

Abstract
Radical-mediated thiol−yne step-growth photopolymerizations are utilized to form highly cross-linked polymer networks. This reaction mechanism is shown to be analogous to the thiol−ene photopolymerization; however, each alkyne functional group is capable of consecutive reaction with two thiol functional groups. The thiol−yne reaction involves the sequential propagation of a thiyl radical with either an alkyne or a vinyl functional group followed by chain transfer of the radical to another thiol. The rate of thiyl radical addition to the alkyne was determined to be approximately one-third of that to the vinyl. Chain-growth polymerization of alkyne and vinyl functionalities was only observed for reactions in which the alkyne was originally in excess. Analysis of initial polymerization rates demonstrated a near first-order dependence on thiol concentration, indicating that chain transfer is the rate-determining step. Further analysis revealed that the polymerization rate scaled with the initiation rate to an exponent of 0.65, deviating from classical square root dependence predicted for termination occurring exclusively by bimolecular reactions. A tetrafunctional thiol was photopolymerized with a difunctional alkyne, forming an inherently higher cross-link density than an analogous thiol−ene resin, displaying a higher glass transition temperature (48.9 vs −22.3 °C) and rubbery modulus (80 vs 13 MPa). Additionally, the versatile nature of this chemistry facilitates postpolymerization modification of residual reactive groups to produce materials with unique physical and chemical properties.
Introduction
Radical-mediated thiol−ene polymerizations have experienced a renaissance in recent years as their advantages over the more common, traditional radical chain-growth polymerizations are exploited.1−7 Thiol−ene photopolymerizations of multifunctional monomers proceed via a radical-mediated, step-growth mechanism, involving alternation between propagation and chain transfer reactions. As such, the molecular weight increases geometrically, leading to a delayed gel-point conversion as compared to chain-growth polymerizations of multifunctional (meth)acrylate monomers. The step-growth mechanism leads to lower shrinkage stress and homogeneous networks containing a relatively low amount of unreacted, leachable compounds.4,5 Moreover, the ability of a peroxy radical to abstract a hydrogen from a thiol imparts thiol−ene polymerizations with exceptional resistance to oxygen inhibition.2,8
Networks formed by chain-growth polymerizations of multifunctional (meth)acrylates have many desirable mechanical and thermal properties, including high strength and high glass transition temperature (Tg),(9) primarily attributable to their high cross-link density. Conversely, thiol−ene photopolymerizations produce networks of relatively low cross-link density. The principal difference in cross-link densities between these two polymerization modes lies in the capacity of the vinyl group to act as either a mono- or difunctional moiety. During a chain-growth polymerization, an actively propagating species adds to a vinyl, which subsequently adds to another vinyl. Thus, each carbon−carbon double bond is replaced by two single bonds, establishing the vinyl group as difunctional.(10) Thiol−ene polymerizations proceed via the addition of a thiyl radical to a vinyl, which subsequently abstracts a hydrogen from a thiol, generating a thioether moiety and regenerating a thiyl radical (Scheme 1). The conversion of a vinyl to a single thioether establishes the vinyl group as monofunctional in a thiol−ene polymerization, limiting the ultimate cross-link densities of these materials.
Scheme 1. Sequential Addition and Hydrogen Abstraction Steps during a Thiol−Ene Polymerization.

Higher cross-link density thiol−ene based polymers are achieved either by increasing monomer functionality or by exploiting mixed-mode polymerizations (i.e., a hybrid step/chain-growth polymerization, where a degree of vinyl homopolymerization occurs).11−13 Nevertheless, increasing the monomer functionality generally leads to a corresponding molecular weight increase, moderating the intended cross-link density increase, whereas mixed-mode polymerizations frequently compromise, rather than maximize, the relative advantages of step- and chain-growth polymerizations. An alternative strategy for increasing the monomer functionality, while avoiding a concomitant molecular weight increase and maintaining the advantages of a pure step-growth polymerization, is to substitute the vinyl with an alkyne moiety, thus establishing a thiol−ynepolymerization.
