

Abstract
Distributed Drug Discovery (D3) proposes solving large drug discovery problems by breaking them into smaller units for processing at multiple sites. A key component of the synthetic and computational stages of D3 is the global rehearsal of prospective reagents and their subsequent use in the creation of virtual catalogs of molecules accessible by simple, inexpensive combinatorial chemistry. The first section of this article documents the feasibility of the synthetic component of Distributed Drug Discovery. Twenty-four alkylating agents were rehearsed in the United States, Poland, Russia, and Spain, for their utility in the synthesis of resin-bound unnatural amino acids 1, key intermediates in many combinatorial chemistry procedures. This global reagent rehearsal, coupled to virtual library generation, increases the likelihood that any member of that virtual library can be made. It facilitates the realistic integration of worldwide virtual D3 catalog computational analysis with synthesis. The second part of this article describes the creation of the first virtual D3 catalog. It reports the enumeration of 24 416 acylated unnatural amino acids 5, assembled from lists of either rehearsed or well-precedented alkylating and acylating reagents, and describes how the resulting catalog can be freely accessed, searched, and downloaded by the scientific community.
Introduction
At Indiana University−Purdue University Indianapolis (IUPUI), we have developed a concept called “Distributed Drug Discovery” (D3).1 Combinatorial chemistry is central to its enablement. Herein is reported the first example of a key component of D3: the distributed rehearsal of reagents and their subsequent use in real and virtual catalog production.
The Distributed Drug Discovery concept is described in full detail in the preceding Perspective.1 Immediately following is an Article providing a specific application to drug discovery.2 The premise of D3 is that the availability of simple, inexpensive equipment and procedures for each of the core scientific drug discovery disciplines (computational chemistry, synthetic chemistry, and biochemical screening) will enable large drug discovery problems to be broken down into manageable smaller units to be solved at multiple sites throughout the developing and developed world. The recombined and coordinated results of these resources can inexpensively accelerate the identification of leads (primarily for developing world and other neglected disease targets) in the early stages of the drug discovery process. In addition, this effort provides educational and job opportunities in both the developed and developing worlds while building cultural and economic bridges for the common good.
Combinatorial virtual catalogs, realistically accessible by globally distributed synthesis, are key to the implementation of D3. In classical combinatorial chemistry, reagent rehearsal increases the likelihood that any individual member of an enumerated virtual library can be synthesized from each unique combination of rehearsed reagents. D3 also incorporates this rehearsal process. However, it imposes additional strong constraints on reagent-rehearsed virtual catalogs: any member of the library must be accessible by rehearsed or precedented synthesis, anywhere in the world, using simple, inexpensive equipment and procedures. This article documents progress toward fulfilling this challenge. Part I reports the first successful global reagent rehearsal, conducted by undergraduate and graduate students in the United States, Poland, Russia, and Spain. In Part II, a virtual D3 catalog based on this work is enumerated and made freely accessible.
Results and Discussion
Part I. Rehearsal of Alkylating Reagents for D3 Combinatorial Chemistry
One of the most common types of intermediates used in combinatorial chemistry is the resin-bound natural or unnatural α-amino acid 1 (Scheme 1). The α-amino acid substructure occurs in 28% of the structures cited in a recent “Comprehensive Survey of Combinatorial Library Synthesis”.3,4 Scheme 1 gives examples of a few of the published libraries based on 1.5−11 With the exception of proline, the proteinogenic α-amino acids differ only in the identity of their α-substituent. When these substituents do not occur in nature they are referred to as “unnatural”. The methodology for the introduction of side chains by carbon−carbon bond construction has expanded considerably the availability of these unnatural α-amino acids.12 Of the existing routes to such molecules, alkylation chemistry13 has played a key role. Thus, introduction of an unnatural side chain onto the α-position of a suitable glycine or higher amino acid derivative by alkylation can provide, respectively, α-substituted or α,α-disubstituted amino acid products.14−18 Because of the central role of 1 in combinatorial chemistry, and the ability to greatly expand its numbers through alkylation chemistry, we chose to focus on rehearsing alkylating agents R1X for our first exemplification of D3 synthesis. In the process, we wished to show the likelihood that undergraduate or graduate students would be able to make any given member of a resulting virtual D3 catalog, further demonstrating the potential of Distributed Drug Discovery. Accordingly, we modified our published synthesis of 1(17c) and incorporated the rehearsal into a student combinatorial chemistry laboratory that transformed 1 to the acylated derivatives 5 (Scheme 2). This generic structure 5 would then become the basis of our first virtual D3 catalog.
Scheme 1. Role of Alkylating Agents R1X as Diversity Reagents for Generating Multiple Combinatorial Libraries from Key Intermediate 1.

Scheme 2. Preparation of Acylated Unnatural Amino Acids.

The time ranges shown in Scheme 2 accommodated the details of laboratory scheduling in academic institutions at different geographic locations. Simple, inexpensive “Bill-Board 6-pack” equipment1 (Figure 1) was used to carry out this procedure, six reactions at a time. Every Bill-Board 6-pack had six reaction vessels in a grid of two rows (A and B) by three columns (1, 2, and 3), and reactions were correspondingly identified by “row, column” (e.g., A1−B3). The equipment was designed to enable the essential steps common to most solid-phase synthetic procedures: cycles of reaction and filtration workup, followed by product cleavage, and collection. One to six students were assigned to each Bill-Board.
Figure 1.

Bill-Board Components and 2 × 3 Experimental Grid.
In this first demonstration of the distributed process, twenty-four alkylating agents R1X were rehearsed, often in quadruplicate, sometimes at geographical locations remote from one another (Indianapolis, IN; Lublin, Poland; Moscow, Russia; and Barcelona, Spain). In all cases the quality of the crude products was assessed by LC/MS to obtain a marker of the suitability of a given reagent in combinatorial library generation. This analytical measurement of quality is not definitive because UV detection can overlook, among other things, poor absolute recovery, the presence of insoluble impurities, or soluble impurities not revealed by the detection system.19 Nevertheless, the LC/MS data permits elimination, from a rehearsed virtual library, products based on alkylating agents that are clearly problematic. Follow up synthesis and purification conducted at IUPUI allowed the isolation and characterization of all the unnatural amino acids reported in this distributed reagent rehearsal study.
Since reagent rehearsal was done by students with little to no prior experience in either organic synthesis or solid-phase chemistry, two key elements were incorporated into the laboratory: (1) Students worldwide conducted the same “control” alkylation reaction, using R1X = benzyl bromide (6) in column 1. (2) The evaluation of new alkylating agents was replicated by at least one other student or student team. This worldwide distributed process permits the assessment of both the quality of the work carried out by any given student/team and the reproducible efficacy of the evaluated reagents.
The first synthetic rehearsals were conducted at IUPUI, utilizing four concurrent laboratory sections, to pilot the process of replicated synthesis in multiple students’ hands. There were up to 20 students in each section. A total of 65 students operated in 32 teams of two per Bill-Board (192 reactions), with one extra Bill-Board used for the solo student.20 The control alkylating agent, benzyl bromide (6), was used by all students in column 1 (Figure 1). The new alkylating agents were rehearsed in columns 2 and 3. In these experiments, the acylating agents used in rows A and B of the Bill-Board were always Fmoc chloride and 2-naphthoyl chloride, respectively.
(1). Role of Starting Resin 2 in Product Quality
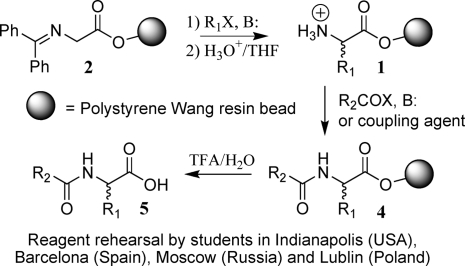
In this first extensive study, two commercial suppliers, I and II, were commissioned to produce the starting benzophenone imine resin 2. For comparison purposes, resin from source I was used in sections one, two, and four (teams 1−8, 10−17, and 26−33), and resin from source II was used in section three (teams 18−25). Since all 32 teams prepared the same product (FmocPhe 5{6}a, see Table 1) in position A1 of their Bill-Boards it was informative to analyze the quality, as a function of resin source, of all 32 of these replicated products. Figure 2 shows a comparison of representative products A1 from teams using resins from these two suppliers.
Table 1. Purity of Products from Alkylation Rehearsal at IUPUI (Using Resin from Sources I and II).
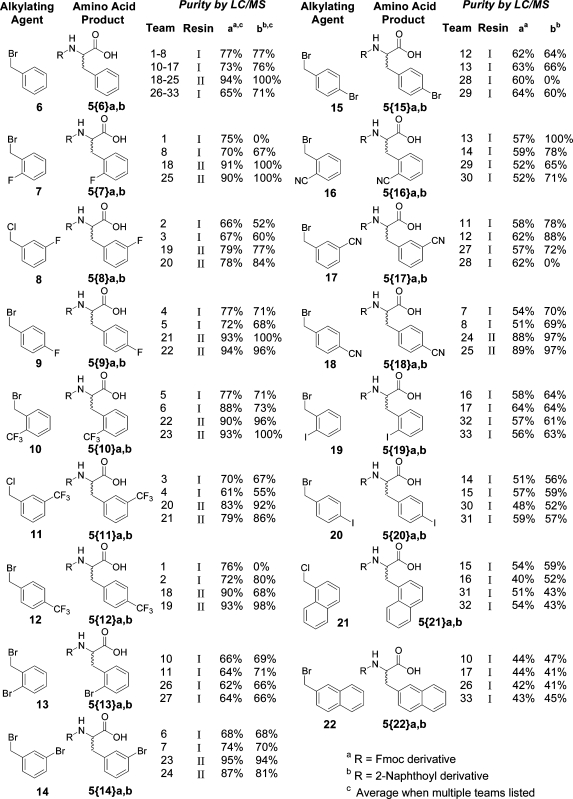 |
Figure 2.

Representative UV trace from LC/MS analysis of A1 from either resin supplier I or II. (a) LC trace of product (team 1, Al) using resin from supplier I. (b) LC trace of product (team 18, Al) using resin from supplier II.
In the products obtained from reactions using resin from I (Figure 2a), there was, in addition to desired product (5.04 min, M + 1 = 388, 77%), the universal presence of three impurities (2.5 min, M + 1 = 256, 4%; 4.8 min, M + 1 = 445, 11%; 5.3 min, M + 1 = 494, 8%). The MS data allowed the tentative assignment of the structures 23, 24, and 25 for these respective peaks (Figure 3). In contrast, there were insignificant levels of the impurities at 2.5 and 4.8 min in samples prepared from resin supplier II (Figure 2b). The Supporting Information contains a more complete discussion of the origin of these proposed impurities 23 and 24 in resin from I,21 and 25 in both resins from either I or II.22
Figure 3.

Proposed structures for impurities produced from resin source I.
Although resins from I and II were used in the initial laboratories at IUPUI, because of its higher quality, only resin from commercial source II was used in subsequent laboratories at IUPUI, and in laboratories conducted in Poland, Russia, and Spain.
