quinone reduction in complex I has been attributed to essentially every conceivable reaction site (see above), but until now, no investigation has aimed specifically at identifying the site or presented substantial experimental support for any proposal.
Ping-pong reactions involving NADH and quinones at flavins are well-known in the diverse class of single-subunit NADH:Q oxidoreductases. Kinetic evidence of a ping-pong mechanism in one of the alternative (non-energy transducing) NADH dehydrogenases which react with extremely hydrophobic quinones such as Q10, and which replace complex I in the respiratory chains of a number of species, has been presented (44). However, related enzymes which are involved in detoxification have been studied more extensively. In contrast to complex I, which has a thermodynamically accessible flavosemiquinone (45), they are obligatory two-electron donors (perhaps because they reduce bound quinones by the transfer of hydride from the flavin), and so they avoid the formation of semiquinone species and redox cycling reactions (46). The structure of duroquinone bound to human NAD(P)H:quinone oxidoreductase shows the quinone ring stacked above the flavin isoalloxazine system (47), providing a visual representation for how quinones may bind and react at the flavin site in complex I.
How Do Phospholipids Affect Quinone Reduction at the Hydrophobic and Hydrophilic Sites in Isolated Complex I?
Phospholipids may affect the reactivity of isolated complex I toward a short-chain ubiquinone in two ways: they may bind to the enzyme, at the active site or as a phospholipid annulus, to stabilize active conformations and promote catalytic activity (the “enzyme effect”), or they may alter the availability of the quinone to the enzyme, by partitioning it into a hydrophobic phase or by dispersing quinone micelles (the “quinone effect”).
The reaction at the hydrophobic site is enhanced strongly by phospholipids in the assay medium (Figure 1, also observed in many previous studies, for example, refs (7), (28), and (48)−(50)), an effect that is not mimicked by similar concentrations of detergents, suggesting the importance of the enzyme effect. However, previous studies have suggested that the quinone effect contributes also. In studies of SMPs (for which the enzyme effect does not apply), detergents or sonicated phospholipids were found to enhance the rate of DQ reduction by complex I, probably by dispersing quinone micelles and aiding quinone−quinol exchange between the membrane and the aqueous phase (9). An earlier study of the effects of phospholipid concentration on Q1 reduction by isolated complex I focused on partitioning of quinone between the aqueous and lipidic phases. However, the rate of the hydrophobic reaction did not follow the concentration of Q1 in the lipidic phase (it followed the total concentration), and it was increased strongly in higher phospholipid concentrations (as observed here) (7). Indeed, if partitioning is the dominant effect, then higher phospholipid concentrations should actually decrease the rate of the hydrophobic reaction, by increasing the volume of the hydrophobic phase and diluting the Q1. We conclude that, in studies of the isolated enzyme, the enzyme effect is dominant in controlling the reaction at the hydrophobic site, although (particularly with the more hydrophobic quinones) the quinone effect may also contribute.
It is unlikely that there is a significant enzyme effect at the hydrophilic binding site, and Figure 1 shows that the rate of the hydrophilic reaction tends to decrease with the addition of phospholipids. The effect is most marked for IDE, observed to a lesser extent for Q1 and not at all for Q0 (for DQ the hydrophilic reaction site is so slow that no conclusions can be drawn). Previously, for isolated complex I and Q1, Ragan proposed that phospholipids partition the Q1 out of the hydrophilic phase, so that it cannot interact with the hydrophilic site (7). The extent of hydrophilic Q1 reduction was decreased by ∼45% in 0.4 mg/mL phospholipids [compared to ∼20% observed here (Figure 1)]; however, the phospholipid dependence was investigated in much greater depth than we have attempted, and a convincing fit, over a range of phospholipid and Q1 concentrations, was described. The fact that IDE reduction at the hydrophilic site is most affected by phospholipids, and Q0 reduction least affected, is consistent with the relative partition coefficients of IDE, Q1, and Q0. However, note that the extent of partitioning may differ when biological membranes are present and that neither partition coefficients or the concept of a hydrophobic phase is physically relevant to experiments on isolated complex I. Instead, the system comprises mixed micelles of quinone, phospholipid, and detergent (plus a large hydrophobic enzyme). Equations for describing mixed micelle formation are available (51,52), but the complexity of our system and the lack of basic information about the micellular properties of its components allow them to provide only general physical insights at present.
