

Abstract
For the successful implementation of Distributed Drug Discovery (D3) (outlined in the accompanying Perspective), students, in the course of their educational laboratories, must be able to reproducibly make new, high quality, molecules with potential for biological activity. This article reports the successful achievement of this goal. Using previously rehearsed alkylating agents, students in a second semester organic chemistry laboratory performed a solid-phase combinatorial chemistry experiment in which they made 38 new analogs of the most potent member of a class of antimelanoma compounds. All compounds were made in duplicate, purified by silica gel chromatography, and characterized by NMR and LC/MS. As a continuing part of the Distributed Drug Discovery program, a virtual D3 catalog based on this work was then enumerated and is made freely available to the global scientific community.
Introduction
Three key components of the chemistry phase of Distributed Drug Discovery (D3)(1) are reagent rehearsal,(2) virtual library enumeration, and targeted library synthesis. In the rehearsal laboratories, diversity reagents are evaluated in duplicate for potential use at each step of a distributed drug discovery combinatorial synthesis. Virtual D3 catalogs are then generated using only those diversity reagents that perform satisfactorily or whose potential is well-precedented from the literature. Computational analysis of these catalogs leads to a “targeted” library subset, which students then synthesize in a distributed fashion.
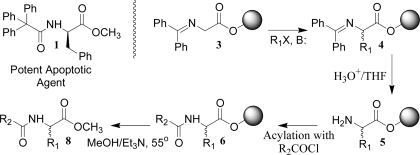
Our first virtual D3 catalog was reported in the previous paper.(2) It is now freely available to the global community for analysis by computational models appropriate for neglected diseases. While waiting for molecular targets to emerge from this process, a disease and class of target molecules was identified that would allow testing the capability of D3 methodologies to make new, high-quality, potentially biologically active molecules. In 2005 researchers at the University of Illinois reported the discovery, from a combinatorial library, of molecules that induced apoptosis in a melanoma cell line.3−5 The most potent molecule (IC50 = 0.5 μM) in their collection was 1 (from the R-isomer of phenylalanine). A close analog, 2, also showed significant activity (Figure 1).
Figure 1.

Compounds inducing apoptosis in a melanoma cell line.
We had already demonstrated a distributed drug discovery synthesis procedure that can provide, from alkylating agents R1X and acylating agents R2COCl, acylated unnatural amino acids of general structure 7 (original route, Scheme 1).2,6 The four-step sequence involves the alkylation of resin-bound glycinate (3 → 4), followed by imine hydrolysis (4 → 5), N-acylation (5 → 6), and final cleavage from the resin (6 → 7). It was apparent that modification of this procedure (modified route, Scheme 1) could produce numerous phenylalanine methyl ester analogs, 8, of 1 and 2.
Scheme 1. General Route to Analogs 8 of Anti-Melanoma Compounds.

In Part I, we report using this adaptation to perform the first successful demonstration of targeted D3 synthesis. In Part II, we enumerate, for open-access analysis, an accompanying virtual D3 catalog of acylated unnatural amino acid methyl esters.
Results and Discussion
Part I. Development and Implementation of a D3 Synthetic Process Providing Analogs of Anti-Melanoma Compounds
The synthesis of 6 was carried out as previously reported.(2) Cleavage of 6 to 8 by transesterification with methanol in triethylamine at 55 °C for 48 h proceeded smoothly and with good conversion. In the student laboratory, this modified route to compounds 8 proceeded uneventfully. In addition to the two known compounds 8a and 8b, 38 new compounds were prepared in duplicate. A single student purified each compound to afford characterized products in high purity. The results for the alkylating agents used and the products formed are shown in Table 1.
Table 1. Alkylating Agents Used and Products Synthesizeda.
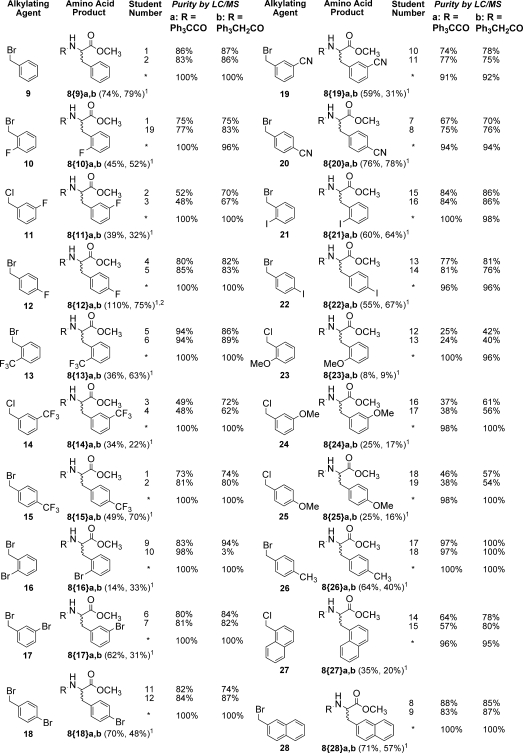 |
Crude LC/MS purities. Asterisk (*) represents the purified product. Superscript one indicates isolated, overall purified yields for four step synthesis to a and b, respectively, and superscript two indicates a result that is greater than the theoretical result based on presumed resin loading (see ref (7)).
As predicted from the database of alkylating agents reported in the previous paper in this Journal,(2) the desired molecules were obtained in all cases, even with the problematic methoxy-substituted benzene derivatives 8{23}a,b, 8{24}a,b, and 8{25}a,b. Each product from this laboratory was subsequently purified by manual silica gel chromatography, six compounds at a time, using a Bill-Board1,8 setup modified for purification rather than synthesis. The final products were, on average, >95% pure by LC/MS. Sufficient material was obtained to provide product for testing, with an average isolated chemical yield of 48% for the four-step, 50 μmol reaction sequence (see Experimental Section). The purified samples were fully characterized by NMR. Figure 2 shows crude and purified product for a representative case where the crude purity was close to the average for all the samples.
Figure 2.

Representative analytical results for the crude and purified analog 8{15}a.
Part II. Virtual D3 Catalog Enumeration of Acylated Unnatural Amino Acid Methyl Esters for Open-Access Availability
An essential component of Distributed Drug Discovery is the open-access availability of databases of molecules accessible through distributed chemistry.(1) Computational chemists can analyze these large databases to identify and make globally available smaller databases of potential drug leads targeted for developing world diseases. Subsequently, the synthesis of the selected molecules will be carried out, in a distributed fashion, at sites across the world. As new D3 compatible synthetic procedures are developed, accompanying virtual D3 catalogs will be enumerated for open-access retrieval and searching. Since the present work demonstrates the D3 route to acylated unnatural amino acid methyl esters, a new virtual D3 catalog based on this chemistry was created. The compounds (including stereoisomers when appropriate) in this virtual D3 catalog were enumerated with commercial software.(9) The complete virtual D3 catalog is freely available online through the Collaborative Drug Discovery interface.(10) Utilizing the same electrophiles and carboxylic acids (and rationale for selection) as in our previous synthesis of acylated unnatural amino acids,(2) 24 192 acylated unnatural amino acid methyl esters were enumerated.