(2). Results and Analysis of IUPUI Reagent Rehearsal Data
These initial laboratories at IUPUI evaluated 17 alkylating agents R1X. Replicated alkylations (2−4 replicates), using the same resin and alkylating agent, gave strikingly similar results (Table 1). Individual results rarely differed from the average by more than 3%. As expected, because of the impurities present in the starting resin from I (see earlier discussion and Supporting Information) the purity of alkylation products from this resin was lower (by ∼15−20%) than those from II (compare, for example, the results for the Fmoc derivatives 5{7}a through 5{12}a from alkylating agents 7 through 12, where the first two results for each compound were from resin source I, and the second two results were from resin source II). Nevertheless, from the same resin source the alkylation results are very reproducible. In rare cases, replicated experiments showed considerable variability. This is to be expected when so many different researchers (students) are performing this work for the first time. For example, team 28 obtained none of the expected products 5{15}b or 5{17}b, whereas three other teams using the same reagent combinations (teams 12, 13, 29 and 11, 12, 27) were successful and had similar results (63% ± 3% and 80% ± 8%, respectively) for each of these compounds in the appropriate Bill-Board positions.23
(3). Distributed Reagent Rehearsal Worldwide (Poland, Russia, and Spain)
Encouraged by the success of the IUPUI student alkylating agent rehearsal, we tested the ability to further distribute this process to students outside of the United States. Using the same procedures (with minor modifications), equipment, and resin from source II, the laboratory was first replicated in Spain. In 36 separate reactions, teams at the University of Barcelona (two or three students per Bill-Board) evaluated control benzyl bromide (6, six replicates) and six additional alkylating agents in duplicate (Table 2).
Table 2. Alkylating Agent Rehearsal Purity Results from University of Barcelona.
 |
With the exclusive use of starting resin purchased from supplier II, the Barcelona laboratories obtained excellent results, providing strong documentation for the ability of distributed discovery to reproducibly synthesize molecules, both within the same site, and around the world. Except for one example (Barcelona, team 3), all the control FmocPhe samples 5{6}a were synthesized in >92% purity as determined by LC/MS (Table 2). This was consistent with the data generated in the United States at IUPUI, where the subset of compounds 5{6}a synthesized used starting resin from the same supplier (II) produced 7 of 8 products in >93% purity (Table 1).
The Barcelona laboratories also rehearsed, in duplicate, six new alkylating agents, 26−31. Several points are worth noting. On the basis of their replicated chemistry, alkylating agents 26, 27, and 30 should be dependable reagents in libraries. The data from teams 2 and 3 for 5{28}b indicates that the alkylating agent 28 is also likely to give desired product but with moderate purity.24
The methoxy-substituted reagents 29 and 31 illustrate the need for a nuanced interpretation of results. The crude material recoveries for the methoxy derivatives 5{29}a,b and 5{31}a,b were notably lower compared to the rest of the products in this laboratory. These molecules, as anisole derivatives, are susceptible to Friedel−Crafts reactions. It is well established in the peptide field that, in the absence of a powerful cation quenching agent (such as anisole itself), products capable of Friedel−Crafts reactions can reattach themselves to the resin cation generated in the cleavage process.25 Accordingly, selective, cleavage-condition dependent removal of desired product (by reattachment to the resin) could mask a successful alkylation prior to cleavage.
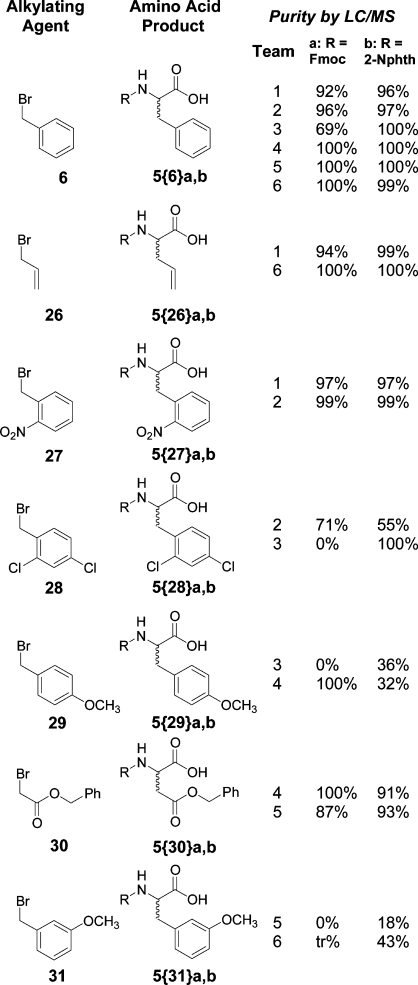
To further demonstrate the ability of the distributed reagent rehearsal project to provide replicated results in student laboratories across the globe, laboratories were carried out in Poland and Russia. The same simple equipment, procedures, and resin from source II were used. Table 3 compares a set of molecules prepared at IUPUI (United States), the University of Barcelona (Spain), Moscow State University (Russia), and the Lublin School of Pharmacy (Poland). These results indicate that D3 syntheses can be carried out at multiple global locations and afford reproducible results.
Table 3. Comparison of Crude Purities of Products Synthesized at Multiple Sites.
 |
Average of six after dropping highest and lowest values.
Average of four after dropping highest and lowest values. (Resin II was used for the products reported at all four sites).
The required reagents were not available.
An additional alkyating agent, 5-(chloromethyl)-2-chloropyridine (32), was evaluated only at the Moscow site. The product 5{32}a (Figure 4) was obtained in 76% purity (LC/MS analysis). Follow up synthesis and purification at IUPUI confirmed the Moscow results.
Figure 4.

Moscow single-site result.
(4). Confirmation of Rehearsed Reagent Results
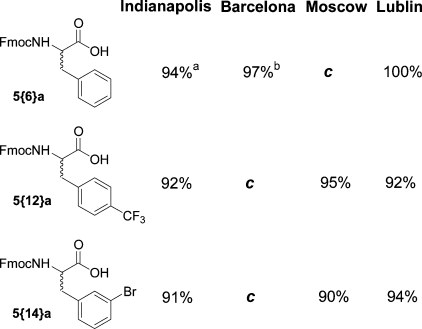
To fully characterize products from the reagent rehearsal and confirm that student-generated results can reliably predict synthetic outcomes, all of the listed alkylating agents were subsequently re-evaluated by a postdoctoral fellow at IUPUI. Using resin from source II, the Fmoc derivative of each alkylated product was resynthesized, purified, and characterized. The experimental procedure used was identical to that employed in the rehearsal laboratories at IUPUI. The results, summarized in Table 4, confirm the ability of student rehearsed chemistry to be replicated by a third party. These results also resolved the questions arising from the large variability of results in the rehearsal of reagents 28 and 29 reported in Table 2. In addition, the single result reported by the Moscow site for reagent 32 was confirmed. The Supporting Information provides LC/MS data for all the crude products from Indianapolis, Lublin, and Moscow, along with NMR data for all the purified, new compounds prepared.
Table 4. Results Obtained by Postdoc (PD) Compared with Previous Compiled Results from Rehearsal Labs (RL) in Indianapolis, Barcelona, Lublin, and Moscow.
 |
Two different resin sources, I or II. See text for discussion.
RL = averaged results from student rehearsal laboratories; PD = result from postdoc synthesis at IUPUI.
Overall purified yield for four step synthesis.
Single value, other value was 0%.
In summary, these first reagent rehearsal laboratories document a key requirement of the Distributed Drug Discovery concept: the global ability of students to evaluate reagents and reproducibly make new potential drug leads, using simple, inexpensive equipment and procedures. These reagents are now acceptable as input in the enumeration of the D3 accessible virtual catalogs to be described in Part II.
Part II. Virtual D3 Catalog Enumeration for Open-Access Availability
An essential component of Distributed Drug Discovery is the open-access availability of databases of molecules accessible through distributed chemistry. Computational chemists can then analyze these large databases to identify and make globally available smaller databases of potential drug leads targeted for developing world diseases. Subsequently, the synthesis of the selected molecules can be carried out, in a distributed fashion, at sites across the world.
The compounds (including stereoisomers when appropriate) in this virtual D3 catalog were enumerated with commercial software.26 To ensure that virtual library generation is not limited by cost we also enumerate libraries with alternative software currently available for free to academia.27 The complete virtual D3 catalog is available online through the Collaborative Drug Discovery interface.28
On the basis of the distributed chemistry developed and reported in this article, two types of library enumeration were envisioned. Each structure made in the reagent rehearsal can be stored in a library. However, a much larger, “virtual” D3 catalog (including the compounds actually made) can be enumerated from all combinations of rehearsed or otherwise well-precedented reagents used at the variable alkylation and acylation steps.
To illustrate this process, we assembled lists of acceptable, commercially available, alkylating and acylating agents (Tables 5 and 6, respectively) to enumerate a representative virtual D3 catalog of acylated unnatural amino acids (Scheme 2). For the alkylation step extensive precedence is available, both from our solution-26 and solid-phase17 studies and those of others,18 to predict the reactivity of Schiff base esters and related systems with these electrophiles under a variety of basic conditions.14 For example, even though Michael addition reactions were not rehearsed in the present study, we have confidence, based on our earlier solid-phase research using resin-bound Schiff bases and Michael acceptors as electrophiles,17e,17j that reactants 93−108 (Table 5) can be utilized successfully. Table 5 also lists references2,16−18,29 that provide precedence for these choices.30 In the future, we plan to incorporate all rehearsed reagent information into a single database to further assist reagent selection.
Table 5. One Hundred Commercially Available Electrophiles (84 Alkyl Halides and 16 Michael Acceptors) for Enumeration of 24 416-Member Library.
 |
Table 6. On Hundred Commercially Available Carboxylic Acids Used for Enumeration of 24 416-Member Library.
 |
Table 5. Continued.
 |
Table 6. Continued.
 |
Table 6. Continued.
 |
In the acylation step reported in this work, acid chlorides were utilized as the second variable reagent. However, we have since modified our D3 laboratories to use carboxylic acids to form the amides by traditional carbodiimide mediated couplings.31 This makes available a much wider repertoire of inexpensive reactants for enumeration at the second variable position. Because the reactivity of carboxylic acids in forming amide bonds is extensively documented, simple chemical intuition and reactivity considerations were used to select the 100 carboxylic acids (Table 6) for enumeration.
On the basis of these considerations, a virtual D3 catalog of 24 416 acylated unnatural amino acids 5 was enumerated. As discussed in the following article,2 an additional 24 192 methyl esters of these acylated unnatural amino acids were enumerated on the basis of the cleavage of resin-bound products 4 by methanolysis. Thus, a total of 48 608 unique products are available for D3 catalog access and searching. The 100 commercially available alkylating agents and Michael acceptors2,14−18,29 and 100 carboxylic acids used to perform these enumerations are shown in Tables 5 and 6, respectively.
The catalog of acylated unnatural amino acids contains more than the 20 000 members that would arise from a simple 100 × 100 combinatorial process. This is caused by the formation of stereoisomers in the alkylation step and their combination with other stereoisomers sometimes present in the alkylating or acylating reagents. Thus, from two to eight stereoisomers appear for each structure in the virtual D3 catalog. The separate listing of each of these anticipated stereoisomeric products allows appropriate computational discrimination and selection when three-dimensional modeling software is employed and chirality is important.