The radical-mediated addition of thiocresol (a monothiol) to phenyl acetylene was observed in the 1930s.14,15 The thiol addition to alkynes has also been exploited as an approach to the synthesis of small vinyl sulfide and dithioether adducts.16−24 The thiol−yne radical addition mechanism has been exploited in the synthesis of linear polymers as well as for postsynthetic modification of polymers with pendant alkyne groups;25−27 however, its implementation in the polymerization of highly cross-linked networks has previously been unexplored. Here, we demonstrate the radical-mediated, step-growth photopolymerization of a thiol−yne resin, exploiting the bireactivity of the terminal alkyne to form a highly cross-linked, high glass transition temperature material. This material is compared to an analogous thiol−ene material. The initiation rate dependence and reaction orders of the photopolymerization are determined, and conditions under which homopolymerization of the unsaturated species occurs are identified.
Experimental Section
1,9-Decadiyne (DDY) was obtained from Acros Organics; 1,4-butanediol divinyl ether (BDDVE), 1-octyne, and butyl 3-mercaptopropionate were obtained from Sigma-Aldrich, and pentaerythritol tetra(3-mercaptopropionate) (PETMP) was obtained from Evans Chemetics. PETMP and DDY were chosen for their low molecular weight per functional group, low volatility, and miscibility. Irgacure 184 (1-hydroxycyclohexylphenyl ketone), an ultraviolet-active radical generating photoinitiator, was obtained from Ciba Specialty Chemicals. Structures of the materials used are shown in 1. All chemicals were used as received.
Figure 1.

Materials used: (1) DDY, (2) BDDVE, (3) PETMP, (4) Irgacure 184, (5) 1-octyne, (6) butyl 3-mercaptopropionate, and (7) BOETP.
Butyl 3-mercaptopropionate (1.505 g, 0.0093 mol) (Sigma-Aldrich) was added to a large excess of 1-octyne (4.305 g, 0.0391 mol) (Sigma-Aldrich) in a 20 mL vial. This solution was formulated with 0.024 g of Irgacure 184, stirred, and irradiated using an EXFO Acticure 4000 high-pressure mercury vapor short arc lamp equipped with a 320−500 nm bandpass filter. After the reaction proceeded to complete conversion (as determined by the disappearance of the thiol peak observed in the IR spectrum at 2570 cm−1), residual 1-octyne was removed under reduced pressure and subsequently bulb-to-bulb distilled under vacuum to yield 1.796 g of butyl 3-(oct-1-enylthio)propanoate (BOETP) (70.9% yield). Structural identification of BOETP (shown in 1) was confirmed using 1H NMR (see Supporting Information).
Fourier transform infrared (FTIR) spectroscopy was performed using a Nicolet Magna-IR 750 Series II FTIR spectrometer to monitor evolution of the functional group concentrations during photopolymerizations. Spectra were collected at a resolution of 2 cm−1 and a rate of 2.5 scans/s. Formulated resins were sandwiched between two NaCl crystals separated by polyester shims of known thickness and irradiated in situ using an EXFO Acticure 4000 high-pressure mercury vapor short arc lamp equipped with a 365 nm narrow bandpass filter. Light intensity was measured using an International Light IL1400A hand-held radiometer equipped with a GaAsP detector (model SEL005), a wide bandpass filter (model WBS320), and a quartz diffuser (model W). All experiments were performed in triplicate.
Alkyne concentrations were determined by integrating the peaks centered at 3288 or 2116 cm−1, corresponding to the propargyl C−H stretch or propargyl C≡C stretch, respectively. The peak used for any particular analysis was chosen by determining which of the two alkyne peaks maximized signal-to-noise while remaining in the linear response region of the detector throughout the polymerization. Thiol concentrations were determined by integrating the peak centered at 2570 cm−1, corresponding to the SH stretch.
The vinyl sulfide C=C stretch molar absorptivity for the peak centered at 1609 cm−1 was determined using BOETP as a model compound (see Supporting Information). The vinyl sulfide concentration throughout the polymerization was subsequently determined by integrating the peak centered at 1609 cm−1 and, given the sample path length, utilizing the Beer−Lambert law.
Dynamic mechanical analyses (DMA) were performed in triplicate using a TA Instruments Q800 scanning at 1 °C/min from −60 to 100 °C. The glass transition temperature (Tg) was determined as the temperature at the peak in the tan δ curve.