As in the previous enumeration, this 24 192-member virtual catalog contains more than the 10 000 members that would arise from a simple 100 × 100 combinatorial process. This is because the alkylation step provides racemic products, and the acylation step can lead to additional stereoisomers, each of which is listed separately. This allows appropriate computational discrimination and selection when three-dimensional molecular modeling software is employed and chirality is important. See the earlier article for a more complete discussion of these stereochemical issues.(2) The compound structures in the virtual catalog were further adjusted to take into consideration possible transformations in the final methanolysis step11−16 (e.g., transesterification of ethyl esters to methyl esters).
Summary and Conclusion
This laboratory exercise demonstrates the ability to carry out a critical component of Distributed Drug Discovery: the targeted, replicated synthesis, by students in their normal undergraduate laboratory, of new molecules selected from a large virtual catalog of molecules accessible from student-rehearsed diversity reagents. Because these reagents had been previously rehearsed globally in Indianapolis, Barcelona, Lublin and Moscow, D3 correctly predicted every combinatorial synthesis would work. All the desired analogs were successfully made and purified. A 24 192-member virtual D3 catalog based on this chemistry was enumerated and is available through open-access.
Experimental Section
General Methods
All resins were purchased from Polymer Laboratories, and all reactions and washes were conducted at ambient temperature unless otherwise noted. Organic solvents were of reagent grade and were used directly without purification. Anhydrous N-methylpyrrolidinone (NMP), chloroform-d1, 2-fluorobenzyl bromide, 4-fluorobenzyl bromide, 2-(trifluoromethyl)benzyl bromide, 4-(trifluoromethyl)benzyl bromide, 2-bromobenzyl bromide, 2-methoxybenzyl chloride, and 4-methylbenzyl bromide were purchased from Acros Organics. Trifluoroacetic acid (TFA) and hydrochloric acid were obtained from Fisher Scientific. BTPP (tert-butyl-imino-tri(pyrrolidino)phosphorane)(17) and Fmoc chloride were purchased from Fluka. N,N-Diisopropylethylamine (DIEA), benzyl bromide, 3-fluorobenzyl chloride, 3-(trifluoromethyl)benzyl chloride, 3-bromobenzyl bromide, 4-bromobenzyl bromide, 3-(bromomethyl)benzonitrile, 4-(bromomethyl)benzonitrile, 2-iodobenzyl bromide, 4-iodobenzyl bromide, 3-methoxybenzyl chloride, 4-methoxybenzyl chloride, 1-(chloromethyl)naphthalene, 2-(bromomethyl)naphthalene, and methanol-d4 were obtained from Aldrich Chemical Co.
Manual solid-phase organic syntheses at ambient temperature were carried out in 3.5 mL fritted glass reaction vessels equipped with polypropylene screw caps with Teflon faced silicon septa on the Bill-Board set, which was designed by one of us (WLS) as inexpensive equipment(8) to simplify and expedite multiple, manual solid-phase syntheses. This set is available from Leads Metal Products Inc. (larry@leadsmetal.com). The Bill-Board reaction vessel components are available from ChemGlass: IUP-0305-270H for 3.5 mL reaction vessel; IUP-0305-280H (polypropylene screw cap); IUP-0305-281H (Teflon faced silicone septa).
Analytical thin layer chromatography (TLC) was performed with Merck silica gel 60 F254, 0.25 mm precoated glass plates. TLC plates were visualized using UV254. Flash column chromatography was performed on silica gel 60 (230−400 mesh) from Silicycle Chemical Division. The yields of the final compounds, after chromatographic purification, were calculated on the basis of the initial loading of the starting resins and are the overall yields of all reaction steps starting from these resins.
1H NMR spectra were recorded at 200 MHz (Varian spectrometer) in chloroform-d1 and methanol-d4 (3−10%), with TMS as standard. Electrospray ionization mass spectrometry was conducted using a PESciex API III triple-stage quadrupole mass spectrometer operated in either positive-ion or negative-ion detection mode.
The composition of crude reaction mixtures was determined based on the integration of NMR spectra, as well as LC-MS results. LC-MS analyses were conducted using an Agilent system, consisting of a 1100 series HPLC connected to a diode array detector and a 1946D mass spectrometer configured for positive-ion/negative-ion electrospray ionization. The LC-MS samples were analyzed as solutions in CH3CN, prepared at 0.08−0.12 mg/mL concentration. The composition of mixtures was determined by LC-MS based on UV integration at 210 nm.
General Procedures for Manual Solid-Phase Organic Synthesis
(a) Alkylation (3 to 4)
Resin 3 was distributed by volume as aliquots from an isopycnic suspension. The Bill-Boards (19, one for each student in the laboratory, a total of 114 reactions) were placed in their drain trays and, from a neutral buoyancy suspension in dichloromethane (DCM)/NMP, 50 μmols of the benzophenone imine of glycine Wang resin (3, resin loading = 0.7 mmol/g) was distributed, via repeated 1 mL aliquots, to each of the 6 reaction vessels in a given Bill-Board 6-pack. During the distribution of the resin, the isopycnic solvent was allowed to drain through the frit in the reaction vessels. When distribution was complete, residual solvent was removed with an “air-push” from a disposable plastic pipet fitted with a pierced septum that could be placed over the open mouth of the reaction vessel. The resin was then washed with NMP (3 × 3 mL).
The bottom of each reaction vessel was capped and a new calibrated disposable plastic pipet was used for each alkylating agent. The first 1R1-X (benzyl bromide) [0.5 mL of a 0.20 M solution in NMP, 100 μmols, 2 equiv] was added to the resin in the two reaction vessels in the first column positions (i.e., A1 and B1). Then the second 2R1-X [0.5 mL of a 0.20 M solution in NMP, 100 μmols, 2 equiv] was added to the resin in the two vessels in the second column positions (i.e., A2 and B2). Finally, the third 3R1-X [0.5 mL of a 0.20 M solution in NMP, 100 μmols, 2 equiv] was added to the resin in the two vessels in the third column positions (i.e., A3 and B3). Base was then added [0.5 mL of a 0.20 M solution of BTPP in NMP, 100 μmols, 2 equiv] to each of the six reaction vessels. The tops of all reaction vessels were capped and the Bill-Board was placed in a rotation apparatus. The alkylation reaction was allowed to proceed for two days with rotation.(18)
(b) Hydrolysis (4 to 5)
The Bill-Board was removed from the rotation apparatus and, after inverting the board, the bottom caps were removed. The Board was then placed, top side up, in the drain tray; the top caps were removed, and the reagents from the alkylation step were allowed to drain, followed by an air-push. The resin-bound alkylated products 4 were washed with THF (1 × 3 mL). Using 6 clean caps, the bottom of each reaction vessel was capped, and a 1 N aqueous HCl-THF solution (1:2, 2.5 mL) was added to each of the six reaction vessels. The caps were then put on the top of each vessel, and the Bill-Board was returned to the rotator for 20 min. The reagents and byproduct from the hydrolysis were then removed from hydrolyzed resin-bound product (5) by filtration and washing with THF (1 × 3 mL) and then NMP (1 × 3 mL).