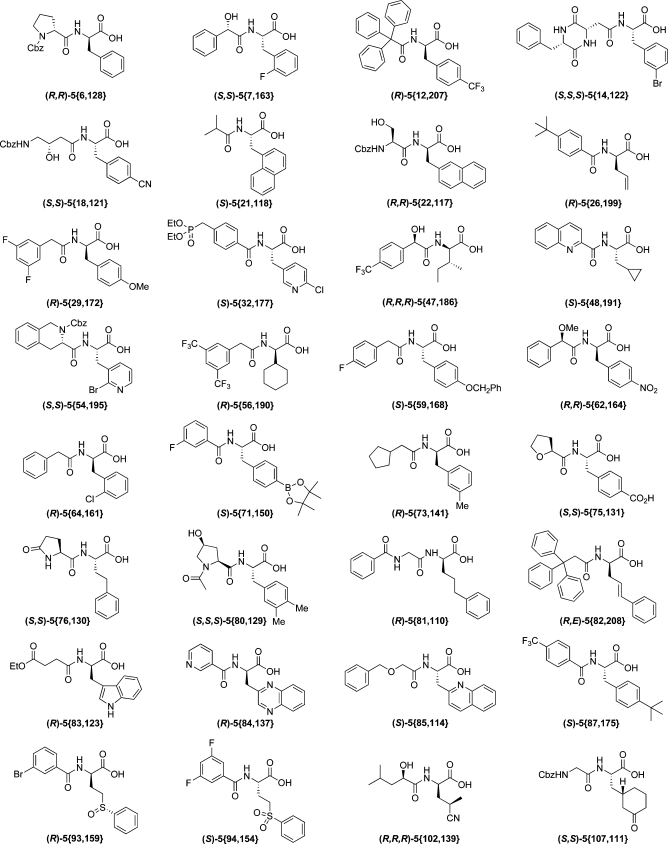
Table 7 contains some examples illustrating the range of this stereochemical complexity. Two stereoisomers arise when there are no other stereogenic elements in either the electrophile or the acid [e.g., (R)-5{12,207} plus enantiomer] or when the electrophile or acid is present as a single enantiomer (R,R)-5{22,117} plus diastereomer] or geometric isomer (R,E)-5{82,208} plus enantiomer]. Four stereoisomers are contained in the library when a racemic or prochiral electrophile or a racemic carboxylic acid is used (S,S)-5{18,121} or (R)-5{93,159} plus three other stereoisomers for each]. Eight stereoisomers arise when both a racemic electrophile and a racemic carboxylic acid are used (R,R,R)-5{47,186} plus seven other stereoisomers].
Table 7. Representative Examples of 24 416 Member Library (Listed in Ascending Order {n}).
 |
The synthesis of stereoisomeric mixtures of unnatural amino acid derivatives has the advantage of providing stereoisomers for evaluation. It is not uncommon to carry out initial biochemical screening with mixtures of stereoisomers. The follow up work to identify the active stereoisomer can be accomplished, without stereoselective synthesis, through bioassay guided fractionation32 or chiral chromatography.33 The active stereoisomer identified by these techniques can be subsequently prepared by known stereoselective synthetic routes,12,14−16,17g,17j,18,34 separation techniques,33 or resolution procedures.35
It is important to consider potential side-reactions during the various steps of the synthesis of acylated unnatural amino acids 5. For example, the tert-butyl esters present in resin-bound products 4 that originated in the reactants used for alkylation, Michael addition, or acylation (36, 75, 96) will be converted to the carboxylic acids during TFA cleavage.36 Likewise, the t-butyl carbamate (N-Boc) group in reactant 83 will be cleaved to the free indole NH on TFA cleavage to products 5.37 A lead reference for each of these subsequent reactions is noted with the respective reactant,38 and this is reflected in the enumerated product structures. Other more subtle side reactions will be incorporated into the enumeration products as they are discovered on the basis of literature precedence or rehearsal of reagents by our group or by others.39
To illustrate the variety of virtual molecules available through this enumeration, 32 examples are shown in Table 7.
The open-access availability of this virtual D3 catalog makes possible the global computational selection of candidates for drug synthesis, while simultaneously enabling the prioritization of future reagent rehearsals. When computational analysis suggests a particular library member may be a drug lead for a developing world disease target, that molecule will be selected for distributed synthesis. If the reagent used as input in its enumeration is unrehearsed, the synthesis will evaluate its suitability as a reagent and enable it to become a part of this expanding rehearsed reagent database, stored electronically and available through open-access.
Summary and Conclusion
This project demonstrates the power and potential of a globally distributed synthetic process. Students carry out, using simple procedures and inexpensive equipment, worldwide replicated solid-phase combinatorial chemistry, rehearsing new reagents and generating new compounds for library production. At the same time they learn and develop important synthetic skills and methodologies. The data generated and reported here allows the construction of a globally accessible D3 database of well-documented alkylating agents and Michael acceptors for the synthesis of resin-bound unnatural amino acids 1. This intermediate can then be incorporated into a wide variety of published combinatorial chemistry procedures. Virtual D3 catalogs based on rehearsed or well-precedented reagents and inexpensive global procedures are essential to the goals of Distributed Drug Discovery. Accordingly, we have enumerated a 24 416-member virtual D3 catalog of acylated unnatural amino acids 5 based on the current work. Freely available virtual D3 catalogs, such as the one reported in this article, can be computationally analyzed to select potential drug leads for developing-world and other neglected diseases. In turn, the distributed reagent rehearsal provides strong precedent and documented procedures to successfully make, through distributed synthesis, any of the molecules selected by this process.
Experimental Section
General Methods
Organic solvents were of reagent grade and were used directly without purification. Anhydrous NMP, chloroform-d1, 2-fluorobenzyl bromide, 4-fluorobenzyl bromide, 2-(trifluoromethyl)benzyl bromide, 4-(trifluoromethyl)benzyl bromide, 2-bromobenzyl bromide, allyl bromide, 2-nitrobenzyl bromide, 2,4-dichlorobenzyl chloride, and benzyl 2-bromoacetate and were purchased from Acros Organics. Trifluoroacetic acid and hydrochloric acid were obtained from Fisher Scientific. BTPP (tert-butyl-imino-tri(pyrrolidino)phosphorane).16r and Fmoc chloride were purchased from Fluka. 5-(Chloromethyl)-2-chloropyridine, N,N-diisopropylethylamine, benzyl bromide, 3-fluorobenzyl chloride, 3-(trifluoromethyl)benzyl chloride, 3-bromobenzyl bromide, 4-bromobenzyl bromide, 2-(bromomethyl)benzonitrile, 3-(bromomethyl)benzonitrile, 4-(bromomethyl)benzonitrile, 2-iodobenzyl bromide, 4-iodobenzyl bromide, 1-(chloromethyl)naphthalene, 2-(bromomethyl)naphthalene, 4-methoxybenzyl bromide, 3-methoxybenzyl bromide, and methanol-d4 were obtained from Aldrich Chemical Co. All resins were purchased from Midwest Bio-Tech Laboratories (resin I) or Polymer Laboratories (resin II) unless otherwise noted. All reactions and washes were conducted at ambient temperature unless otherwise noted.
Manual solid-phase organic syntheses at ambient temperature were carried out in 3.5 mL fritted glass reaction vessels equipped with polypropylene screw caps with Teflon-faced silicon septa on the Bill-Board set, which was designed by one of us (WLS) as inexpensive equipment40 to simplify and expedite multiple, manual solid-phase syntheses. The Bill-Board reaction vessel components are available from ChemGlass: IUP-0305−270H for 3.5 mL reaction vessel; IUP-0305−280H (polypropylene screw cap); IUP-0305−281H (Teflon faced silicone septa).
Analytical thin layer chromatography (TLC) was performed with Merck silica gel 60 F254, 0.25 mm precoated glass plates. TLC plates were visualized using UV254. Flash column chromatography was performed on silica gel 60 (230−400 mesh) from Silicycle Chemical Division. The yields of the final compounds, after chromatographic purification, were calculated on the basis of the initial loading of the starting resins and are the overall yields of all reaction steps starting from these resins.
1H NMR spectra were recorded at 200 MHz (Varian spectrometer) using TMS (0.00 ppm) and chloroform-d1 mixed with methanol-d4 (3−10%). Electrospray ionization mass spectrometry was conducted using a PESciex API III triple stage quadrupole mass spectrometer operated in either positive-ion or negative-ion detection mode.
The composition of reaction mixtures was determined based on the integration of NMR spectra as well as LC-MS results. LC-MS analyses were conducted using an Agilent system, consisting of a 1100 series HPLC connected to a diode array detector and a 1946D mass spectrometer configured for positive-ion/negative-ion electrospray ionization. The LC-MS samples were analyzed as solutions in CH3CN, prepared at 0.08−0.12 mg/mL concentration. The LC-MS derived composition of mixtures was determined based on UV integration at 210 nm.
General Procedures for Manual Solid-Phase Organic Synthesis
(a). Distribution of Resin and Alkylation (2 to 3)
Depending on the number of reactions to be performed, resin 2 was distributed either by weight or as aliquots from an isopycnic suspension. In the case of distribution by volume from an isopycnic suspension, the Bill-Boards were placed in their drain trays, and from a neutral buoyancy suspension in CH2Cl2/NMP, 50 μmols of the benzophenone imine of glycine Wang resin (2, the resin loading is 0.7 − 0.9 mmol/g) was distributed, via repeated 1 mL aliquots, to each of the six reaction vessels in a given Bill-Board 6-pack. During the distribution of the resin, the isopycnic solvent was allowed to drain through the frit in the reaction vessels. When distribution was complete, residual solvent was removed with an “air-push” from a disposable plastic pipet (Fisher, 13−711−23) fitted with a pierced septum (Aldrich, Z 12743−4). The resin was washed with NMP (3 × 3 mL) (this NMP wash was also done when resin was weighed out into reaction vessels). The bottom of each reaction vessel was then capped, and a new calibrated pipet (Fisher, 13-711-24) was used for adding each alkylation reagent in the following step.
The first 1R1-X (benzyl bromide) [0.5 mL of a 0.20 M solution in NMP, 100 μmols, 2 equiv] was added to the resin in the two reaction vessels in the first column positions (i.e., A1 and B1). Then the second 2R1-X [0.5 mL of a 0.20 M solution in NMP, 100 μmols, 2 equiv] was added to the resin in the two vessels in the second column positions (i.e., A2 and B2). Finally, the third 3R1-X [0.5 mL of a 0.20 M solution in NMP, 100 μmols, 2 equiv] was added to the resin in the two vessels in the third column positions (i.e., A3 and B3). Base was then added [0.5 mL of a 0.20 M solution of BTPP in NMP, 100 μmols, 2 equiv] to each of the six reaction vessels. The tops of all reaction vessels were capped, and the Bill-Board was placed in a rotation apparatus. Depending on the school location and laboratory schedule, the reaction was allowed to proceed from 24 h to 2 days with rotation. This and all subsequent reactions were performed at room temperature.
(b). Hydrolysis (3 to 1)
The Bill-Board was removed from the rotation apparatus and, after inverting the board, the bottom caps were removed. The Board was then placed, top side up, in the drain tray, the top caps were removed, and the reagents from the alkylation step were allowed to drain, followed by an air-push. The alkylated resin-bound resin products 3 were washed with THF (1 × 3 mL). Using 6 clean caps, the bottom of each reaction vessel was capped, and a 1 N aqueous HCl/THF solution (1:2, 2.5 mL) was added to each of the six reaction vessels. The caps were then put on the top of each vessel, and the Bill-Board was returned to the rotator for 20 min. The reagents and byproduct from the hydrolysis were then removed from hydrolyzed resin-bound product (1) by filtration and washing with THF (1 × 3 mL) and then NMP (1 × 3 mL).