The Identity of the Ubiquinone Determines Its Rate of Reaction at both the Hydrophobic and Hydrophilic Sites
The relative rates of reaction of the four ubiquinones at the hydrophobic and hydrophilic sites do not correlate in a simple fashion with their hydrophobicity. At the flavin site, the ubiquinone ring probably stacks against the flavin isoalloxazine ring system, with the hydrophobic tail lying along the channel which binds the ADP-ribose moiety of NADH. The release of bound waters upon binding will tend to increase the affinity as the hydrophobic tail lengthens, explaining the low rate of reaction of Q0. For the more hydrophobic quinones, such as DQ, the entropic effect is opposed by their decreasing solubility in the external solution, so the maximal activity is observed for Q1 and IDE. It is possible that the binding of IDE is enhanced by hydrogen bonding between its terminal hydroxyl group and a nearby charged residue in the site. For the hydrophobic site, Sakamoto and co-workers studied the reduction of a range of coenzyme Q2 derivatives by complex I (53). They concluded that the role of the tail is not simply to enhance the hydrophobicity and attributed the high affinity of Q2 itself to molecular interactions between the binding site and the methyl branch and π-system of the first isoprene unit. The fact that Q2 reacts more slowly than DQ, for example, was attributed to slow dissociation of the ubiquinol product. The characteristics of reduction of Q2 and IDE by complex I are similar (24), and our results are consistent with IDE reduction being limited by product dissociation also. Thus, reactivity at the hydrophobic site requires an “intermediate affinity”, high enough for binding and reaction but not high enough to cause inhibition. DQ and Q1 react rapidly because they have intermediate affinities; IDE reacts slowly because it has an overly high affinity, and Q0 probably reacts slowly because it has an overly low affinity. Importantly, the affinities are again affected by how the quinone interacts with the external environment, and this may differ considerably between a membrane and a micellar system.
Here, we suggest that DQ is the most appropriate substrate for complex I, in systems which lack endogenous quinone. Under appropriate conditions, it displays a substantial rate at the hydrophobic (physiological) quinone binding site and a minimal rate at the flavin site. For some experiments, in which the effects of the reaction at the flavin site can be subtracted out, Q1 also displays a substantial rate at the hydrophobic site. However, Q1 is clearly very unsuitable for any investigation of reactive oxygen species generation or semiquinone formation by complex I.
Implications for Complex I in Mitochondria
IDE has shown some promise in counteracting the cardiomyopathy associated with Friedreich’s Ataxia, a neurodegenerative disease (27). Although IDE undoubtedly reacts in several different ways in mitochondria (24,32), we are concerned here with only complex I. IDE is reduced by the flavin in complex I, to produce semiquinone species which result in an increased level of reactive oxygen species formation, and it is reduced slowly at the physiological quinone binding site. The rates of both reactions are affected by how IDE partitions between the membrane and the aqueous phases. The “negative” effects of IDE are thus reactive oxygen species production, and the inhibition of physiological quinone reduction, caused by slow IDE product release. The “positive” effects of IDE are that, if Q10 is not sufficiently available, it increases the rate of NADH oxidation and regenerates NAD+, and its high affinity means that it may be more effective at outcompeting an inhibitor or reacting with a damaged binding site.