(c) Acylation (5 to 6)
Using clean caps, the bottom of each reaction vessel was capped, and the first acylating agent, triphenylacetyl chloride [0.5 mL of a 0.30 M solution of R2COCl in NMP, 150 μmols, 3 equiv] was added to the resin 5 in each of the three vessels in row A (A1, A2, and A3). Then the second acylating agent, triphenylpropionyl chloride [0.5 mL of a 0.30 M solution of R2COCl in NMP, 150 μmols, 3 equiv] was added to the resin 5 in each of the three vessels in row B (B1, B2, and B3). This was followed by addition of DIEA [0.5 mL of a 0.60 M solution in NMP, 300 μmols, 6 equiv] to each of the six reaction vessels. The tops of the reaction vessels were capped and the reaction was allowed to proceed with rotation for 30 min.
(d) Cleavage of Products from Resin (6 to 8)
The Bill-Board was removed from the rotation apparatus, followed by the usual filtration/washing procedure. Acylated resin-bound product 6 was washed with NMP (2 × 3 mL), CH2Cl2 (2 × 3 mL), THF (2 × 3 mL), and MeOH (2 × 3 mL). The bottom of each reaction vessel was capped with a clean cap and Et3N/MeOH (3:7, 2.5 mL) was added to each vessel. After securely capping the top of each reaction vessel with a clean cap, the reaction vessels were placed in an oven for two days at 55 °C. The products 8 were then collected in tared tool-necked vials. An air-push apparatus was used to finish draining the reaction vessels. The resin was rinsed with THF (1 × 2 mL), and this rinse was also collected. After each collection vial was swirled to thoroughly mix the cleavage and rinse solutions, a sample of each solution (100 μL) containing product 8 was transferred to an autosampler vial for LC/MS analysis. The cleavage solvent and rinses were evaporated with a simple, inexpensive apparatus designed to speed up the evaporation with a stream of nitrogen.
Purification of Products
One student purified all the products from this laboratory. To enable an efficient selection of gradient conditions, the products were gathered in groups of 6 according to retention times from the LC/MS data. The purifications were done six at a time, using a modified Bill-Board in which the reaction vessel plate was suspended above the collection vial rack and silica gel cartridges (6 × 3 mL/500 mg, Fisher Cat. No. 11-131-1) replaced the reaction vessels.
a Series: 8, R2 = N-(Triphenylacetyl)
Methyl N-(Triphenylacetyl)phenylalaninate [8{9}a]:
16.7 mg (74% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 100%, tR = 6.01 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.92 (dd, J = 14.0 Hz, J = 7.6 Hz, 1H), 3.13 (dd, J = 14.2 Hz, J = 5.2 Hz, 1H), 3.71 (s, 3H), 4.90 − 5.00 (m, 1H), 6.17 (d, J = 7.2 Hz, 1H), 7.11 − 7.25 (m, 20H); LC/MS calcd for C30H27NO3 [M + H]+ 450.2; found 450.2.
Methyl 2-Fluoro-N-(triphenylacetyl)phenylalaninate [8{10}a]:
10.6 mg (45% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 77%, tR = 5.99 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 3.01 (dd, J = 14.1 Hz, J = 8.1 Hz, 1H), 3.15 (dd, J = 13.9 Hz, J = 5.5 Hz, 1H), 3.72 (s, 3H), 4.87−4.97 (m, 1H), 6.28 (d, J = 7.4 Hz, 1H), 6.90−7.02 (m, 4H), 7.03−7.25 (m, 15H); LC/MS calcd for C30H26FNO3 [M + H]+ 468.2; found 468.2.
Methyl 3-Fluoro-N-(triphenylacetyl)phenylalaninate [8{11}a]:
9.2 mg (39% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 52%, tR = 6.00 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.93 (dd, J = 14.1 Hz, J = 7.5 Hz, 1H), 3.12 (dd, J = 14.2 Hz, J = 5.2 Hz, 1H), 3.72 (s, 3H), 4.90−5.02 (m, 1H), 6.16 (d, J = 7.1 Hz, 1H), 6.58−6.95 (m, 4H), 7.09−7.30 (m, 15H); LC/MS calcd for C30H26FNO3 [M + H]+ 468.2; found 468.2.
Methyl 4-Fluoro-N-(triphenylacetyl)phenylalaninate [8{12}a]:
25.6 mg (110% isolated yield(7)) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 85%, tR = 6.01 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.91 (dd, J = 14.1 Hz, J = 7.5 Hz, 1H), 3.09 (dd, J = 14.1 Hz, J = 5.3 Hz, 1H), 3.71 (s, 3H), 4.89−4.99 (m, 1H), 6.17 (d, J = 7.6 Hz, 1H), 6.78−6.97 (m, 4H), 7.08−7.30 (m, 15H); LC/MS calcd for C30H26FNO3 [M + H]+ 468.2; found 468.2.
Methyl 2-(Trifluoromethyl)-N-(triphenylacetyl)phenylalaninate [8{13}a]:
9.3 mg (36% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 94%, tR = 6.21 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 3.01 (m, 1H), 3.30 (dd, J = 15.0 Hz, J = 5.4 Hz, 1H), 3.72 (s, 3H), 4.96−5.08 (m, 1H), 6.21 (d, J = 7.8 Hz, 1H), 7.04−7.06 (m, 18H), 7.61 (d, J = 7.8 Hz, 1H); LC/MS calcd for C31H26F3NO3 [M + H]+ 518.2; found 518.2.
Methyl 3-(Trifluoromethyl)-N-(triphenylacetyl)phenylalaninate [8{14}a]:
8.9 mg (34% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 49%, tR = 6.24 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 3.02 (dd, J = 14.1 Hz, J = 7.3 Hz, 1H), 3.21 (dd, J = 14.1 Hz, J = 5.3 Hz, 1H), 3.70 (s, 3H), 4.99 (dd, J = 12.7 Hz, J = 7.3 Hz, 1H), 6.21 (d, J = 7.0 Hz, 1H), 7.09 − 7.35 (m, 18H), 7.49 (d, J = 7.6 Hz, 1H); LC/MS calcd for C31H26F3NO3 [M + H]+ 518.2; found 518.2.
Methyl 4-(Trifluoromethyl)-N-(triphenylacetyl)phenylalaninate [8{15}a]:
12.7 mg (49% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 81%, tR = 6.28 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 3.00 (dd, J = 14.2 Hz, J = 7.2 Hz, 1H), 3.20 (dd, J = 14.1 Hz, J = 5.3 Hz, 1H), 3.72 (s, 3H), 4.94−5.05 (m, 1H), 6.16 (d, J = 7.4 Hz, 1H), 7.02 (d, J = 8.0 Hz, 2H), 7.12−7.26 (m, 15H), 7.44 (d, J = 8.0 Hz, 2H); LC/MS calcd for C31H26F3NO3 [M + H]+ 518.2; found 518.2.