(c). Acylation (1 to 4)
The bottom of each reaction vessel was capped, and the acylating agent, 1R2COCl [0.5 mL of a 0.30 M solution of R2COCl in NMP, 150 μmols, 3 equiv] was added to the resin 1 in each of the three vessels in row A (A1, A2, and A3). Then the second acylating agent, 2R2COCl [0.5 mL of a 0.30 M solution of R2COCl in NMP, 150 μmols, 3 equiv] was added to the resin 1 in each of the three vessels in row B (B1, B2, and B3). This was followed by addition of DIEA [0.5 mL of a 0.30 M solution in NMP, 150 μmols, 3 equiv] to each of the six reaction vessels. The tops of the reaction vessels were capped, and the reaction was allowed to rotate overnight or up to 5 days (again, dependent on school laboratory schedules).
(d). Cleavage of Products from Resin (4 to 5)
Acylated resin product 4 was washed with NMP (2 × 3 mL), THF (2 × 3 mL), and CH2Cl2 (3 × 3 mL). The product was cleaved from resin with TFA/H2O (95:5, 2 mL) for 30 min. The filtrate from the cleavage reaction was collected, and the resin was washed with TFA/H2O (95:5, 2 mL) and CH2Cl2 (1 × 2 mL). After each collection vial was swirled to thoroughly mix the cleavage and rinse solutions, a sample of each solution (100 μL) containing product 5 was transferred to an autosampler vial for LC/MS analysis. The cleavage solvent and rinses were evaporated with a simple, inexpensive apparatus.40 designed to speed up the evaporation with a stream of nitrogen, in a contained system, while trapping the evaporating TFA in 2 N NaOH.
General Procedure for Purification of Crude Products
Purification of the crude product was performed via flash column chromatography, using the glass fritted reaction vessel (3.5 mL) loaded with bulk silica gel (650−700 mg). The crude product was dissolved in CH2Cl2 and, if necessary, MeOH (total solution volume ≤0.5 mL) and applied to the column. Then a gradient of eluting solvent was applied starting with CH2Cl2 to CH2Cl2/MeOH/H2O (300:10:0.5). The purified product was dried in the evaporation equipment described above at ambient temperature under a flow of nitrogen.
Variations on General Procedure
(a). Indianapolis
Each of the four separate laboratories operated on a Monday/Wednesday cycle. The alkylations were begun on the first day. Forty eight hours later (Wednesday), the reactions were worked-up; the imine was hydrolyzed and worked-up, and the acylation reactions were begun. Five days later (the following Monday), the acylation reactions were worked-up; the product was cleaved from the resin, and samples taken for TLC and LC/MS analysis. All samples were analyzed by LC/MS in the analytical department at Eli Lilly & Company (Indianapolis).
(b). Barcelona, Moscow, and Lublin
At each of these locations, the complete laboratory was conducted over a three consecutive day period. The alkylations were begun on the first day. The next day the reactions were worked-up; the imine was hydrolyzed and worked-up, and the acylation reactions were begun. The next (and last) day the acylation reactions were worked-up; the product was cleaved from the resin, and samples taken for TLC and LC/MS analysis. The Barcelona samples were analyzed by an LC/MS available on site. The samples from Moscow and Lublin were taken back to Indianapolis where they were analyzed by LC/MS in the analytical department at Eli Lilly.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]phenylalanine [5{6}a]:
6.9 mg (35% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 82%, tR = 5.11 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.00−3.14 (m, 1H), 3.20 (dd, J = 13.9 Hz, J = 5.5 Hz, 1H), 4.14−4.27 (m, 1H), 4.28−4.49 (m, 2H), 4.59 (t, J = 5.8 Hz, 1H), 7.16 (m, 2H), 7.23−7.46 (m, 7H), 7.52−7.62 (m, 2H), 7.78 (d, J = 6.8 Hz, 2H); LC/MS calcd for C24H22NO4 [M + H]+ 388.1; found 388.1.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-2-fluorophenylalanine [5{7}a]:
8.3 mg (41% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 83%, tR = 5.12 min;1H NMR (200 MHz, CDCl3/CD3OD) δ 3.09 (dd, J = 14.2 Hz, J = 7.7 Hz, 1H), 3.32 (dd, J = 14.2 Hz, J = 4.9 Hz, 1H), 4.12−4.23 (m, 1H), 4.24−4.44 (m, 2H), 4.61 (t, J = 5.5 Hz, 1H), 6.96−7.10 (m, 2H), 7.14−7.45 (m, 6H), 7.51−7.60 (m, 2H), 7.76 (d, J = 7.2 Hz, 2H); LC/MS calcd for C24H21FNO4 [M + H]+ 406.1; found 406.2.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-3-fluorophenylalanine [5{8}a]:
10.0 mg (49% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 82%, tR = 5.17 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.08 (dd, J = 13.8 Hz, J = 6.2 Hz, 1H), 3.19 (dd, J = 13.9 Hz, J = 5.5 Hz, 1H), 4.20 (t, J = 7.0 Hz, 1H), 4.28−4.51 (m, 2H), 4.60 (t, J = 5.5 Hz, 1H), 6.85−7.00 (m, 2H), 7.17−7.45 (m, 6H), 7.51−7.63 (m, 2H), 7.77 (d, J = 7.4 Hz, 2H); LC/MS calcd for C24H21FNO4 [M + H]+ 406.1; found 406.3.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-4-fluorophenylalanine [5{9}a]:
7.0 mg (34% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 97%, tR = 5.18 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.02 (dd, J = 13.8 Hz, J = 6.0 Hz, 1H), 3.15 (dd, J = 14.1 Hz, J = 5.3 Hz, 1H), 4.19 (t, J = 6.7 Hz, 1H), 4.28−4.52 (m, 2H), 4.56 (m, 1H), 6.95 (m, 2H), 7.05−7.15 (m, 2H), 7.28−7.46 (m, 4H), 7.52−7.61 (m, 2H), 7.77 (d, J = 7.2 Hz, 2H); LC/MS calcd for C24H21FNO4 [M + H]+ 406.1; found 405.8.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-2-(trifluoromethyl)phenylalanine [5{10}a]:
12.2 mg (54% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 94%, tR = 5.46 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.10 (dd, J = 14.2 Hz, J = 9.6 Hz, 1H), 3.46 (dd, J = 14.7 Hz, J = 5.1 Hz, 1H), 4.07−4.20 (m, 1H), 4.21−4.36 (m, 2H), 4.64 (dd, J = 9.5 Hz, J = 5.1 Hz, 1H), 7.24−7.48 (m, 7H), 7.54 (t, J = 6.6 Hz, 2H), 7.64 (d, J = 7.4 Hz, 1H), 7.75 (d, J = 7.4 Hz, 2H); LC/MS calcd for C25H19F3NO4 [M + H]+ 456.1; found 456.2.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-3-(trifluoromethyl)phenylalanine [5{11}a]:
9.4 mg (41% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 90%, tR = 5.53 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.14 (dd, J = 13.8 Hz, J = 6.0 Hz, 1H), 3.26 (dd, J = 13.7 Hz, J = 5.3 Hz, 1H), 4.19 (t, J = 6.6 Hz, 1H), 4.27−4.51 (m, 2H), 4.61 (t, J = 5.5 Hz), 7.28−7.50 (m, 8H), 7.51−7.62 (m, 2H), 7.77 (dd, J = 7.4 Hz, 2H); LC/MS calcd for C25H19F3NO4 [M + H]+ 456.1; found 456.1.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-4-(trifluoromethyl)phenylalanine [5{12}a]:
9.4 mg (41% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 88%, tR = 5.55 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.12 (dd, J = 13.7 Hz, J = 5.9 Hz, 1H), 3.26 (dd, J = 14 Hz, J = 5.6 Hz, 1H), 4.19 (t, J = 6.6 Hz, 1H), 4.30−4.54 (m, 2H), 4.63 (t, J = 5.3 Hz, 1H), 7.21−7.46 (m, 6H), 7.47−7.61 (m, 4H), 7.77 (d, J = 7.2 Hz, 2H); LC/MS calcd for C25H19F3NO4 [M − H]− 454.1; found 454.2.
2-Bromo-N-[(9H-fluoren-9-ylmethoxy)carbonyl]phenylalanine [5{13}a]:
11.2 mg (48% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5): initial LC/MS purity 90%, tR = 5.39 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.12 (dd, J = 13.8 Hz, 9.2 Hz, 1H), 3.44 (dd, J = 14.1 Hz, 5.3 Hz, 1H), 4.05−4.40 (m, 3H), 4.68 (dd, J = 8.8 Hz, 5.1 Hz, 1H), 7.03−7.16 (m, 1H), 7.18−7.45 (m, 6H), 7.55 (d, J = 7.6 Hz, 3H), 7.76 (d, J = 7.4 Hz, 2H); LC/MS calcd for C24H21BrNO4 [M + H]+ 466.1; found 468.0.
3-Bromo-N-[(9H-fluoren-9-ylmethoxy)carbonyl]phenylalanine [5{14}a]:
10.7 mg (46% isolated yield) following chromatographic purification (CH2Cl/MeOH/H2O, 300:10:0.5); initial LC/MS purity 97%, tR = 5.49 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.05 (dd, J = 13.8 Hz, 6.6 Hz, 1H), 3.17 (dd, J = 13.9 Hz, 5.5 Hz, 1H), 4.15−4.51 (m, 3H), 4.59 (t, J = 5.5 Hz, 1H), 7.06−7.21 (m, 2H), 7.30−7.46 (m, 6H), 7.52−7.63 (m, 2H), 7.77 (d, J = 7.4 Hz, 2H); LC/MS calcd for C24H21BrNO4 [M + H]+ 466.1; found 466.1.
4-Bromo-N-[(9H-fluoren-9-ylmethoxy)carbonyl]phenylalanine [5{15}a]:
9.8 mg (42% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 98%, tR = 5.52 min; 1H NMR (200 MHz, CDCl3/CD3OD): δ 3.03 (dd, J = 13.6 Hz, J = 6.1 Hz, 1H), 3.15 (dd, J = 14.4 Hz, J = 5.8 Hz, 1H), 4.19 (t, J = 6.2 Hz, 1H), 4.28−4.51 (m, 2H), 4.52−4.62 (m, 1H), 7.01 (d, J = 8.4 Hz, 2H), 7.31−7.46 (m, 6H), 7.53−7.62 (m, 2H), 7.78 (d, J = 7.0 Hz, 2H); LC/MS calcd for C24H21BrNO4 [M + H]+ 466.1; found 467.8.
2-Cyano-N-[(9H-fluoren-9-ylmethoxy)carbonyl]phenylalanine [5{16}a]:
7.8 mg (38% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 94%, tR = 4.87 min; 1H NMR (200 MHz, CDCl3/CD3OD): δ 3.25 (dd, J = 14.3 Hz, J = 7.7 Hz, 1H), 3.48 (dd, J = 14.3 Hz, J = 5.5 Hz, 1H), 4.16 (t, J = 6.7 Hz, 1H), 4.22−4.44 (m, 2H), 4.60−4.76 (m, 1H), 7.26−7.64 (m, 10H), 7.76 (d, J = 7.0 Hz, 2H); LC/MS calcd for C25H19N2O4 [M + H]+ 413.1; found 413.2.