It is clear that the extremely hydrophobic, physiological quinone cannot react at the flavin site in complex I, because the site is spatially separated from the membrane and must be entered from the aqueous phase (Q10 is unable to dissociate from the membrane); the flavin-mediated reaction is only relevant for relatively hydrophilic quinones. However, the ability of the flavin site to reduce quinones at all suggests that it is more promiscuous than previously considered: it may be capable of binding and reacting with many different compounds. Indeed, a number of compounds which are used, for example, as drugs or pesticides have been reported to undergo redox cycling reactions at complex I. Important examples are anthracyclines such as adriamycin (a chemotherapeutic agent with cardiotoxic side effects) (54) and paraquat (a herbicide, which is used to increase the level of superoxide production in biochemical studies of oxidative stress also) (55). Furthermore, in Parkinson’s disease, there is considerable evidence of oxidative damage to dopaminergic neurons, and of the involvement of complex I (56). It is possible that quinoid compounds, from the environment or produced as metabolic byproducts or in the metabolism of dopamine, contribute to the production of reactive oxygen species by reacting by the mechanism described. Thus, the role of the complex I flavin site in promoting redox cycling reactions and reactive oxygen species production, mediated by a variety of compounds and via a universal reaction mechanism, is worthy of further consideration.
Acknowledgments
We thank Kenneth R. Pryde (Medical Research Council) for providing submitochondrial particles from bovine heart mitochondria and Dr. Michael P. Murphy for advice on the reactivity of hydrophilic quinone species in mitochondria.
Footnotes
Abbreviations: cyt c, partially acetylated cytochrome c; DQ, decylubiquinone; HRP, horseradish peroxidase; IDE, idebenone; Qn, coenzyme Qn or ubiquinone-n; LSQE, least-squares error; SOD, superoxide dismutase; SMP, submitochondrial particle.
References
- Brandt U. (2006) Energy converting NADH:quinone oxidoreductase (complex I). Annu. Rev. Biochem. 75, 69–92. [DOI] [PubMed] [Google Scholar]
- Hirst J. (2005) Energy transduction by respiratory complex I: An evaluation of current knowledge. Biochem. Soc. Trans. 33, 525–529. [DOI] [PubMed] [Google Scholar]
- Carroll J.; Fearnley I. M.; Skehel M.; Shannon R. J.; Hirst J.; Walker J. E. (2006) Bovine complex I is a complex of 45 different subunits. J. Biol. Chem. 281, 32724–32727. [DOI] [PubMed] [Google Scholar]
- Hirst J.; Carroll J.; Fearnley I. M.; Shannon R. J.; Walker J. E. (2003) The nuclear encoded subunits of complex I from bovine heart mitochondria. Biochim. Biophys. Acta 1604, 135–150. [DOI] [PubMed] [Google Scholar]
- Yakovlev G.; Reda T.; Hirst J. (2007) Reevaluating the relationship between EPR spectra and enzyme structure for the iron-sulfur clusters in NADH:quinone oxidoreductase. Proc. Natl. Acad. Sci. U.S.A. 104, 12720–12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sazanov L. A.; Hinchliffe P. (2006) Structure of the hydrophilic domain of respiratory complex I from Thermus thermophilus. Science 311, 1430–1436. [DOI] [PubMed] [Google Scholar]
- Ragan C. I. (1978) The role of phospholipids in the reduction of ubiquinone analogues by the mitochondrial reduced nicotinamide-adenine dinucleotide-ubiquinone oxidoreductase complex. Biochem. J. 172, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio F.; Azzone G. F. (1982) Activation of site I redox-driven H+ pump by exogenous quinones in intact mitochondria. J. Biol. Chem. 257, 4106–4113. [PubMed] [Google Scholar]