Results and Discussion
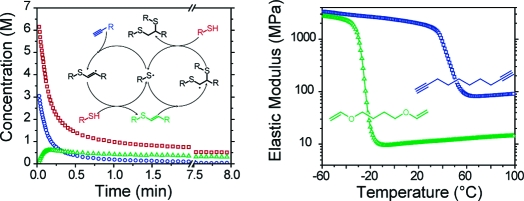
Alkyne, vinyl sulfide, and thiol concentrations were determined by IR spectroscopy during the photopolymerization of a stoichiometrically balanced thiol−yne monomer mixture, where two thiols are present for every alkyne (shown in 2). Rapid consumption of both the alkyne and thiol functional groups is observed in the first several minutes of the reaction while the intermediate vinyl sulfide species is formed and subsequently consumed during the polymerization. As the reaction proceeds, the vinyl sulfide concentration achieves a maximum at ∼0.2 min of irradiation, beyond which time its consumption exceeds its formation resulting from the thiol addition to alkyne.
Figure 2.

Concentrations of the various reactive species, thiols (◻), alkyne (○), and vinyl sulfide (Δ), during curing of a stoichiometrically balanced (2 thiols/alkyne) PETMP/DDY sample formulated with 3 wt % Irgacure 184 and irradiated at 365 nm, 30 mW/cm2.
An idealized thiol−yne step-growth mechanism, paralleling that of the thiol−ene mechanism (Scheme 1), is presented in Scheme 2. Similar to the thiol−ene reaction, the thiol−yne polymerization proceeds by the addition of a thiyl radical to an alkyne with the resulting carbon-centered radical subsequently abstracting a hydrogen from another thiol, generating a vinyl sulfide moiety and regenerating a thiyl radical. However, unlike the thioether generated by the thiol−ene reaction, the vinyl sulfide is capable of undergoing further reaction through addition of a second thiyl radical to the vinyl sulfide.(28) Thus, after complete reaction, each alkyne functional group has combined with two thiols to generate a dithioether, thus establishing the alkyne as difunctional in thiol−yne step-growth polymerizations. The thiol−yne polymerization is analogous to the epoxy−amine polymerization, where a primary amine undergoes an addition reaction with an epoxide, producing a secondary amine that is able to undergo a subsequent addition reaction with a second epoxide.
Scheme 2. Sequential Addition and Hydrogen Abstraction Steps of (1) Primary Alkyne and the (2) Subsequent Vinyl Sulfide during a Thiol−Yne Polymerization.

Measurements of the functional group concentrations during stoichiometrically unbalanced reactions demonstrate that when the alkyne is in excess, the accumulation of vinyl sulfide is more pronounced than in stoichiometrically balanced reactions (3), whereas no vinyl sulfide accumulation is observed when the thiol is initially in excess (3).
Figure 3.

Concentrations of the various reactive species, thiol (◻), alkyne (○), and vinyl sulfide (Δ), during curing of two different initial stoichiometric ratios of reactive groups including (A) 6:5 thiol:alkyne (excess alkyne) and (B) 10:3 thiol:alkyne (excess thiol). Samples were formulated with 3 wt % Irgacure 184 and irradiated at 365 nm, 30 mW/cm2.
The addition of thiol to alkyne and the subsequent addition of thiol to vinyl sulfide, as described in Scheme 2, can be expressed by the reactive species balances
and
where kP,1 and kP,2 are the propagation rate constants for addition of a thiyl radical to alkyne and vinyl sulfide, respectively. Division of eq 2 by eq 1 yields
For kP,2/kP,1 ≪ 1, the rate of alkyne disappearance is equal to the rates of thiol loss and vinyl creation. Conversely, for kP,2/kP,1 ≫ 1, vinyl sulfides are consumed immediately upon creation; thus, a significant vinyl sulfide concentration would not be observed. For the stoichiometrically balanced PETMP/DDY monomer mixture (as shown in 2), the rate constant ratio (kP,2/kP,1) was determined to be ∼3 by fitting the numerical integration of eq 3 up to 30% thiol conversion (see Supporting Information).