3-Cyano-N-[(9H-fluoren-9-ylmethoxy)carbonyl]phenylalanine [5{17}a]:
7.8 mg (38% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 82%, tR = 4.81 min;1H NMR (200 MHz, CDCl3/CD3OD) δ 3.09 (dd, J = 13.8 Hz, J = 6.2 Hz, 1H), 3.22 (dd, J = 13.5 Hz, J = 5.1 Hz, 1H), 4.19 (t, J = 6.4 Hz, 1H), 4.29−4.54 (m, 2H), 4.58 (m, 1H), 7.27−7.62 (m, 10H), 7.77 (d, J = 7.4 Hz, 2H); LC/MS calcd for C25H19N2O4 [M − H]− 412.1; found 411.3.
4-Cyano-N-[(9H-fluoren-9-ylmethoxy)carbonyl]phenylalanine [5{18}a]:
7.5 mg (36% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 91%, tR = 4.79 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.10 (dd, J = 13.7 Hz, J = 7.2 Hz, 1H), 3.19−3.33 (m, 1H), 4.19 (t, J = 6.4 Hz, 1H), 4.31−4.55 (m, 2H), 4.56−4.64 (m, 1H), 7.22−7.46 (m, 6H), 7.52−7.62 (m, 4H), 7.78 (d, J = 7.4 Hz, 2H); LC/MS calcd for C25H19N2O4 [M − H]− 412.1; found 412.5.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-2-iodophenylalanine [5{19}a]:
11.4 mg (44% isolated yield) following chromatographic purification (CH2Cl2-MeOH-H2O, 300:10:0.5); initial LC/MS purity 97%, tR = 5.51 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.11 (dd, J = 14.4 Hz, J = 9.2 Hz, 1H), 3.40 (dd, J = 13.9 Hz, J = 5.1 Hz, 1H), 4.09−4.21 (m, 1H), 4.22−4.39 (m, 2H), 4.67 (dd, J = 9.2 Hz, J = 5.1 Hz, 1H), 6.86−6.97 (m, 1H), 7.22−7.44 (m, 6H), 7.52−7.61 (m, 2H), 7.75 (d, J = 7.4 Hz, 2H), 7.83 (d, J = 7.8 Hz, 1H); LC/MS: calcd for C24H21INO4 [M + H]+ 514.0; found 514.0.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-4-iodophenylalanine [5{20}a]:
7.1 mg (28% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 97%, tR = 5.61 min; 1H NMR (200 MHz, CDCl3/CD3OD): δ 3.01 (dd, J = 14.2 Hz, J = 6.4 Hz, 1H), 3.14 (dd, J = 13.7 Hz, J = 5.3 Hz, 1H), 4.19 (t, J = 6.6 Hz, 1H), 4.27−4.49 (m, 2H), 4.50−4.62 (m, 1H), 5.82 (d, J = 8.4 Hz, 1H), 6.89 (d, J = 8.2 Hz, 2H), 7.27−7.47 (m, 4H), 7.51−7.64 (m, 4H), 7.78 (d, J = 7.4 Hz, 2H); LC/MS calcd for C24H21INO4 [M + H]+ 514.0; found 514.0.
α-[[(9H-Fluoren-9-ylmethoxy)carbonyl]amino]-1-naphthalenepropanoic acid [5{21}a]:
5.9 mg (27% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 86%, tR = 5.57 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.38−3.50 (m, 1H), 3.75 (dd, J = 14.2 Hz, 5.4 Hz, 1H), 4.08−4.22 (m, 1H), 4.23−4.39 (m, 2H), 4.73 (dd, J = 7.4 Hz, 5.6 Hz, 1H), 7.23−7.55 (m, 10H), 7.70−7.89 (m, 4H), 8.16 (d, J = 7.8 Hz, 1H); LC/MS calcd for C28H24NO4 [M + H]+ 438.2; found 438.2.
α-[[(9H-Fluoren-9-ylmethoxy)carbonyl]amino]-2-naphthalenepropanoic acid [5{22}a]:
8.8 mg (40% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 85%, tR = 5.58 min;1H NMR (200 MHz, CDCl3/CD3OD) δ 3.24 (dd, J = 14 Hz, J = 6.8 Hz, 1H), 3.31−3.45 (m, 1H), 4.12−4.45 (m, 1H), 4.27−4.46 (m, 2H), 4.69 (t, J = 5.6 Hz, 1H), 7.19−7.56 (m, 10H), 7.64 (m, 1H), 7.71−7.83 (m, 4H); LC/MS calcd for C28H24NO4 [M + H]+ 438.2; found 438.2.
2-[[(9H-Fluoren-9-ylmethoxy)carbonyl]amino]-4-pentenoic acid [5{26}a]:
10.2 mg (60% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 93%, tR = 4.74 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 2.45−2.71 (m, 2H), 4.23 (t, J = 6.7 Hz, 1H), 4.30−4.49 (m, 3H), 5.05−5.25 (m, 2H), 5.73 (m, 1H), 7.29−7.46 (m, 4H), 7.61 (d, J = 7.2 Hz, 2H), 7.77 (d, J = 6.6 Hz, 2H); LC/MS calcd for C20H20NO4 [M + Na]+ 360.1; found 360.1.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-2-nitrophenylalanine [5{27}a]:
9.6 mg (44% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 91%, tR = 5.00 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.25 (dd, J = 13.2 Hz, 9.4 Hz, 1H), 3.63 (dd, J = 13.9 Hz, 5.5 Hz, 1H), 4.06−4.20 (m, 1H), 4.20−4.37 (m, 2H), 4.70 (dd, J = 8.6 Hz, 5.4 Hz, 1H), 7.27−7.59 (m, 9H), 7.75 (d, J = 7.2 Hz, 2H), 7.94 (d, J = 8.0 Hz, 1H); LC/MS calcd for C24H21N2O6 [M + H]+ 433.1; found 433.2.
2,4-Dichloro-N-[(9H-fluoren-9-ylmethoxy)carbonyl]phenylalanine [5{28}a]:
8.9 mg (39% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 89%, tR = 5.71 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.07 (dd, J = 13.6 Hz, 8.8 Hz, 1H), 3.32−3.46 (m, 1H), 4.16 (m, 1H), 4.22−4.42 (m, 2H), 4.62 (dd, J = 8.4 Hz, 5.4 Hz, 1H), 7.16 (s, 2H), 7.28−7.45 (m, 5H), 7.49−7.61 (m, 2H), 7.76 (d, J = 6.8 Hz, 2H); LC/MS calcd for C24H20Cl2NO4 [M + H]+ 456.1; found 456.2.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-O-methyltyrosine [5{29}a]:
10.3 mg (49% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 94%, tR = 5.08 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.03 (dd, J = 13.9 Hz, 6.7 Hz, 1H), 3.34 (dd, J = 13.9 Hz, 5.5 Hz, 1H), 3.77 (s, 3H), 4.19 (t, J = 6.9 Hz, 1H), 4.25−4.49 (m, 2H), 4.56 (t, J = 5.5 Hz, 1H), 6.82 (d, J = 7.6 Hz, 2H), 7.07 (d, J = 8.0 Hz, 2H), 7.29−7.48 (m, 4H), 7.52−7.64 (m, 2H), 7.77 (d, J = 7.2 Hz, 2H); LC/MS calcd for C25H24NO5 [M + H]+ 418.2; found 418.2.
4-(Phenylmethyl)-N-[(9H-fluoren-9-ylmethoxy)carbonyl]aspartic Acid [5{30}a]:
7.9 mg (35% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 84%, tR = 5.25 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 2.94 (dd, J = 16.7 Hz, 4.5 Hz, 1H), 3.05 (dd, J = 17.2 Hz, 5.2 Hz, 1H), 4.22 (t, J = 7.1 Hz, 1H), 4.29−4.45 (m, 2H), 4.62 (m, 1H), 5.15 (s, 2H), 6.18 (d, J = 8.0 Hz, 1H), 7.26−7.46 (m, 9H), 7.60 (d, J = 7.4 Hz, 2H), 7.76 (d, J = 7.4 Hz, 2H); LC/MS calcd for C26H24NO6 [M + H]+ 446.2; found 446.2.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-3-methoxyphenylalanine [5{31}a]:
5.6 mg (27% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5) initial LC/MS purity 89%, tR = 5.14 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 3.00−3.26 (m, 2H), 3.76 (s, 3H), 4.14−4.46 (m, 3H), 4.57 (m, 1H), 6.73−6.83 (m, 2H), 7.14−7.46 (m, 6H), 7.52−7.62 (m, 2H), 7.76 (d, J = 7.0 Hz, 2H); LC/MS calcd for C25H24NO5 [M + H]+ 418.2; found 418.2.
N-[(9H-Fluoren-9-ylmethoxy)carbonyl]-2-(chloropyridin-5-yl)alanine [5{32}a]:
5.5 mg (26% isolated yield) following chromatographic purification (CH2Cl2/MeOH/H2O, 300:10:0.5); initial LC/MS purity 80%, tR = 2.53 min; 1H NMR (200 MHz, CDCl3/CD3OD) δ 2.75−3.00 (m, 2 H), 3.95 (t, J = 6.4 Hz, 1 H), 4.06−4.38 (m, 3 H), 6.94−7.22 (m, 6 H), 7.32 (d, J = 7.2 Hz, 2 H), 7.52 (d, J = 7.0 Hz, 2 H), 7.90 (s, 1 H); LC/MS calcd for C23H19ClN2O4 [M − H]− 421.1; found 421.0.
Acknowledgments
This article is dedicated to the 97 students and international participants41 who carried out the combinatorial syntheses described in this paper. We wish to thank Jordi Alsina, Donald B. Boyd, Guillermo Morales, Daniel H. Robertson, Richard T. Taylor, and James H. Wikel for helpful discussions and Ralph Mazitschek for assistance in the enumeration. We gratefully acknowledge Frank Jasper for the photograph of the Bill-Board equipment. We acknowledge the National Institutes of Health (R01 GM028193), The National Science Foundation (MRI CHE-0619254), The Camille and Henry Dreyfus Foundation, and the Lilly Research Laboratories for their financial support.
Supporting Information Available
IUPUI-D3 virtual catalog tutorial, analysis of resins from sources I and II and interpretation of results, and a selection of LC/MS and NMR data.This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- See previous Perspective:Scott W. L.; O’Donnell M. J. J. Comb. Chem. 2009, 11, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See following article:Scott W. L.; Audu C. O.; Dage J. L.; Goodwin L. A.; Martynow J. G.; Platt L. K.; Smith J. G.; Strong A. T.; Wickizer K.; Woerly E. M.; O’Donnell M. J. J. Comb. Chem. 2009, 11, 34–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Search strategy: nitrogen-carbon-carbonyl substructure contained in 103 of the 365 structures in Tables 1−10 in ref (4i).