Methyl 2-Bromo-N-(triphenylacetyl)phenylalaninate [8{16}a]:
3.6 mg (14% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 98%, tR = 6.20 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 3.05 (dd, J = 13.9 Hz, J = 9.9 Hz, 1H), 3.28 (dd, J = 14.2 Hz, J = 5.4 Hz, 1H), 3.73 (s, 3H), 4.91−5.03 (m, 1H), 6.34 (d, J = 7.8 Hz, 1H), 7.04−7.26 (m,18H), 7.49 (d, J = 4.5 Hz, 1H); LC/MS calcd for C31H26BrNO3 [M + H]+ 528.1; found 528.1.
Methyl 3-Bromo-N-(triphenylacetyl)phenylalaninate [8{17}a]:
16.2 mg (62% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 81%, tR = 6.26 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.89 (dd, J = 13.9 Hz, J = 7.7 Hz, 1H), 3.13 (dd, J = 14.1 Hz, J = 5.3 Hz, 1H), 3.72 (s, 3H), 4.88−4.99 (m, 1H), 6.17 (d, J = 7.4 Hz, 1H), 6.85 (d, J = 7.6 Hz, 1H), 7.02−7.38 (m,18H); LC/MS calcd for C31H26BrNO3 [M + H]+ 528.1; found 528.1.
Methyl 4-Bromo-N-(triphenylacetyl)phenylalaninate [8{18}a]:
18.5 mg (70% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 84%, tR = 6.30 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.89 (dd, J = 14 Hz, J = 7.8 Hz, 1H), 3.08 (dd, J = 14.2 Hz, J = 5.4 Hz, 1H), 3.71 (s, 3H), 4.94 (dd, J = 12.9 Hz, J = 7.6 Hz, 1H), 6.14 (d, J = 7.2 Hz, 1H), 6.76 (d, J = 8.2 Hz, 2H), 7.12−7.32 (m, 17H); LC/MS calcd for C30H26BrNO3 [M + H]+ 528.1; found 528.1.
Methyl 3-Cyano-N-(triphenylacetyl)phenylalaninate [8{19}a]:
14.0 mg (59% isolated yield) following chromatographic purification (EtOAc/CH2Cl2, 20:80); initial LC/MS purity 77%, tR = 5.66 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.99 (dd, J = 14.2 Hz, J = 7.4 Hz, 1H), 3.16 (dd, J = 14 Hz, J = 5.2 Hz, 1H), 3.72 (s, 3H), 4.99 (dd, J = 12.0 Hz, J = 7.2 Hz, 1H), 6.19 (d, J = 7.6 Hz, 1H), 7.14−7.35 (m, 18H), 7.52 (d, J = 7.6 Hz, 1H); LC/MS calcd for C31H26N2O3 [M + H]+ 475.2; found 475.2.
Methyl 4-Cyano-N-(triphenylacetyl)phenylalaninate [8{20}a]:
17.9 mg (76% isolated yield) following chromatographic purification (EtOAc/CH2Cl2, 20:80); initial LC/MS purity 75%, tR = 5.66 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 3.02 (dd, J = 14.1 Hz, J = 7.3 Hz, 1H), 3.19 (dd, J = 13.9 Hz, J = 5.3 Hz, 1H), 3.72 (s, 3H), 5.01 (dd, J = 14.0 Hz, J = 7.2 Hz, 1H), 6.20 (d, J = 7.6 Hz, 1H), 7.02 (d, J = 8.2 Hz, 2H), 7.15−7.28 (m, 15H), 7.47 (d, J = 8.4 Hz, 2H); LC/MS calcd for C31H26N2O3 [M + H]+ 475.2; found 475.2.
Methyl 2-Iodo-N-(triphenylacetyl)phenylalaninate [8{21}a]:
17.3 mg (60% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 84%, tR = 6.27 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.95−3.10 (m, 1H), 3.29 (dd, J = 14.2 Hz, J = 7.4 Hz, 1H), 3.74 (s, 3H), 4.93 (m, 1H), 6.32 (d, J = 7.6 Hz, 1H), 6.91−7.25 (m, 18H), 7.79 (d, J = 7.0 Hz, 1H); LC/MS calcd for C30H26INO3 [M + H]+ 576.1; found 576.1.
Methyl 4-Iodo-N-(triphenylacetyl)phenylalaninate [8{22}a]:
15.9 mg (55% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 81%, tR = 6.38 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.87 (dd, J = 14 Hz, J = 7.6 Hz, 1H), 3.07 (dd, J = 14.1 Hz, J = 5.3 Hz, 1H), 3.71 (s, 3H), 4.94 (dd, J = 14 Hz, J = 7.6 Hz, 1H), 6.13 (d, J = 7.4 Hz, 1H), 6.63 (d, J = 8.2 Hz, 2H), 7.12 − 7.27 (m, 15H), 7.50 (d, J = 8.2 Hz, 2H); LC/MS calcd for C30H26INO3 [M + H]+ 576.1; found 576.1.
Methyl 2-Methoxy-N-(triphenylacetyl)phenylalaninate [8{23}a]:
3.9 mg (8% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85) of pooled duplicate samples; initial LC/MS purity 24%−25%, tR = 6.07 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.91−3.12 (m, 2H), 3.43 (s, 3H), 3.72 (s, 3H), 4.75−4.85 (m, 1H), 6.59 (d, J = 6.6 Hz, 1H), 6.72−7.10 (m, 4H), 7.11−7.29 (m,15H); LC/MS calcd for C31H29NO4 [M + H]+ 480.2; found 480.2.
Methyl 3-Methoxy-N-(triphenylacetyl)phenylalaninate [8{24}a]:
12.0 mg (25% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85) of pooled duplicate samples; initial LC/MS purity 37%−38%, tR = 5.97 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.88 (dd, J = 13.9 Hz, J = 8.1 Hz, 1H), 3.12 (dd, J = 14.1 Hz, J = 5.3 Hz, 1H), 3.69 (s, 3H), 3.72 (s,3H), 4.87−4.97 (m, 1H), 6.17 (d, J = 7.2 Hz, 1H), 6.45−6.78 (m, 4H), 7.06−7.29 (m,15H). LC/MS calcd for C31H29NO4 [M + H]+ 480.2; found 480.2.
Methyl O-Methyl-N-(triphenylacetyl)tyrosinate [8{25}a]:
12.0 mg (25% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85) of pooled duplicate samples; initial LC/MS purity 38%−46%, tR = 5.94 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.86 (dd, J = 14.2 Hz, J = 7.6 Hz, 1H), 3.05 (dd, J = 14.1 Hz, J = 5.1 Hz, 1H), 3.71 (s, 3H), 3.78 (s, 3H), 4.85−4.96 (m, 1H), 6.15 (d, J = 7.6 Hz, 1H), 6.68−6.82 (m, 4H), 7.12−7.26 (m,15H); LC/MS calcd for C31H29NO4 [M + H]+ 480.2; found 480.2.