- Estornell E.; Fato R.; Pallotti F.; Lenaz G. (1993) Assay conditions for the mitochondrial NADH:coenzyme Q oxidoreductase. FEBS Lett. 332, 127–131. [DOI] [PubMed] [Google Scholar]
- Degli Esposti M.; Ngo A.; McMullen G. L.; Ghelli A.; Sparla F.; Benelli B.; Ratta M.; Linnane A. W. (1996) The specificity of mitochondrial complex I for ubiquinones. Biochem. J. 313, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fato R.; Estornell E.; Di Bernardo S.; Pallotti F.; Castelli G. P.; Lenaz G. (1996) Steady-state kinetics of the reduction of coenzyme Q analogues by complex I (NADH:ubiquinone oxidoreductase) in bovine heart mitochondria and submitochondrial particles. Biochemistry 35, 2705–2716. [DOI] [PubMed] [Google Scholar]
- Lenaz G. (1998) Quinone specificity of complex I. Biochim. Biophys. Acta 1364, 207–221. [DOI] [PubMed] [Google Scholar]
- Tocilescu M. A.; Fendel U.; Zwicker K.; Kerscher S.; Brandt U. (2007) Exploring the ubiquinone binding cavity of respiratory complex I. J. Biol. Chem. 282, 29514–29520. [DOI] [PubMed] [Google Scholar]
- Miyoshi H. (1998) Structure-activity relationships of some complex I inhibitors. Biochim. Biophys. Acta 1364, 236–244. [DOI] [PubMed] [Google Scholar]
- Degli Esposti M. (1998) Inhibitors of NADH-ubiquinone reductase: An overview. Biochim. Biophys. Acta 1364, 222–235. [DOI] [PubMed] [Google Scholar]
- Darrouzet E.; Issartel J. P.; Lunardi J.; Dupuis A. (1998) The 49-kDa subunit of NADH-ubiquinone oxidoreductase (complex I) is involved in the binding of piericidin and rotenone, two quinone-related inhibitors. FEBS Lett. 431, 34–38. [DOI] [PubMed] [Google Scholar]
- Schuler F.; Yano T.; Di Bernardo S.; Yagi T.; Yankovskaya V.; Singer T. P.; Casida J. E. (1999) NADH-quinone oxidoreductase: PSST subunit couples electron transfer from iron-sulfur cluster N2 to quinone. Proc. Natl. Acad. Sci. U.S.A. 96, 4149–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley F. G. P.; Patel S. D.; Ragan C. I.; Attardi G. (1987) Photolabelling of a mitochondrially encoded subunit of NADH dehydrogenase with [3H]dihydrorotenone. FEBS Lett. 219, 108–113. [DOI] [PubMed] [Google Scholar]
- Murai M.; Ishihara A.; Nishioka T.; Yagi T.; Miyoshi H. (2007) The ND1 subunit constructs the inhibitor binding domain in bovine heart mitochondrial complex I. Biochemistry 46, 6409–6416. [DOI] [PubMed] [Google Scholar]
- Nakamaru-Ogiso E.; Sakamoto K.; Matsuno-Yagi A.; Miyoshi H.; Yagi T. (2003) The ND5 subunit was labeled by a photoaffinity analogue of fenpyroximate in bovine mitochondrial complex I. Biochemistry 42, 746–754. [DOI] [PubMed] [Google Scholar]
- Schatz G.; Racker E. (1966) Partial resolution of the enzymes catalyzing oxidative phosphorylation. VII. Oxidative phosphorylation in the diphosphopyridine nucleotide-cytochrome b segments of the respiratory chain: Assay and properties in submitochondrial particles. J. Biol. Chem. 241, 1429–1438. [PubMed] [Google Scholar]
- Zickermann V.; Bostina M.; Hunte C.; Ruiz T.; Radermacher M.; Brandt U. (2003) Functional implications from an unexpected position of the 49-kDa subunit of NADH:ubiquinone oxidoreductase. J. Biol. Chem. 278, 29072–29078. [DOI] [PubMed] [Google Scholar]
- Tormo J. R.; Estornell E. (2000) New evidence for the multiplicity of ubiquinone- and inhibitor-binding sites in the mitochondrial complex I. Arch. Biochem. Biophys. 381, 241–246. [DOI] [PubMed] [Google Scholar]