- For an excellent series of structurally-based comprehensive reviews on combinatorial library syntheses, see; a Dolle R. E. Mol. Diversity 1998, 4, 233–256. [DOI] [PubMed] [Google Scholar]; b Dolle R. E.; Nelson K. H. Jr. J. Comb. Chem. 1999, 1, 235–282. [DOI] [PubMed] [Google Scholar]; c Dolle R. E. J. Comb. Chem. 2000, 2, 383–433. [DOI] [PubMed] [Google Scholar]; d Dolle R. E. J. Comb. Chem. 2001, 3, 477–517. [DOI] [PubMed] [Google Scholar]; e Dolle R. E. J. Comb. Chem. 2002, 4, 369–418. [DOI] [PubMed] [Google Scholar]; f Dolle R. E. J. Comb. Chem. 2003, 5, 693–753. [DOI] [PubMed] [Google Scholar]; g Dolle R. E. J. Comb. Chem. 2004, 6, 623–679. [DOI] [PubMed] [Google Scholar]; h Dolle R. E. J. Comb. Chem. 2005, 7, 739–798. [DOI] [PubMed] [Google Scholar]; i Dolle R. E.; Le Bourdonnec B.; Morales G. A.; Moriarty K. J.; Salvino J. M. J. Comb. Chem. 2006, 8, 597–635. [DOI] [PubMed] [Google Scholar]; j Dolle R. E.; Le Bourdonnec B.; Goodman A. J.; Morales G. A.; Salvino J. M.; Zhang W. J. Comb. Chem. 2007, 9, 855–902. [DOI] [PubMed] [Google Scholar]; k Dolle R. E.; Le Bourdonnec B.; Goodman A. J.; Morales G. A.; Thomas C. J.; Zhang W. J. Comb. Chem. 2008, 10, 753–802. [DOI] [PubMed] [Google Scholar]
- Peptides; a Merrifield R. B. J. Am. Chem. Soc. 1963, 85, 2149–2154. [Google Scholar]; b Merrifield B. Science 1986, 232, 341–347. [DOI] [PubMed] [Google Scholar]
- Benzodiazepines; a Bunin B. A.; Ellman J. A. J. Am. Chem. Soc. 1992, 114, 10997–10998. [Google Scholar]; b DeWitt S. H.; Kiely J. S.; Stankovic C. J.; Schroeder M. C.; Reynolds Cody D. M.; Pavia M. R. Proc. Natl. Acad. Sci. U. S. A. 1993, 90, 6909–6913. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kamal A.; Reddy K. L.; Devaiah V.; Shankaraiah N.; Reddy D. R. Mini-Rev. Med. Chem. 2006, 6, 53–69. [DOI] [PubMed] [Google Scholar]
- α-Alkyl-α-amino-γ-lactams; a Scott W. L.; Alsina J.; Kennedy J. H.; O’Donnell M. J. Org. Lett. 2004, 6, 1629–1632. [DOI] [PubMed] [Google Scholar]; b Scott W. L.; Martynow J. G.; Huffman J. C.; O’Donnell M. J. J. Am. Chem. Soc. 2007, 129, 7077–7088. [DOI] [PubMed] [Google Scholar]
- 1,4-Benzodiazepine-2,5-diones; a Mayer J. P.; Zhang J.; Bjergarde K.; Lenz D. M.; Gaudino J. J. Tetrahedron Lett. 1996, 37, 8081–8084. [Google Scholar]; b Boojamra C. G.; Burow K. M.; Thompson L. A.; Ellman J. A. J. Org. Chem. 1997, 62, 1240–1256. [Google Scholar]
- Hydantoins:Meusel M.; Gütschow M. Org. Prep. Proced. Int. 2004, 36, 391–443. [Google Scholar]
- α-Substituted Prolines:Murphy M. M.; Schullek J. R.; Gordon E. M.; Gallop M. A. J. Am. Chem. Soc. 1995, 117, 7029–7030. [Google Scholar]
- Diketopiperazines; a Gordon D. W.; Steele J. Bioorg. Med. Chem. Lett. 1995, 5, 47–50. [Google Scholar]; b Scott B. O.; Siegmund A. C.; Marlowe C. K.; Pei Y.; Spear K. L. Mol. Diversity 1995, 1, 125–134. [DOI] [PubMed] [Google Scholar]; c Dinsmore C. J.; Beshore D. C. Tetrahedron 2002, 58, 3297–3312. [Google Scholar]; d Fischer P. M. J. Peptide Sci. 2003, 9, 9–35. [DOI] [PubMed] [Google Scholar]; e Martins M. B.; Carvalho I. Tetrahedron 2007, 63, 9923–9932. [Google Scholar]
- Selected monographs and reviews related to amino acid synthesis and utilization; a Amino Acids, Peptides and Proteins; Specialist Periodical Reports; The Royal Society of Chemistry: London, 1969−2007; Vols. 1-36. [Google Scholar]; b Barrett G. C.Chemistry and Biochemistry of the Amino Acids; Chapman and Hall: London, 1985. [Google Scholar]; c For the α-Amino Acid Synthesis Symposium-in-Print, see:O’Donnell M. J., Ed. Tetrahedron 1988, 44, 5253−5614. [Google Scholar]; d Williams R. M. In Synthesis of Optically Active a-Amino Acids; Organic Chemistry Series; Baldwin J. E., Ed.; Pergamon Press: Oxford, U.K., 1989. [Google Scholar]; e Duthaler R. O. Tetrahedron 1994, 50, 1539–1650. [Google Scholar]; f Waldmann H. Synlett 1995, 133–141. [Google Scholar]; g Studer A. Synthesis 1996, 793–815. [Google Scholar]; h Seebach D.; Sting A. R.; Hoffmann M. Angew. Chem., Int. Ed. Engl. 1996, 35, 2708–2748. [Google Scholar]; i Obrecht D.; Altorfer M.; Bohdal U.; Daly J.; Huber W.; Labhardt A.; Lehmann C.; Müller K.; Ruffieux R.; Schönholzer P.; Spiegler C.; Zumbrunn C. Biopolymers 1997, 42, 575–626. [Google Scholar]; j Amino Acid Derivatives: A Practical Approach; Barrett G. C., Ed.; Oxford University Press: Oxford, U.K., 1999. [Google Scholar]; k Calmes M.; Daunis J. Amino Acids 1999, 16, 215–250. [DOI] [PubMed] [Google Scholar]; l Bouifraden S.; Drouot C.; El Hadrami M.; Guenon F.; Lecointe L.; Mai N.; Paris M.; Pothion C.; Sadoune M.; Sauvagnat B.; Amblard M.; Aubagnac J. L.; Calmes M.; Chevallet P.; Daunis J.; Enjalbal C.; Fehrentz J. A.; Lamaty F.; Lavergne J. P.; Lazaro R.; Rolland V.; Roumestant M. L.; Viallefont P.; Vidal Y.; Martinez J. Amino Acids 1999, 16, 345–379. [DOI] [PubMed] [Google Scholar]; m Gibson S. E.; Guillo N.; Tozer M. J. Tetrahedron 1999, 55, 585–615. [Google Scholar]; n For the Asymmetric Synthesis of Novel Sterically Constrained Amino Acids Symposium-in-Print. see:Hruby V. J.; Soloshonok V. A., Eds. Tetrahedron 2001, 57, 6329−6650. [Google Scholar]; o Park K.-H.; Kurth M. J. Tetrahedron 2002, 58, 8629–8659. [Google Scholar]; p Ma J.-A. Angew. Chem., Int. Ed. 2003, 42, 4290–4299. [DOI] [PubMed] [Google Scholar]; q Sagan S.; Karoyan P.; Lequin O.; Chassaing G.; Lavielle S. Curr. Med. Chem. 2004, 11, 2799–2822. [DOI] [PubMed] [Google Scholar]; r Vogt H.; Bräse S. Org. Biomol. Chem. 2007, 5, 406–430. [DOI] [PubMed] [Google Scholar]; s Cativiela C.; Díaz-de-Villegas M. D. Tetrahedron: Asymmetry 2007, 18, 569–623. [Google Scholar]; t Nájera C.; Sansano J. M. Chem. Rev. 2007, 107, 4584–4671. [DOI] [PubMed] [Google Scholar]
- For a recent general discussion, with extensive reviews and primary references, concerning aliphatic nucleophilic substitution [mechanisms, reactivity, and reactions (classified by attacking atom of the nucleophile: O, S, N, halogen, H, C)], see:Smith M. B.; March J.. March’s Advanced Organic Chemistry: Reactions, Mechanisms, and Structure; Wiley-Interscience: Hoboken, NJ, 2007; Chapter 10, pp 425−656. [Google Scholar]
- Reviews from the authors’ laboratory on the use of Schiff base derivatives for the synthesis of amino acids, peptides and peptidomimetics; a O’Donnell M. J.Asymmetric Phase-Transfer Reactions. In Catalytic Asymmetric Synthesis; Ojima I., Ed.; VCH: New York, 1993; Chapter 8, pp 389−411. [Google Scholar]; b O’Donnell M. J.; Esikova I. A.; Mi A.; Shullenberger D. F.; Wu S.. Amino Acid and Peptide Synthesis Using Phase Transfer Catalysis. In Phase Transfer Catalysis; ACS Symposium Series; Halpern M. E., Ed.; American Chemical Society: Washington, D.C., 1997; Chapter 10, pp 124−135. [Google Scholar]; c O’Donnell M. J.Asymmetric Phase-Transfer Reactions. In Catalytic Asymmetric Synthesis, 2nd ed.; Ojima I., Ed.; Wiley-VCH: New York, 2000; Chapter 10, pp 727−755. [Google Scholar]; d O’Donnell M. J. Aldrichim. Acta 2001, 34, 3–15. [Google Scholar]; e O’Donnell M. J. Acc. Chem. Res. 2004, 37, 506–517. [DOI] [PubMed] [Google Scholar]
- Reviews by others on the use of Schiff base derivatives for the synthesis of amino acids, peptides and peptidomimetics; a Lygo B.Phase-Transfer Reactions. In Rodd’s Chemistry of Carbon Compounds, 2nd ed.; Elsevier: Oxford, U.K., 2001; Vol. 5, pp 101−149. [Google Scholar]; b Maruoka K.; Ooi T. Chem. Rev. 2003, 103, 3013–3028. [DOI] [PubMed] [Google Scholar]; c Lygo B.; Andrews B. I. Acc. Chem. Res. 2004, 37, 518–525. [DOI] [PubMed] [Google Scholar]; d Vachon J.; Lacour J. Chimia 2006, 60, 266–275. [Google Scholar]; e Vicario J. L.; Badía D.; Carrillo L. Synthesis 2007, 2065–2092. [Google Scholar]; f Ooi T.; Maruoka K. Angew. Chem., Int. Ed. 2007, 46, 4222–4266. [DOI] [PubMed] [Google Scholar]; g Hashimoto T.; Maruoka K. Chem. Rev. 2007, 107, 5656–5682. [DOI] [PubMed] [Google Scholar]