Methyl 4-Methyl-N-(triphenylacetyl)phenylalaninate [8{26}a]:
14.7 mg (64% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 97%, tR = 6.21 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.31 (s, 3H), 2.87 (dd, J = 14.0 Hz, J = 7.6 Hz, 1H), 3.07 (dd, J = 14.0 Hz, J = 5.0 Hz, 1H), 3.71 (s, 3H), 4.86−4.96 (m, 1H), 6.15 (d, J = 7.2 Hz, 1H), 6.76 (d, J = 7.8 Hz, 2H), 6.99 (d, J = 7.8 Hz, 2H), 7.12−7.25 (m, 15H); LC/MS calcd for C34H29NO3 [M + H]+ 464.2; found 464.2.
Methyl α-[(Triphenylacetyl)amino]-1-naphthalenepropanoate [8{27}a]:
8.8 mg (35% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 64%, tR = 6.31 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 3.26−3.74 (m, 2H), 3.67 (s, 3H), 5.02 (dd, J = 15.0 Hz, J = 7.0 Hz, 1H), 6.23 (d, J = 6.8 Hz, 1H), 6.95−7.51 (m, 19H), 7.80 (m, 2H), 8.00 (d, J = 7.6 Hz, 1H); LC/MS calcd for C30H26BrNO3 [M + H]+ 500.2; found 500.2.
Methyl α-[(Triphenylacetyl)amino]-2-naphthalenepropanoate [8{28}a]:
17.7 mg (71% isolated yield) following chromatographic purification (EtOAc/hexanes, 15:85); initial LC/MS purity 88%, tR = 6.32 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 3.05 (dd, J = 14.1 Hz, J = 8.1 Hz, 1H), 3.33 (dd, J = 13.9 Hz, J = 5.1 Hz, 1H), 3.74 (s, 3H), 4.97−5.07 (m, 1H), 6.17 (d, J = 7.0 Hz, 1H), 7.07−7.33 (m, 15H), 7.44−7.83 (m, 7H); LC/MS calcd for C34H29NO3 [M + H]+ 500.2; found 500.2.
b = Series: 8, R2 = N-(1-Oxo-3,3,3-triphenylpropyl) = N-(Triphenylpropionyl)
Methyl N-(1-Oxo-3,3,3-triphenylpropyl)phenylalaninate [8{9}b]:
18.3 mg (79% isolated yield) following chromatographic purification (CH2Cl2/EtOAc/hexanes, 10:15:75 to 20:20:60) of pooled duplicate samples; initial LC/MS purity 86, 87%, tR = 5.92 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.74−2.78 (m, 2H), 3.52 (d, J = 15.0 Hz, 1H), 3.56 (s, 3H), 3.66 (d, J = 15.0 Hz, 1H), 4.59 (dd, J = 14 Hz, J = 6.2 Hz, 1H), 5.40 (d, J = 7.4 Hz, 1H), 6.82−6.87 (m, 2H), 7.18−7.29 (m, 18H); LC/MS calcd for C31H29NO3 [M + H]+ 464.2; found 464.2.
Methyl 2-Fluoro-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{10}b]:
12.6 mg (52% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 83%, tR = 5.91 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.79−2.84 (m, 2H), 3.57 (d, J = 14.8 Hz, 1H), 3.58 (s, 3H), 3.63 (d, J = 14.8 Hz, 1H), 4.56 (dd, J = 13.5 Hz, J = 6.4 Hz, 1H), 5.44 (d, J = 7.0 Hz, 1H), 6.85−7.26 (m, 19H); LC/MS calcd for C31H28FNO3 [M + H]+ 482.2; found 482.2.
Methyl 3-Fluoro-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{11}b]:
7.7 mg (32% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 70%, tR = 5.95 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.73−2.79 (m, 2H), 3.54 (d, J = 15.2 Hz, 1H), 3.58 (s, 3H), 3.67 (d, J = 15.2 Hz, 1H), 4.60 (dd, J = 13.4 Hz, J = 6.2 Hz, 1H), 5.40 (d, J = 7.0 Hz, 1H), 6.54−6.93 (m, 3H), 7.09−7.30 (m, 16H); LC/MS: calcd for C31H28FNO3 [M + H]+ 482.2; found 482.2.
Methyl 4-Fluoro-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{12}b]:
18.1 mg (75% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 83%, tR = 5.93 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.71−2.76 (m, 2H), 3.53 (d, J = 14.6 Hz, 1H), 3.57 (s, 3H), 3.67 (d, J = 14.6 Hz, 1H), 4.58 (dd, J = 13.4 Hz, J = 6.0 Hz, 1H), 5.39 (d, J = 7.2 Hz, 1H), 6.75−6.92 (m, 4H), 7.16−7.30 (m, 15H); LC/MS: calcd for C31H28FNO3 [M + H]+ 482.2; found 482.2.
Methyl 2-(Trifluoromethyl)-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{13}b]:
16.6 mg (63% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92); initial LC/MS purity 89%, tR = 6.13 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.66−2.93 (m, 2H), 3.51 (d, J = 15.0 Hz, 1H), 3.52 (s, 3H), 3.66 (d, J = 15.2 Hz, 1H), 4.60 (dd, J = 15.0 Hz, J = 7.6 Hz, 1H), 5.41 (d, J = 7.6 Hz, 1H), 7.12−7.43 (m, 18H), 7.59 (d, J = 7.2 Hz, 1H); LC/MS calcd for C32H29F3NO3 [M + H]+ 532.2; found 532.2.
Methyl 3-(Trifluoromethyl)-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{14}b]:
5.7 mg (22% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92); initial LC/MS purity 72%, tR = 6.21 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.70−2.91 (m, 2H), 3.54 (d, J = 14.8 Hz, 1H), 3.56 (s, 3H), 3.66 (d, J = 15.0 Hz, 1H), 4.61 (dd, J = 13.3 Hz, J = 6.3 Hz, 1H), 5.39 (d, J = 7.2 Hz, 1H), 7.05 (d, J = 7.6 Hz, 1H), 7.14−7.35 (m, 17H), 7.47 (d, J = 7.6 Hz, 1H); LC/MS calcd for C32H29F3NO3 [M + H]+ 532.2; found 532.2.
Methyl 4-(Trifluoromethyl)-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{15}b]:
18.5 mg (70% isolated yield) following chromatographic purification (EtOAc/hexanes in CH2Cl2, 4:46:50); initial LC/MS purity 80%, tR = 6.22 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.70−2.97 (m, 2H), 3.54 (d, J = 15.0 Hz, 1H), 3.58 (s, 3H), 3.68 (d, J = 15.0 Hz, 1H), 4.64 (dd, J = 13.4 Hz, J = 6.0 Hz, 1H), 5.39 (d, J = 7.4 Hz, 1H), 6.95 (d, J = 8.0 Hz, 2H), 7.10−7.35 (m, 15H), 7.43 (d, J = 8.0 Hz, 2H); LC/MS calcd for C32H29F3NO3 [M + H]+ 532.2; found 532.2.