The gel-point conversion for a step-growth polymerization between two monomers with degrees of functionality fA and fB is predicted by the Flory−Stockmayer (FS) equation
where pg is the gel-point conversion and r is the stoichiometric ratio.29,30 Assuming equal reactivity of the alkyne and subsequent vinyl sulfide, the gel-point conversion for a stoichiometrically balanced, step-growth polymerization between a tetrathiol and a dialkyne (here, the diyne would behave as a tetrafunctional monomer) is 33%. Dušek, however, demonstrated deviation from the gel-point conversion predicted by the FS equation for the step-growth mechanism where consecutive additions to a polymerizable moiety are of unequal reactivity.(31) Systems in which the rate of initial addition is faster than the subsequent addition (i.e., kP,2/kP,1 < 1) have a higher gel-point conversion than predicted by the FS equation; the opposite trend is observed when the rate of the subsequent addition exceeds that of the initial (i.e., kP,2/kP,1 > 1). Thus, the observed rate constant ratio for PETMP/DDY (i.e., kP,2/kP,1 ∼ 3) indicates that a gel-point conversion somewhat lower than the Flory−Stockmayer prediction will occur; however, as kP,2/kP,1 is still of the same order of magnitude, the deviation from this prediction is likely small. The predicted gel-point conversion of 33% is significantly higher relative to multifunctional (meth)acrylate networks, which have been seen to gel below 5% conversion.9,32,33 Thiol−ene polymerizations have demonstrated considerably reduced shrinkage stress in comparison with (meth)acrylate networks, attributed in part to their delayed gelation; thus, thiol−ynes are likely to exhibit similarly low shrinkage stress behavior.
Scheme 2 represents an idealized step-growth mechanism and neglects potential chain-growth addition reactions between unsaturated species. To understand better the extent to which such behavior is occurring, the total concentration of polymerizable π-bonds, defined as twice the alkyne concentration plus the vinyl concentration, is plotted against the thiol concentration for polymerizations of various PETMP/DDY ratios (4). At high thiol concentrations, the functional group concentrations follow ideal step-growth evolution (as represented by the solid line); however, in “thiol-limited” samples, where the thiol is the limiting reactant, the π-bond concentration deviates from this ideal behavior. The greater than expected disappearance of unsaturated species during polymerization at low thiol concentration is attributed to consumption of alkyne or vinyl sulfide functional groups by chain-growth addition mechanisms, i.e., homo- or copolymerization. Moreover, a higher degree of homo- or copolymerization is observed at raised relative alkyne concentrations. The tendency toward homo- or copolymerization is thus influenced by the increased ratio of unsaturated species to the thiol, as the chain-growth addition reaction dominates over the step-growth hydrogen abstraction. Conversely, the stoichiometrically balanced and alkyne-limited samples (data not shown) correspond well with those predicted for an ideal step-growth mechanism.
Figure 4.

Total π-bond concentration (twice the alkyne concentration plus the vinyl concentration) versus thiol concentration for various initial ratios of thiols to alkynes: 2:1 (Δ), 6:5 (○), and 2:3 (◻). Solid lines represent ideal, equal consumption of thiols and π-bonds. Samples were formulated with 3 wt % Irgacure 184 and irradiated at 365 nm, 30 mW/cm2.
In the absence of homo- or copolymerization of unsaturated species, such as observed for stoichiometrically balanced reactions, the thiol consumption rate can be equated to the polymerization rate, Rp. The polymerization rate scales with the initial reactant concentrations according to
where α and β are the reaction orders relative to alkyne and thiol concentrations, respectively. To avoid diffusion limitations associated with cross-linking during polymerization of multifunctional monomers, reaction orders for the thiol−yne system were determined using solutions of 1-octyne and butyl 3-mercaptopropionate in ethylene glycol diacetate. Performing experiments using ethylene glycol diacetate as an inert solvent enables the alkyne and thiol concentrations to be varied independently and over broader ranges than achievable in bulk. Regression analysis (5) demonstrates near zero-order reaction with respect to the alkyne (i.e., α = 0.10 ± 0.02) and near first-order with respect to the thiol (i.e., β = 0.80 ± 0.02), indicating that the kinetics of the initial thiol addition to an alkyne are limited by the chain transfer step.(34) For radical thiol−ene reactions, it has been suggested that the propagation rate is positively correlated to the electron density of the vinyl group while the chain transfer rate is negatively correlated to the intermediate radical stability.(34) Radical thiol−ene polymerizations limited by chain transfer, such as the thiol−allyl ether polymerization, generally involve a relatively stable radical that is slow to abstract a thiol hydrogen. Sulfide substituents are believed to have a stabilizing effect on neighboring radicals,35,36 which may explain the rate-limiting chain transfer step in the thiol−yne polymerization.
Figure 5.

Log−log plot of initial polymerization rate versus initial concentration of thiol (◻, at 1.0 M alkyne) and alkyne (○, at 2.0 M thiol). Samples were formulated with 0.0490 M Irgacure 184 and irradiated at 365 nm, 30 mW/cm2. The respective reaction orders are denoted by the slope of the lines of best fit.