- Degli Esposti M.; Ngo A.; Ghelli A.; Benelli B.; Carelli V.; McLennan H.; Linnane A. W. (1996) The interaction of Q analogs, particularly hydroxydecyl benzoquinone (idebenone), with the respiratory complexes of heart mitochondria. Arch. Biochem. Biophys. 330, 395–400. [DOI] [PubMed] [Google Scholar]
- Genova M. L.; Ventura B.; Guiuliano G.; Bovina C.; Formiggini G.; Castelli G. P.; Lenaz G. (2001) The site of production of superoxide radical in mitochondrial complex I is not a bound semiquinone but presumably iron-sulfur cluster N2. FEBS Lett. 505, 364–368. [DOI] [PubMed] [Google Scholar]
- Čénas N. K.; Bironaité D. A.; Kulys J. J. (1991) On the mechanism of rotenone-insensitive reduction of quinones by mitochondrial NADH:ubiquinone reductase. FEBS Lett. 284, 192–194. [DOI] [PubMed] [Google Scholar]
- Pandolfo M. (2008) Drug insight: Antioxidant therapy in inherited ataxias. Nat. Clin. Pract. Neurol. 4, 86–96. [DOI] [PubMed] [Google Scholar]
- Sharpley M. S.; Shannon R. J.; Draghi F.; Hirst J. (2006) Interactions between phospholipids and NADH:ubiquinone oxidoreductase (complex I) from bovine mitochondria. Biochemistry 45, 241–248. [DOI] [PubMed] [Google Scholar]
- Fearnley I. M.; Carroll J.; Shannon R. J.; Runswick M. J.; Walker J. E.; Hirst J. (2001) GRIM-19, a cell death regulatory gene product, is a subunit of bovine mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Biol. Chem. 276, 38345–38348. [DOI] [PubMed] [Google Scholar]
- Reda T.; Barker C. D.; Hirst J. (2008) Reduction of the iron-sulfur clusters in mitochondrial NADH:ubiquinone oxidoreductase (complex I) by EuII-DTPA, a very low potential reductant. Biochemistry 47, 8885–8893. [DOI] [PubMed] [Google Scholar]
- Kotlyar A. B.; Vinogradov A. D. (1990) Slow active/inactive transition of the mitochondrial NADH-ubiquinone reductase. Biochim. Biophys. Acta 1019, 151–158. [DOI] [PubMed] [Google Scholar]
- Rauchová H.; Drahota Z.; Bergamini C.; Fato R.; Lenaz G. (2008) Modification of respiratory-chain enzyme activities in brown adipose tissue mitochondria by idebenone (hydroxydecyl-ubiquinone). J. Bioenerg. Biomembr. 40, 85–93. [DOI] [PubMed] [Google Scholar]
- Yakovlev G.; Hirst J. (2007) Transhydrogenation reactions catalyzed by mitochondrial NADH-ubiquinone oxidoreductase (complex I). Biochemistry 46, 14250–14258. [DOI] [PubMed] [Google Scholar]
- Kushnareva Y.; Murphy A. N.; Andreyev A. (2002) Complex I-mediated reactive oxygen species generation: Modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 368, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kussmaul L.; Hirst J. (2006) The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U.S.A. 103, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Votyakova T. V.; Reynolds I. J. (2004) Detection of hydrogen peroxide with amplex red: Interference by NADH and reduced glutathione auto-oxidation. Arch. Biochem. Biophys. 431, 138–144. [DOI] [PubMed] [Google Scholar]
- Azzi A.; Montecucco C.; Richter C. (1975) The use of acetylated ferricytochrome c for the detection of superoxide radicals produced in biological membranes. Biochem. Biophys. Res. Commun. 65, 597–603. [DOI] [PubMed] [Google Scholar]
- Nakashima Y.; Shinzawa-Itoh K.; Watanabe K.; Naoki K.; Hano N.; Yoshikawa S. (2002) The second coenzyme Q1 binding site of bovine heart NADH:coenzyme Q oxidoreductase. J. Bioenerg. Biomembr. 34, 89–94. [DOI] [PubMed] [Google Scholar]
- Jauslin M. L.; Wirth T.; Meier T.; Schoumacher F. (2002) A cellular model for Friedreich Ataxia reveals small-molecule glutathione peroxidase mimetics as novel treatment strategy. Hum. Mol. Genet. 11, 3055–3063. [DOI] [PubMed] [Google Scholar]