Methyl 2-Bromo-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{16}b]:
8.8 mg (33% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92); initial LC/MS purity 94%, tR = 6.11 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.77 (dd, J = 14.1 Hz, J = 7.5 Hz, 1H), 2.92 (dd, J = 14.1 Hz, J = 6.7 Hz, 1H), 3.54 (d, J = 14.8 Hz, 1H), 3.57 (s, 3H), 3.63 (d, J = 14.8 Hz, 1H), 4.60 (dd, J = 14.5 Hz, J = 7.3 Hz, 1H), 5.39 (d, J = 7.2 Hz, 1H), 6.93 (d, J = 7.0 Hz, 1H), 7.00−7.30 (m, 17H), 7.50 (d, J = 7.8 Hz, 1H); LC/MS: calcd for C31H29BrNO3 [M + H]+ 542.1; found 542.2.
Methyl 3-Bromo-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{17}b]:
8.5 mg (31% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92); initial LC/MS purity 84%, tR = 6.21 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.60−2.85 (m, 2H), 3.54 (d, J = 15.2 Hz, 1H), 3.58 (s, 3H), 3.67 (d, J = 15.2 Hz, 1H), 4.59 (dd, J = 13.3 Hz, J = 6.3 Hz, 1H), 5.38 (d, J = 7.2 Hz, 1H), 6.79 (d, J = 7.8 Hz, 1H), 6.95−7.40 (m, 18H); LC/MS calcd for C31H29BrNO3 [M + H]+ 542.1; found 542.2.
Methyl 4-Bromo-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{18}b]:
12.9 mg (48% isolated yield) following chromatographic purification (EtOAc/hexanes in CH2Cl2, 4:46:50); initial LC/MS purity 87%, tR = 6.23 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.60−2.90 (m, 2H), 3.53 (d, J = 15.0 Hz, 1H), 3.57 (s, 3H), 3.67 (d, J = 15.0 Hz, 1H), 4.59 (dd, J = 13.3 Hz, J = 6.3 Hz, 1H), 5.38 (d, J = 7.4 Hz, 1H), 6.70 (d, J = 8.4 Hz, 2H), 7.10−7.38 (m, 17H); LC/MS calcd for C31H29BrNO3 [M + H]+ 542.1; found 542.2.
Methyl 3-Cyano-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{19}b]:
7.5 mg (31% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 78%, tR = 5.67 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.62−2.91 (m, 2H), 3.55 (d, J = 15.2 Hz, 1H), 3.58 (s, 3H), 3.67 (d, J = 15.2 Hz, 1H), 4.62 (dd, J = 12.9 Hz, J = 6.1 Hz, 1H), 5.42 (d, J = 7.2 Hz, 1H), 7.05−7.39 (m, 18H), 7.50 (d, J = 7.8 Hz, 1H); LC/MS calcd for C32H29N2O3 [M + H]+ 489.2; found 489.2.
Methyl 4-Cyano-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{20}b]:
19.1 mg (78% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 76%, tR = 5.65 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.77 (dd, J = 13.9 Hz, J = 6.1 Hz, 1H), 2.87 (dd, J = 13.9 Hz, J = 6.1 Hz, 1H), 3.54 (d, J = 15.0 Hz, 1H), 3.57 (s, 3H), 3.68 (d, J = 15.0 Hz, 1H), 4.63 (dd, J = 13.3 Hz, J = 6.3 Hz, 1H), 5.41 (d, J = 7.4 Hz, 1H), 6.96 (d, J = 8.4 Hz, 2H), 7.10−7.38 (m, 15H), 7.46 (d, J = 8.4 Hz, 2H); LC/MS calcd for C32H29N2O3 [M + H]+: 489.2; found 489.3.
Methyl 2-Iodo-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{21}b]:
18.9 mg (64% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92); initial LC/MS purity 86%, tR = 6.18 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.74 (dd, J = 14.2 Hz, J = 7.6 Hz, 1H), 2.89 (dd, J = 14.1 Hz, J = 6.9 Hz, 1H), 3.54 (d, J = 15.2 Hz, 1H), 3.57 (s, 3H), 3.64 (d, J = 15.2 Hz, 1H), 4.60 (dd, J = 14.7, Hz, J = 7.3 Hz, 1H), 5.37 (d, J = 7.2 Hz, 1H), 6.92 (d, J = 7.6 Hz, 2H), 7.08−7.40 (m, 16H), 7.78 (d, J = 7.8 Hz, 1H); LC/MS calcd for C31H29INO3 [M + H]+ 590.1; found 590.1.
Methyl 4-Iodo-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{22}b]:
19.6 mg (67% isolated yield) following chromatographic purification (EtOAc/hexanes in CH2Cl2, 4:46:50); initial LC/MS purity 81%, tR = 6.31 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.59−2.82 (m, 2H), 3.53 (d, J = 15.2 Hz, 1H), 3.57 (s, 3H), 3.67 (d, J = 15.2 Hz, 1H), 4.59 (dd, J = 13.4 Hz, J = 6.0 Hz, 1H), 5.38 (d, J = 7.4 Hz, 1H), 6.58 (d, J = 8.2 Hz, 2H), 7.10−7.32 (m, 15H), 7.49 (d, J = 8.2 Hz, 2H); LC/MS calcd for C31H29INO3 [M + H]+ 590.1; found 590.1.
Methyl 2-Methoxy-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{23}b]:
4.3 mg (9% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85) of pooled duplicate samples; initial LC/MS purity 40%−42%, tR = 5.96 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.73−2.76 (m, 2H), 3.43 (d, J = 15.0 Hz, 1H), 3.56 (s, 3H), 3.63 (d, J = 15.0 Hz, 1H), 3.73 (s, 3H), 4.47 (dd, J = 13.6 Hz, J = 6.8 Hz, 1H), 5.58 (d, J = 6.6 Hz, 1H), 6.79−6.81 (m, 3H), 7.14−7.26 (m, 16H); LC/MS calcd for C32H31NO4 [M + H]+ 494.2; found 494.3.
Methyl 3-Methoxy-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{24}b]:
4.3 mg (17% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 61%, tR = 5.90 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.70−2.75 (m, 2H), 3.53 (d, J = 15.2 Hz, 1H), 3.57 (s, 3H), 3.65 (d, J = 15.2 Hz, 1H), 3.74 (s, 3H), 4.58 (dd, J = 14.0 Hz, J = 6.2 Hz, 1H), 5.39 (d, J = 7.4 Hz, 1H), 6.41−6.45 (m, 2H), 6.73−6.76 (m, 1H), 7.06−7.63 (m, 16H); LC/MS calcd for C31H29NO3 [M + H]+ 494.2; found 494.3.
Methyl O-Methyl-N-(1-oxo-3,3,3-triphenylpropyl)tyrosinate [8{25}b]:
4.0 mg (16% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92 to 15:85); initial LC/MS purity 57%, tR = 5.86 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.69−2.71 (m, 2H), 3.52 (d, J = 15.2 Hz, 1H), 3.57 (s, 3H), 3.66 (d, J = 15.2 Hz, 1H), 3.77 (s, 3H), 4.55 (dd, J = 13.4 Hz, J = 6.2 Hz, 1H), 5.38 (d, J = 7.4 Hz, 1H), 6.74 (m, 4H), 7.18−7.26 (m, 15H); LC/MS: calcd for C31H29NO3 [M + H]+ 494.2; found 494.3.