- References from the authors’ laboratory concerning various types of alkylations in solution involving Schiff base ester or amide derivatives of α-amino acids, see; a Monoalkylation by ion-pair extraction:O’Donnell M. J.; Boniece J. M.; Earp S. E. Tetrahedron Lett. 1978, 2641–2644. [Google Scholar]; b Monoalkylation by phase-transfer catalysis:O’Donnell M. J.; Eckrich T. M. Tetrahedron Lett. 1978, 4625–4628. [Google Scholar]; c Monoalkylation of an aldimine ester:Ghosez L.; Antoine J.-P.; Deffense E.; Navarro M.; Libert V.; O’Donnell M. J.; Bruder W. A.; Willey K.; Wojciechowski K. Tetrahedron Lett. 1982, 23, 4255–4258. [Google Scholar]; d Dialkylation:O’Donnell M. J.; LeClef B.; Rusterholz D. B.; Ghosez L.; Antoine J.-P.; Navarro M. Tetrahedron Lett. 1982, 23, 4259–4262. [Google Scholar]; e PTC alkylation with K2CO2:O’Donnell M. J.; Bruder W.; Wojciechowski K.; Ghosez L.; Navarro M.; Sainte F.; Antoine J.-P.. The Synthesis of Amino Acids Using Phase-Transfer Catalyzed Alkylations with Potassium Carbonate. In Peptides: Structure and Function; Proceedings of the 8th American Peptide Symposium; Hruby V. J., Rich D. H., Eds.; Pierce Chemical Co.: Rockford, IL, 1983; pp 151−154. [Google Scholar]; f Alkylation with α,ω-dihaloalkanes:O’Donnell M. J.; Bruder W. A.; Eckrich T. M.; Shullenberger D. F.; Staten G. S. Synthesis 1984, 127–128. [Google Scholar]; g PTC alkylation with K2CO2:O’Donnell M. J.; Wojciechowski K.; Ghosez L.; Navarro M.; Sainte F.; Antoine J.-P. Synthesis 1984, 313–315. [Google Scholar]; h O’Donnell M. J.; Bruder W. A.; Daugherty B. W.; Liu D.; Wojciechowski K. Tetrahedron Lett. 1984, 25, 3651–3654. [Google Scholar]; i Organoborane alkylation:O’Donnell M. J.; Falmagne J.-B. Chem. Commun. 1985, 1168–1169. [Google Scholar]; j Alkylation with α-bromo-α-fluorotoluene:O’Donnell M. J.; Barney C. L.; McCarthy J. R. Tetrahedron Lett. 1985, 26, 3067–3070. [Google Scholar]; k pKa of Schiff bases and rate studies of mono- and dialkylation:O’Donnell M. J.; Bennett W. D.; Bruder W. A.; Jacobsen W. N.; Knuth K.; LeClef B.; Polt R. L.; Bordwell F. G.; Mrozack S. R.; Cripe T. R. J. Am. Chem. Soc. 1988, 110, 8520–8525. [Google Scholar]; l Synthesis of α-methylhistidine by alkylation with a benzylic acetate:O’Donnell M. J.; Rusterholz D. B. Synth. Commun. 1989, 19, 1157–1165. [Google Scholar]; m Enantioselective PTC monoalkylation:O’Donnell M. J.; Bennett W. D.; Wu S. J. Am. Chem. Soc. 1989, 111, 2353–2355. [Google Scholar]; n Oxygen alkylation:O’Donnell M. J.; Cook G. K.; Rusterholz D. B. Synthesis 1991, 989–993. [Google Scholar]; o Enantioselective PTC dialkylation:O’Donnell M. J.; Wu S. Tetrahedron: Asymmetry 1992, 3, 591–594. [Google Scholar]; p Solution-phase peptide alkylation:O’Donnell M. J.; Burkholder T. P.; Khau V. V.; Roeske R. W.; Tian Z. Pol. J. Chem. 1994, 68, 2477–2488. [Google Scholar]; q New enantioselective PTC catalysts:O’Donnell M. J.; Wu S.; Huffman J. C. Tetrahedron 1994, 50, 4507–4518. [Google Scholar]; r Homogeneous catalytic enantioselective alkylations:O’Donnell M. J.; Delgado F.; Hostettler C.; Schwesinger R. Tetrahedron Lett. 1998, 39, 8775–8778. [Google Scholar]; s Enantiomeric enrichment by recrystallization of optically enriched alkylation products:O’Donnell M. J.; Delgado F. Tetrahedron 2001, 57, 6641–6650. [Google Scholar]; t Enantioselective boron alkylation:O’Donnell M. J.; Drew M. D.; Cooper J. T.; Delgado F.; Zhou C. J. Am. Chem. Soc. 2002, 124, 9348–9349. [DOI] [PubMed] [Google Scholar]; u Diastereoselective boron alkylation:O’Donnell M. J.; Cooper J. T.; Mader M. J. Am. Chem. Soc. 2003, 125, 2370–2371. [DOI] [PubMed] [Google Scholar]; v Catalytic enantioselective SN2′-like alkylations with allylic acetates:Ramachandran P. V.; Madhi S.; Bland-Berry L.; Reddy M. V. R.; O’Donnell M. J. J. Am. Chem. Soc. 2005, 127, 13450–13451. [DOI] [PubMed] [Google Scholar]; w Glycinamide and alaninamide alkylations:O’Donnell M. J.; Keeton J. D.; Khau V. V.; Bollinger J. C. Can. J. Chem. 2006, 84, 1301–1312. [Google Scholar]; x Fluorinated Glu derivatives by SN2′-like alkylation with allylic acetates:Ramachandran P. V.; Madhi S.; O’Donnell M. J. J. Fluorine Chem. 2007, 128, 78–83. [Google Scholar]
- References from the authors’ laboratory concerning the solid-phase synthesis of unnatural amino acids, peptides, and peptidomimetics; a Monoalkylation:O’Donnell M. J.; Zhou C.; Scott W. L. J. Am. Chem. Soc. 1996, 118, 6070–6071. [Google Scholar]; b Dialkylation:Scott W. L.; Zhou C.; Fang Z.; O’Donnell M. J. Tetrahedron Lett. 1997, 38, 3695–3698. [Google Scholar]; c Unactivated alkyl halides:O’Donnell M. J.; Lugar C. W.; Pottorf R. S.; Zhou C.; Scott W. L.; Cwi C. L. Tetrahedron Lett. 1997, 38, 7163–7166. [Google Scholar]; d Sequential mono- and dialkylation:Griffith D. L.; O’Donnell M. J.; Pottorf R. S.; Scott W. L.; Porco J. A. Jr. Tetrahedron Lett. 1997, 38, 8821–8824. [Google Scholar]; e Michael additions:Domínguez E.; O’Donnell M. J.; Scott W. L. Tetrahedron Lett. 1998, 39, 2167–2170. [Google Scholar]; f Glycine cation equivalentO’Donnell M. J.; Delgado F.; Drew M. D.; Pottorf R. S.; Zhou C.; Scott W. L. Tetrahedron Lett. 1999, 40, 5831–5835. [Google Scholar]; g Enantioselective alkylations:O’Donnell M. J.; Delgado F.; Pottorf R. S. Tetrahedron 1999, 55, 6347–6362. [Google Scholar]; h Preparation of amino or peptide aldehyde and amino or peptide ketone derivatives:O’Donnell M. J.; Drew M. D.; Pottorf R. S.; Scott W. L. J. Comb. Chem. 2000, 2, 172–181. [DOI] [PubMed] [Google Scholar]; i Review of unnatural amino acid and peptide synthesis:O’Donnell M. J.; Scott W. L.. Unnatural Amino Acid and Peptide Synthesis (UPS) In Peptides 2000. Martinez J., Fehrentz J.-A., Eds.; EDK: Paris, 2001; pp 31−36. [Google Scholar]; j Enantioselective Michael additions:O’Donnell M. J.; Delgado F.; Domínguez E.; de Blas J.; Scott W. L. Tetrahedron: Asymmetry 2001, 12, 821–828. [Google Scholar]; k Preparation of amino amides and peptide amides:Scott W. L.; Delgado F.; Lobb K.; Pottorf R. S.; O’Donnell M. J. Tetrahedron Lett. 2001, 42, 2073–2076. [Google Scholar]; l α,ω-Dihalides for the preparation of α,α-disubstituted side chain derivatives:Scott W. L.; O’Donnell M. J.; Delgado F.; Alsina J. J. Org. Chem. 2002, 67, 2960–2969. [DOI] [PubMed] [Google Scholar]; m Preparation of α-substituted prolines and homologues:Scott W. L.; Alsina J.; O’Donnell M. J. J. Comb. Chem. 2003, 5, 684–692. [Google Scholar]; n α,ω-Dihalides for the preparation of α-substituted side-chain derivatives:O’Donnell M. J.; Alsina J.; Scott W. L. Tetrahedron Lett. 2003, 44, 8403–8406. [Google Scholar]; o Preparation of terminal and internal lactam peptidomimetics: ref (7a).; p Preparation of α-substituted proline hydantoins and analogs:Alsina J.; Scott W. L.; O’Donnell M. J. Tetrahedron Lett. 2005, 46, 3131–3135. [Google Scholar]; q Resin-bound aldehyde intermediates for the synthesis of multiple classes of highly-substituted lactam peptidomimetics: ref (7b).
- Selected references from other laboratories concerning alkylations or Michael additions of Schiff base ester derivatives of α-amino acids:; a Chenault H. K.; Dahmer J.; Whitesides G. M. J. Am. Chem. Soc. 1989, 111, 6354–6364. [Google Scholar]; b Tilley J. W.; Danho W.; Lovey K.; Wagner R.; Swistok J.; Makofske R.; Michalewsky J.; Triscari J.; Nelson D.; Weatherford S. J. Med. Chem. 1991, 34, 1125–1136. [DOI] [PubMed] [Google Scholar]; c Josien H.; Martin A.; Chassaing G. Tetrahedron Lett. 1991, 32, 6547–6550. [Google Scholar]; d Chauvel E. N.; Coric P.; Llorens-Cortès C.; Wilk S.; Roques B. P.; Fournié-Zaluski M.-C. J. Med. Chem. 1994, 37, 1339–1346. [DOI] [PubMed] [Google Scholar]; e Huang X.; Long E. C. Biorg. Med. Chem. Lett. 1995, 5, 1937–1940. [Google Scholar]; f Sagan S.; Josien H.; Karoyan P.; Brunissen A.; Chassaing G.; Lavielle S. Bioorg. Med. Chem. 1996, 4, 2167–2178. [DOI] [PubMed] [Google Scholar]; g Rao A. V. R.; Reddy K. L.; Rao A. S.; Vittal T. V. S. K.; Reddy M. M.; Pathi P. L. Tetrahedron Lett. 1996, 37, 3023–3026. [Google Scholar]; h Satoh Y.; Gude C.; Chan K.; Firooznia F. Tetrahedron Lett. 1997, 38, 7645–7648. [Google Scholar]; i Corey E. J.; Xu F.; Noe M. C. J. Am. Chem. Soc. 1997, 119, 12414–12415. [Google Scholar]; j Guillena G.; Nájera C. Tetrahedron: Asymmetry 1998, 9, 3935–3938. [Google Scholar]; k Corey E. J.; Noe M. C.; Xu F. Tetrahedron Lett. 1998, 39, 5347–5350. [Google Scholar]; l Ooi T.; Takeuchi M.; Kameda M.; Maruoka K. J. Am. Chem. Soc. 2000, 122, 5228–5229. [Google Scholar]; m Jew S.-s.; Jeong B.-S.; Yoo M.-S.; Huh H.; Park H.-g. Chem. Commun. 2001, 1244–1245. [Google Scholar]; n Jew S.-s.; Yoo M.-S.; Jeong B.-S.; Park I. Y.; Park H.-g. Org. Lett. 2002, 4, 4245–4248. [DOI] [PubMed] [Google Scholar]; o Lygo B.; Andrews B. I.; Crosby J.; Peterson J. A. Tetrahedron Lett. 2002, 43, 8015–8018. [Google Scholar]; p Shibuguchi T.; Fukuta Y.; Akachi Y.; Sekine A.; Ohshima T.; Shibasaki M. Tetrahedron Lett. 2002, 43, 9539–9543. [Google Scholar]; q Viswanathan R.; Prabhakaran E. N.; Plotkin M. A.; Johnston J. N. J. Am. Chem. Soc. 2003, 125, 163–168. [DOI] [PubMed] [Google Scholar]; r Ooi T.; Kameda M.; Maruoka K. J. Am. Chem. Soc. 2003, 125, 5139–5151. [DOI] [PubMed] [Google Scholar]; s Lygo B.; Allbutt B.; James S. R. Tetrahedron Lett. 2003, 44, 5629–5632. [Google Scholar]; t Lee J.; Kim S. Y.; Park S.; Lim J.-O.; Kim J.-M.; Kang M.; Lee J.; Kang S.-U.; Choi H.-K.; Jin M.-K.; Welter J. D.; Szabo T.; Tran R.; Pearce L. V.; Toth A.; Blumberg P. M. Biorg. Med. Chem. 2004, 12, 1055–1069. [DOI] [PubMed] [Google Scholar]; u Danner P.; Bauer M.; Phukan P.; Maier M. E. Eur. J. Org. Chem. 2005, 317–325. [Google Scholar]; v Grover G. N.; Kowtoniuk W. E.; MacFarland D. K. Tetrahedron Lett. 2006, 47, 57–60. [Google Scholar]; w Ooi T.; Uematsu Y.; Kameda M.; Maruoka K. Tetrahedron 2006, 62, 11425–11436. [Google Scholar]; x Wang X.; Yin L.; Yang T.; Wang Y. Tetrahedron: Asymmetry 2007, 18, 108–114. [Google Scholar]
- Yan B.; Fang L.; Irving M.; Zhang S.; Boldi A. M.; Woolard F.; Johnson C. R.; Kshirsagar T.; Figliozzi G. M.; Krueger C. A.; Collins N. J. Comb. Chem. 2003, 5, 547–559. [DOI] [PubMed] [Google Scholar]
- The odd student, designated “team 9”, prepared a replicate of team 8’s assignment. For symmetry and ease of data presentation, that student’s data is not included in this report.