- Minakami S.; Ringler R. L.; Singer T. P. (1962) Studies on the respiratory chain-linked dihydrodiphosphopyridine nucleotide dehydrogenase. J. Biol. Chem. 237, 569–576. [PubMed] [Google Scholar]
- Helfenbaum L.; Ngo A.; Ghelli A.; Linnane A. W.; Degli Esposti M. (1997) Proton pumping of mitochondrial complex I: Differential activation by analogs of ubiquinone. J. Bioenerg. Biomembr. 29, 71–80. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C. (1981) Cytochrome c reduction by semiquinone radicals can be directly inhibited by superoxide dismutase. Arch. Biochem. Biophys. 209, 159–167. [DOI] [PubMed] [Google Scholar]
- Galkin A.; Brandt U. (2005) Superoxide radical formation by pure complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica. J. Biol. Chem. 280, 30129–30135. [DOI] [PubMed] [Google Scholar]
- Eschemann A.; Galkin A.; Oettmeier W.; Brandt U.; Kerscher S. (2005) HDQ (1-hydroxy-2-dodecyl-4(1H)quinolone), a high affinity inhibitor for mitochondrial alternative NADH dehydrogenase: Evidence for a ping-pong mechanism. J. Biol. Chem. 280, 3138–3142. [DOI] [PubMed] [Google Scholar]
- Sled V. D.; Rudnitzky N. I.; Hatefi Y.; Ohnishi T. (1994) Thermodynamic analysis of flavin in mitochondrial NADH:ubiquinone oxidoreductase (complex I). Biochemistry 33, 10069–10075. [DOI] [PubMed] [Google Scholar]
- Deller S.; Macheroux P.; Sollner S. (2008) Flavin-dependent quinone reductases. Cell. Mol. Life Sci. 65, 141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faig M.; Bianchet M. A.; Talalay P.; Chen S.; Winski S.; Ross D.; Amzel L. M. (2000) Structures of recombinant human and mouse NAD(P)H:quinone oxidoreductases: Species comparison and structural changes with substrate binding and release. Proc. Natl. Acad. Sci. U.S.A. 97, 3177–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragan C. I.; Racker E. (1973) Resolution and reconstitution of the mitochondrial electron transfer system. IV. The reconstitution of rotenone-sensitive reduced nicotinamide adenine dinucleotide-ubiquinone reductase from reduced nicotinamide adenine dinucleotide dehydrogenase and phospholipids. J. Biol. Chem. 248, 6876–6884. [PubMed] [Google Scholar]
- Fry M.; Green D. E. (1981) Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem. 256, 1874–1880. [PubMed] [Google Scholar]
- Dröse S.; Zwicker K.; Brandt U. (2002) Full recovery of the NADH:ubiquinone activity of complex I (NADH:ubiquinone oxidoreductase) from Yarrowia lipolytica by the addition of phospholipids. Biochim. Biophys. Acta 1556, 65–72. [DOI] [PubMed] [Google Scholar]
- Clint J. H. (1974) Micellization of mixed nonionic surface active agents. J. Chem. Soc. 71, 1327–1334. [Google Scholar]
- Holland P. M.; Rubingh D. N. (1983) Nonideal multicomponent mixed micelle model. J. Phys. Chem. 87, 1984–1990. [Google Scholar]
- Sakamoto K.; Miyoshi H.; Ohshima M.; Kuwabara K.; Kano K.; Akagi T.; Mogi T.; Iwamura H. (1998) Role of the isoprenyl tail of ubiquinone in reaction with respiratory enzymes: Studies with bovine heart mitochondrial complex I and Escherichia coli bo-type ubiquinol oxidase. Biochemistry 37, 15106–15113. [DOI] [PubMed] [Google Scholar]
- Berthiaume J. M.; Wallace K. B. (2007) Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol. Toxicol. 23, 15–25. [DOI] [PubMed] [Google Scholar]
- Cochemé H. M.; Murphy M. P. (2008) Complex I is the major site of mitochondrial superoxide production by paraquat. J. Biol. Chem. 283, 1786–1798. [DOI] [PubMed] [Google Scholar]
- Greenamyre J. T.; Hastings T. G. (2004) Parkinson’s: Divergent causes, convergent mechanisms. Science 304, 1120–1122. [DOI] [PubMed] [Google Scholar]