Methyl 4-Methyl-N-(1-oxo-3,3,3-triphenylpropyl)phenylalaninate [8{26}b]:
9.5 mg (40% isolated yield) following chromatographic purification (EtOAc/hexanes, 8:92); initial LC/MS purity 100%, tR = 6.12 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.30 (s, 3H), 2.60−2.81 (m, 2H), 3.52 (d, J = 15.2 Hz, 1H), 3.57 (s, 3H), 3.65 (d, J = 15.2 Hz, 1H), 4.56 (dd, J = 13.2 Hz, J = 6.2 Hz, 1H), 5.39 (d, J = 7.6 Hz, 1H), 6.73 (d, J = 7.8 Hz, 2H), 6.99 (d, J = 7.8 Hz, 2H), 7.10−7.32 (m, 15H); LC/MS calcd for C32H32NO3 [M + H]+ 478.2; found 478.3.
Methyl α-[(1-Oxo-3,3,3-triphenylpropyl)amino]-1-naphthalenepropanoate [8{27}b]:
5.0 mg (20% isolated yield) following chromatographic purification (EtOAc/hexanes in CH2Cl2, 4:46:50); initial LC/MS purity 80%, tR = 6.23 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 3.03−3.31 (m, 2H), 3.45 (s, 3H), 3.57 (s, 2H), 4.68 (dd, J = 14.0 Hz, J = 7.0 Hz, 1H), 5.43 (d, J = 7.0 Hz, 1H), 6.94 (d, J = 6.8 Hz, 1H), 7.07−8.00 (m, 21H); LC/MS calcd for C35H32NO3 [M + H]+ 514.2; found 514.3.
Methyl α-[(1-Oxo-3,3,3-triphenylpropyl)amino]-2-naphthalenepropanoate [8{28}b]:
14.7 mg (57% isolated yield) following chromatographic purification (EtOAc/hexanes in CH2Cl2, 4:46:50); initial LC/MS purity 87%, tR = 6.26 min; 1H NMR (200 MHz, 10% CD3OD/CDCl3) δ 2.80−3.21 (m, 2H), 3.52 (d, J = 15.2 Hz, 1H), 3.56 (s, 3H), 3.66 (d, J = 15.2 Hz, 1H), 4.71 (dd, J = 13.4 Hz, J = 6.2 Hz, 1H), 5.39 (d, J = 7.4 Hz, 1H), 7.00 (d, J = 6.8 Hz, 1H), 7.02 − 7.82 (m, 21H); LC/MS calcd for C35H32NO3 [M + H]+ 514.2; found 514.3.
Acknowledgments
This article is dedicated to the 19 IUPUI students who carried out the combinatorial syntheses described in this paper.(19) We gratefully acknowledge Mr. Christopher Reutter for operating the LC/MS and reporting the results in a readily understandable format. We wish to thank Donald B. Boyd, Guillermo Morales, Daniel H. Robertson, Richard T. Taylor, and James H. Wikel for helpful discussions and Ralph Mazitschek for assistance in the enumeration. We acknowledge the National Institutes of Health (R01 GM028193), The National Science Foundation (MRI CHE-0619254), The Camille and Henry Dreyfus Foundation, and the Lilly Research Laboratories for their financial support.
Supporting Information Available
Tutorial for using the Distributed Drug Discovery (D3) database and proton NMR spectra of products. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- See preceding Perspective:; Scott W. L.; O’Donnell M. J. J. Comb. Chem. 2009, 11, 3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See preceding article:; Scott W. L.; Alsina J.; Audu C. O.; Babaev E.; Cook L.; Dage J. L.; Goodwin L. A.; Martynow J. G.; Matosiuk D.; Royo M.; Smith J. G.; Strong A. T.; Wickizer K.; Woerly E. M.; Zhou Z.; O’Donnell M. J. J. Comb. Chem. 2009, 11, 14–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dothager R. S.; Putt K. S.; Allen B. J.; Leslie B. J.; Nesterenko V.; Hergenrother P. J. J. Am. Chem. Soc. 2005, 127, 8686–8696. [DOI] [PubMed] [Google Scholar]
- Gao M.; Kong D.; Clearfield A.; Miller K. D.; Sledge G. W.; Zheng Q.-H. Synthesis 2006, 2301–2304. [Google Scholar]
- For recent melanoma reviews, see:; a Markovic S. N.; Erickson L. A.; Rao R. D.; Weenig R. H.; Pockaj B. A.; Bardia A.; Vachon C. M.; Schild S. E.; McWilliams R. R.; Hand J. L.; Laman S. D.; Kottschade L. A.; Maples W. J.; Pittelkow M. R.; Pulido J. S.; Cameron J. D.; Creagan E. T. Mayo Clinic Proc. 2007, 82, 364–380. [DOI] [PubMed] [Google Scholar]; b Markovic S. N.; Erickson L. A.; Rao R. D.; Weenig R. H.; Pockaj B. A.; Bardia A.; Vachon C. M.; Schild S. E.; McWilliams R. R.; Hand J. L.; Laman S. D.; Kottschade L. A.; Maples W. J.; Pittelkow M. R.; Pulido J. S.; Cameron J. D.; Creagan E. T. Mayo Clinic Proc. 2007, 82, 490–513. [DOI] [PubMed] [Google Scholar]
- References from the authors’ laboratory concerning the solid-phase synthesis of unnatural amino acids, peptides, and peptidomimetics:; a O’Donnell M. J.; Zhou C.; Scott W. L. J. Am. Chem. Soc. 1996, 118, 6070–6071. [Google Scholar]; b Scott W. L.; Zhou C.; Fang Z.; O’Donnell M. J. Tetrahedron Lett. 1997, 38, 3695–3698. [Google Scholar]; c O’Donnell M. J.; Lugar C. W.; Pottorf R. S.; Zhou C.; Scott W. L.; Cwi C. L. Tetrahedron Lett. 1997, 38, 7163–7166. [Google Scholar]; d Griffith D. L.; O’Donnell M. J.; Pottorf R. S.; Scott W. L.; Porco J. A. Jr. Tetrahedron Lett. 1997, 38, 8821–8824. [Google Scholar]; e Domínguez E.; O’Donnell M. J.; Scott W. L. Tetrahedron Lett. 1998, 39, 2167–2170. [Google Scholar]; f O’Donnell M. J.; Delgado F.; Drew M. D.; Pottorf R. S.; Zhou C.; Scott W. L. Tetrahedron Lett. 1999, 40, 5831–5835. [Google Scholar]; g O’Donnell M. J.; Delgado F.; Pottorf R. S. Tetrahedron 1999, 55, 6347–6362. [Google Scholar]; h O’Donnell M. J.; Drew M. D.; Pottorf R. S.; Scott W. L. J. Comb. Chem. 2000, 2, 172–181. [DOI] [PubMed] [Google Scholar]; i O’Donnell M. J.; Scott W. L.. Unnatural Amino Acid and Peptide Synthesis (UPS). In Peptides 2000; Martinez J., Fehrentz J.-A., Eds.; EDK: Paris, 2001; pp 31−36. [Google Scholar]; j O’Donnell M. J.; Delgado F.; Domínguez E.; de Blas J.; Scott W. L. Tetrahedron: Asymmetry 2001, 12, 821–828. [Google Scholar]; k Scott W. L.; Delgado F.; Lobb K.; Pottorf R. S.; O’Donnell M. J. Tetrahedron Lett. 2001, 42, 2073–2076. [Google Scholar]; l Scott W. L.; O’Donnell M. J.; Delgado F.; Alsina J. J. Org. Chem. 2002, 67, 2960–2969. [DOI] [PubMed] [Google Scholar]; m Scott W. L.; Alsina J.; O’Donnell M. J. J. Comb. Chem. 2003, 5, 684–692. [Google Scholar]; n O’Donnell M. J.; Alsina J.; Scott W. L. Tetrahedron Lett. 2003, 44, 8403–8406. [Google Scholar]; o Scott W. L.; Alsina J.; Kennedy J. H.; O’Donnell M. J. Org. Lett. 2004, 6, 1629–1632. [DOI] [PubMed] [Google Scholar]; p Alsina J.; Scott W. L.; O’Donnell M. J. Tetrahedron Lett. 2005, 46, 3131–3135. [Google Scholar]; q Scott W. L.; Martynow J. G.; Huffman J. C.; O’Donnell M. J. J. Am. Chem. Soc. 2007, 129, 7077–7088. [DOI] [PubMed] [Google Scholar]
- The greater than theoretical yield could be attributed to uneven distribution of starting resin at the initial isopycnic resin dispensing step, giving rise to >50 μmols of starting material.