- a Atherton E.; Gait M. J.; Sheppard R. C.; Williams B. J. Bioorg. Chem. 1979, 8, 351–370. [Google Scholar]; b Grandas A.; Jorba X.; Giralt E.; Pedroso E. Int. J. Pept. Protein Res. 1989, 33, 386–390. [Google Scholar]
- a Stanger K. J.; Krchňák V. J. Comb. Chem. 2006, 8, 652–654. [DOI] [PubMed] [Google Scholar]; b Colombo A.; De la Figuera N.; Fernàndez J. C.; Fernández-Forner D.; Albericio F.; Forns P. Org. Lett. 2007, 9, 4319–4322. [DOI] [PubMed] [Google Scholar]
- Interestingly the LC/MS data for each of the aberrant team 28 row B samples showed retention time and mass data consistent with product 5{6}b, the 2-naphthoyl amide derivative of phenylalanine. This should have only been produced in Bill-Board position B1. It is likely that in this case team 28 either inadvertently took their B2 and B3 LC/MS samples from B1 or, in the alkylation step, they used, by mistake, benzyl bromide in the B2 and B3 positions, which should have received the two new alkylating agents. The problematic nature of the work performed by team 28 was also indicated by the analysis of the control reaction to form 5{6}a done in position A1 (see Supporting Information for results). Their purity was only 16% compared to the consistent purity of 65−80% for all other teams using resin I. In fact, this single low result significantly depressed the average purity for team 26−33 samples of 5{6}a (Table 1).
- The replicated rehearsal of 28 by team 3 gave conflicting results. One acylated derivative, 5{28}b, was the only product detected by LC/MS, while paradoxically, there was no desired product for the other acylated derivative, 5{28}a. In other reactions (e.g. to control 5{6}a and production of 5{29}a), team 3 also had inconsistent results.
- a Barany G.; Merrifield R. B. In The Peptides; Gross E., Meienhofer J., Eds.; Academic Press: New York, 1979; Vol. 2, pp 211−223. [Google Scholar]; b Pearson D. A.; Blanchette M.; Baker M. L.; Guindon C. A. Tetrahedron Lett. 1989, 30, 2739–2742. [Google Scholar]; c Mehta A.; Jaouhari R.; Benson T. J.; Douglas K. T. Tetrahedron Lett. 1992, 33, 5441–5444. [Google Scholar]
- CombiChem/Excel from CambridgeSoft, 100 CambridgePark Drive Cambridge, MA 02140 (http://www.cambridgesoft.com/ (accessed November 2, 2008)).
- ChemAxon (free academic license available) from ChemAxon Kft., Máramaros köz 3/a, Budapest, 1037 Hungary. Marvin was used for drawing, displaying, and characterizing chemical structures, substructures and reactions. Marvin 5.1.02, ChemAxon http://www.chemaxon.com (accessed November 2, 2008).
- a For access to the IUPUI−Distributed Drug Discovery (D3) database, register for a free read-download account with Collaborative Drug Discovery (CDD) at https://www.collaborativedrug.com/register/iupui-d3 (accessed November 2, 2008) by completing the “Sign up for IUPUI−Distributed Drug Discovery (D3)” information; b A tutorial for use of the IUPUI−Distributed Drug Discovery (D3) database on CDD is provided: (i) in the Supporting Information for this article and the following article,2 (ii) from the D3 database on the CDD website, and (iii) on the IUPUI Department of Chemistry and Chemical Biology website [http://chem.iupui.edu/ (accessed November 2, 2008)] under the faculty & staff directory for O’Donnell or Scott. The tutorial available at ii and iii will be updated periodically
- The 100 electrophiles in Table 5 consist of 84 alkyl halides and 16 Michael acceptors. These electrophiles represent selected examples from the authors’ laboratory (69 electrophiles Table 5, refs (2,14,16), and (17)) and from other groups (31 electrophiles Table 5, refs (15) and (18)).
- To allow synthetic and practical flexibility, sometimes more than one alkylating agent leading to the same product is shown. For example, 2-iodopropane (41, X = I) and 2-bromopropane (41, X = Br) each produce valine. Selected references will help the researcher decide, on a case by case basis, which reagent to employ to obtain the desired product.
- Standard amide coupling conditions: 5 equiv each of the carboxylic acid, dicyclohexylcarbodiimide, and 1-hydroxybenzotriazole in NMP, rt, 48 h.
- Bio-assay guided fractionation; a Phillipson D. W.; Milgram K. E.; Yanovsky A. I.; Rusnak L. S.; Haggerty D. A.; Farrell W. P.; Greig M. J.; Xiong X.; Proefke M. L. J. Comb. Chem. 2002, 4, 591–599. [DOI] [PubMed] [Google Scholar]; b Peake D. A.; Duckworth D. C.; Perun T. J.; Scott W. L.; Kulanthaivel P.; Strege M. A. Comb. Chem. High Throughput Screening 2005, 8, 477–487. [DOI] [PubMed] [Google Scholar]; c Inglese J.; Auld D. S.; Jadhav A.; Johnson R. L.; Simeonov A.; Yasgar A.; Zheng W.; Austin C. P. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 11473–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiral chromatography of racemic or optically enriched samples; a Maier N. M.; Franco P.; Lindner W. J. Chrom. A 2001, 906, 3–33. [DOI] [PubMed] [Google Scholar]; b Kennedy J. H.; Belvo M. D.; Sharp V. S.; Williams J. D. J. Chrom. A 2004, 1046, 55–60. [DOI] [PubMed] [Google Scholar]
- Recrystallization of optically enriched samples; a Ref (16m).; b O’Donnell M. J.; Chen N.; Zhou C.; Murray A.; Kubiak C. P.; Yang F.; Stanley G. G. J. Org. Chem. 1997, 62, 3962–3975. [Google Scholar]; c Ref (16s).
- Resolution; a Fogassy E.; Nógrádi M.; Kozma D.; Egri G.; Pálovics E.; Kiss V. Org. Biomol. Chem. 2006, 4, 3011–3030. [DOI] [PubMed] [Google Scholar]; b Novel Optical Resolution Technologies;Sakai K., Hirayama N., Tamura R., Eds.; Topics in Current Chemistry 269; Springer-Verlag: Berlin, 2007. [Google Scholar]; c Pellissier H. Tetrahedron 2008, 64, 1563–1601. [Google Scholar]
- t-Butyl ester cleavage with TFA:Wuts P. G. M.; Greene T. W.. Greene’s Protective Groups in Organic Synthesis, 4th ed.; Wiley-Interscience: Hoboken, NJ, 2007; pp 582−588. [Google Scholar]
- t-Butyl carbamate (N-Boc) cleavage with TFA:Wuts P. G. M.; Greene T. W.. Greene’s Protective Groups in Organic Synthesis, 4th ed.; Wiley-Interscience; Hoboken, NJ: 2007; pp 725−735. [Google Scholar]
- Compound numbers of reactants expected, when incorporated into products 5, to undergo side reaction upon TFA cleavage: 36, 75, 83, and 96.
- We welcome communication from the scientific community regarding potential or actual side reactions. These will be incorporated into the database as they become available.
- Available from Leads Metal Products, PO Box 441186, Indianapolis, IN 46244−1186 (larry@leadsmetal.com
- Dedicated to the following participants who carried out the combinatorial syntheses described in this paper; a IUPUI students from Chemistry C344 (Organic Chemistry II Laboratory): Abdul Moid M.; Abid F.; Al-Mahrouq E. H.; Amerman T. W.; Andoh G. P.; Applewhite A. O.; Audu C. O.; Baird J. P.; Baker R. M.; Balvich J. C.; Behbahani K.; Bordley J.; Bryan J. D.; Burrus M. A.; Chase S. D.; Fairfield N. D.; Fallowfield G. R.; Fisher K.; Fouad Y. M.; Francis M. M.; Gebrewold S. H.; Hall L. M.; Hamilton J. A.; Haqqani A. A.; Heitman C. J.; Hiatt S. A.; Hutson C. M.; Ingold M. L.; Jeffries P. R.; Johnson H. M.; Johnson D. E.; Kaup C. P.; Keith L. E.; Kheradia P. M.; Kim D. S.; Kominiak J. A.; Kreger A. M.; Lieland J. J.; May M. C.; McAfee B. E.; McCreary G. R.; Mullen P. K.; Musselman H. N.; Nawrocki R. M.; Ndiangang P. L.; Nguyen H. V.; Omery B. M.; Ozdemir H.; Pierce M. R.; Platt L. K.; Pollack N.; Rau N.; Reed M. J.; Salehi S.; Schneider A. L.; Sevier N. G.; Siegel A. P.; Simpson A. T.; Smith A. L.; Smith E. E.; Swanson C. K.; Terew P. D.; Turner J. A.; Wagner J. L.; Weisel T. J.; White C. D.; Whitehair A. N.; Woerly E. M.; Woubeshet K.. ; b Lublin, Poland participants: Aletanska-Kozak M.; Golembiowska A.; Hus M.; Jozwiak M.; Gosniak A.; Kaczor A.; Kijkowska-Murak U.; Panecka E.; Rzadkowska M.; Stefanczyk J.; Szacon E.; Sztanke K.. ; c Moscow, Russia participants: Belyaev S.; Belykh E.; Dlinnykh I.; Gormay P.; Kuznetsov A.; Nevskaya A.; Sadovoy A.; Shvetcov G.. ; d Barcelona, Spain participants: Colombo A.; Garcia M.; Gómez L.; Hosta L.; Mir M.; Morales B.; Vera P..
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.