An examination of the irradiation intensity effect on the conversion rate for the photopolymerization of PETMP/DDY is presented in 6. According to classical bimolecular termination (by either recombination or disproportionation), the polymerization rate is predicted to be proportional to the square root of the initiation rate (Ri) and, by extension, the irradiation intensity. As shown in 6, for photopolymerizations of PETMP/DDY at a stoichiometically balanced ratio, the polymerization rate scales with the initiation rate to the 0.65 ± 0.03 power, demonstrating significant divergence from the classical behavior. Nonclassical exponents greater than 0.5 have been observed in cross-linking chain-growth polymerizations where they were attributed to unimolecular termination associated with trapping of the reactive center in the polymer matrix.37,38 Interestingly, the molecular weight increases geometrically in step-growth polymerizations, likely negating any entrapment as a potential avenue for unimolecular termination, particularly for these initial rates measured at low conversions. Deviation from the classical model has similarly been observed for various thiol−ene formulations(39) and may simply be characteristic of radical-mediated step-growth polymerizations, particularly those involving thiols.
Figure 6.

(A) Thiol conversion versus time and (B) versus time scaled assuming Rp ∼ Ri0.65 for PETMP/DDY, formulated with 3.0 wt % Irgacure 184 and irradiated at various light intensities: 3.14 (∇), 10.2 (Δ), 29.7 (○), and 97.2 mW/cm2 (◻). For clarity, not all data points are plotted.
Dynamic mechanical analysis (DMA) was performed on PETMP/DDY and, for comparison, a tetrathiol/divinyl ether formulation using a divinyl ether monomer of similar molecular weight to DDY (i.e., PETMP/BDDVE), as shown in 7 and Table 1. BDDVE was used as a difunctional analogue for DDY as 1,9-decadiene is immiscible with PETMP; however, the mechanical properties of fully cured PETMP/BDDVE are representative of those generally found in flexible thiol−ene polymer networks.12,13 Each sample exhibits a similar transition from a glassy regime, where the elastic modulus is greater than 1 GPa, to a rubbery regime, where the modulus is proportional to the cross-link density, marked by a characteristic glass transition temperature, Tg. The elastic modulus of PETMP/DDY cured at room temperature displays a glass transition beginning at 25 °C; as seen in 2, this sample contains residual thiol and vinyl sulfide after the cessation of polymerization. As the polymerization of PETMP/DDY proceeds, there is a concomitant increase in its Tg; when the Tg reaches the polymerization temperature, the mobility of unreacted functional groups is significantly impaired, i.e., vitrification, which limits the final conversion. To overcome vitrification and reach higher functional group conversions, photopolymerizations of a PETMP/DDY sample were performed at an elevated temperature of 80 °C, and polymer properties are summarized in Table 1.
Figure 7.

(A) Elastic modulus and (B) tan δ versus temperature for stoichiometrically balanced polymerization of PETMP/DDY reacted at 25 (○) and 80 °C (◻) and PETMP/BDDVE reacted at 25 °C (Δ). Samples were formulated with 3 wt % Irgacure 184 and irradiated at 365 nm, 10 mW/cm2, for 60 min.
Table 1. Summary of Material Properties for PETMP/DDY and PETMP/BDDVE Samples Used in Figure 7.
| system | Tg (°C) | tan δ fwhm (°C) | E′ at 65 °C (MPa) | calculated cross-link density (M) |
|---|---|---|---|---|
| PETMP/DDY,80 °C | 48.9 ± 0.9 | 17.7 ± 0.2 | 80 ± 1 | 9.6 ± 0.2 |
| PETMP/DDY,25 °C | 40.7 ± 0.2 | 17.2 ± 0.5 | 69 ± 2 | 8.4 ± 0.2 |
| PETMP/BDDVE | −22.3 ± 0.5 | 9.3 ± 0.3 | 13 ± 1 | 1.5 ± 0.1 |
A marked difference is observed in the rubbery modulus that is obtained from the traditional thiol−ene network, PETMP/BDDVE, as compared with the thiol−yne network, PETMP/DDY. The cross-link density, ρx, is estimated from the rubbery modulus using the theory of rubber elasticity
where ν is Poisson’s ratio, R is the gas constant, T is the temperature, and E is the elastic modulus.(10) The estimated cross-link densities, assuming the materials are incompressible (ν = 1/2), are presented in Table 1. The cross-link density of fully cured PETMP/DDY is more than 6 times that of the fully cured PETMP/BDDVE, which is attributed to the difunctional alkyne in the thiol−yne mechanism (Scheme 2) as compared to the monofunctional vinyl in the thiol−ene mechanism (Scheme 1).