- Available from Leads Metal Products, PO Box 441186, Indianapolis, IN 46244−1186 (larry@leadsmetal.com).
- a ChemAxon (free academic license available) from ChemAxon Kft., Máramaros köz 3/a, Budapest, 1037 Hungary. Marvin was used for drawing, displaying and characterizing chemical structures, substructures and reactions, Marvin 5.1.02, 2008, ChemAxon (http://www.chemaxon.com (accessed November 2, 2008)).; b CombiChem/Excel from CambridgeSoft, 100 CambridgePark Drive Cambridge, MA 02140 USA (http://www.cambridgesoft.com/ (accessed November 2, 2008)).
- a For access to the IUPUI −Distributed Drug Discovery (D3) database, register for a free read-download account with Collaborative Drug Discovery (CDD) at https://www.collaborativedrug.com/register/iupui-d3 (accessed November 2, 2008) by completing the “Sign up for IUPUI−Distributed Drug Discovery (D3)” information.; b A ttutorial for use of the IUPUI−Distributed Drug Discovery (D3) database on CDD is provided: (i) in the Supporting Information for this article and the preceding article,(2) (ii) from the D3 database on the CDD website, and (iii) on the IUPUI Department of Chemistry and Chemical Biology website [http://chem.iupui.edu/ (accessed November 2, 2008)] under the faculty & staff directory for O’Donnell or Scott. The tutorial available at ii and iii will be updated periodically.
- Lead reference for possible side reaction (transesterification) during cleavage of resin-bound products 6 to product methyl esters of acylated unnatural amino acids 8 by methanolysis (MeOH, Et3N, 55 °C, 48 h):; a Otera J. Chem. Rev. 1993, 93, 1449–1470. [Google Scholar]; b This side reaction is reflected in D3 database products arising from the following reactants in Tables 5 and 6, ref (2): 30, 35, 43, 99, 100, 103, 104, 108, 115, 123, 147, 148.
- Lead reference for possible side reaction (nucleophilic aromatic substitution) during cleavage of resin-bound products 6 to product methyl esters of acylated unnatural amino acids 8 by methanolysis (MeOH, Et3N, 55 °C, 48 h):; a Plante J. P.; Jones P. D.; Powell D. R.; Glass T. E. Chem. Commun. 2003, 336–337. [DOI] [PubMed] [Google Scholar]; b This side reaction is reflected in D3 database products arising from the following reactants in Table 5, ref (2): 32, 54.
- Lead reference for possible side reaction (nucleophilic aromatic substitution) during cleavage of resin-bound products 6 to product methyl esters of acylated unnatural amino acids 8 by methanolysis (MeOH, Et3N, 55 °C, 48 h):; a Hamper B. C.; Leschinsky K. L.; Massey S. S.; Bell C. L.; Brannigan L. H.; Prosch S. D. J. Agric. Food Chem. 1995, 43, 219–228. [Google Scholar]; b This side reaction is reflected in D3 database products arising from the following reactant in Table 5, ref (2): 63.
- Lead reference for possible side reaction (boronate transesterification) during cleavage of resin-bound products 6 to product methyl esters of acylated unnatural amino acids 8 by methanolysis (MeOH, Et3N, 55 °C, 48 h):; a Carboni B.; Pourbaix C.; Carreaux F.; Deleuze H.; Maillard B. Tetrahedron Lett. 1999, 40, 7979–7983. [Google Scholar]; b This side reaction is reflected in D3 database products arising from the following reactant in Table 5, ref (2): 71.
- Lead reference for possible side reaction (lactone opening/closure) during cleavage of resin-bound products 6 to product methyl esters of acylated unnatural amino acids 8 by methanolysis (MeOH, Et3N, 55 °C, 48 h):; a Abe H.; Nishioka K.; Takeda S.; Arai M.; Takeuchi Y.; Harayama T. Tetrahedron Lett. 2005, 46, 3197–3200. [Google Scholar]; b This side reaction is reflected in D3 database products arising from the following reactant in Table 5, ref (2): 86.
- Lead reference for possible side reaction (phosphonate transesterification) during cleavage of resin-bound products 6 to product methyl esters of acylated unnatural amino acids 8 by methanolysis (MeOH, Et3N, 55 °C, 48 h):; a Wróblewski A. E.; Bak-Sypien I. I. Tetrahedron: Asymmetry 2007, 18, 520–526. [Google Scholar]; b This side reaction is reflected in D3 database products arising from the following reactant in Table 5, ref (2): 177.
- O’Donnell M. J.; Delgado F.; Hostettler C.; Schwesinger R. Tetrahedron Lett. 1998, 39, 8775–8778. [Google Scholar]
- The two-day alkylation was used to accommodate the student laboratory schedule.
- Dedicated to the following IUPUI students from Chemistry C344 (Organic Chemistry II Laboratory) who carried out the combinatorial syntheses described in this paper: Bunkowfst L. L.; Bwititi T.; Byun S.; Chang J. N.; Cox C. E.; de Oliveira C. D.; Farmer M. D.; Grunden K. A.; Hodgson W. D.; Kebede M. S.; Martin A. J.; Martin J. R.; Neeb Z. P.; Richey J. M.; Schroering A. M.; Skiles J. C.; Walsh J. L.; Weber J. M.; Williams V. C..
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.