The Tg of the more completely reacted PETMP/DDY network is ∼70 °C higher than the PETMP/BDDVE network. The Tg in polymer networks is primarily determined by the stiffness of the polymer backbone and the cross-link density.(40) Both DDY and BDDVE possess similarly nonrigid, saturated, nonfunctionalized backbones, which are presumably of similar stiffness. The observed increase in glass transition temperature is thus attributed to the higher cross-link density of PETMP/DDY.
The breadth of tan δ, as determined by the full width at half-maximum (fwhm), for the PETMP/DDY network is approximately twice that for the PETMP/BDDVE network; however, this breadth is still very narrow compared to chain-growth networks, indicative of a much more uniform network structure.13,41 While highly cross-linked networks are readily produced via chain-growth polymerizations, such materials typically exhibit a spatially heterogeneous cross-link density.42−44 Epoxy−amine networks are typically homogeneous owing to their step-growth polymerization mechanism. Furthermore, they display a high cross-link density as primary amines are difunctional when polymerized with epoxy functional groups. Networks formed by the thiol−yne step-growth polymerization demonstrated here are analogous to those formed by the epoxy−amine mechanism, simultaneously exhibiting homogeneity and high cross-link density, while their capacity for photoinitiation allows for spatial and temporal control over the polymerization.
Thiol−yne polymerizations produce homogeneous, high-Tg networks. Moreover, residual functionalities in fully cured networks are readily introduced through an imbalance in the initial monomer ratio (see 3), allowing for highly tailorable materials through subsequent chemical modification of combinations of residual alkyne, vinyl, and thiol reactive groups. Thiols participate in a wide variety of reactions including addition to epoxides, Michael addition, and radical-mediated thiol−ene reactions, while the copper-catalyzed 1,3-cycloaddition “click” reaction proceeds rapidly and robustly between azides and alkynes. Thus, various hybrid polymerization schemes or postcuring modifications to thiol−yne networks using any combination of such semiorthogonal reactions can be used to produce materials with unique physical and chemical properties, potentially establishing a new class of functional and structurally tailored materials.
Conclusions
Photopolymerizations of thiol−yne systems yield high cross-link density networks, much like multifunctional chain-growth polymerizations, while largely maintaining a step-growth mechanism that results in a delayed gel-point and network homogeneity, as commonly observed in thiol−ene polymerizations. During photopolymerization, the emergence of a vinyl sulfide intermediate functionality was observed, and the rate of thiol addition to this vinyl sulfide was ∼3 times that of the thiol addition to the alkyne. This ratio of reaction rates is expected to produce a slightly reduced gel-point conversion as compared to that predicted by the Flory−Stockmayer equation but is still significantly delayed relative to typical multifunctional chain-growth polymerizations. The thiol−yne reaction rate was found to be relatively independent of alkyne concentration and dependent on the thiol concentration to an exponent of 0.8 as indicative of chain transfer being the rate-determining step in the reaction. Furthermore, when the polymerization became thiol-limited, deviation from the step-growth mechanism was observed where homo- or copolymerization of the unsaturated species occurred, further indicating the thiol-controlled nature of the polymerization.
The polymerization rate scaled with the initiation rate with an exponent of 0.65, demonstrating a departure from classical bimolecular termination kinetics, which may be characteristic of radical mediated step-growth polymerizations. Thiol−yne and thiol−ene networks were polymerized using dialkyne and divinyl monomers of similar molecular weight and demonstrated dramatically different thermomechanical behavior. The cross-link density of the fully cured thiol−yne network was over 6 times greater than the equivalent thiol−ene network, leading to an increase of 70 °C in the glass transition temperature and demonstrating the utility for thiol−yne networks as structural materials. Moreover, residual functionalities, introduced via off-stoichiometric polymerizations of the monomers, can be exploited for postpolymerization network modification to produce materials with unique physical and chemical properties, establishing a new class of functional and structurally tailored materials.
Acknowledgments
This work was supported by NSF Grants CTS 0626023 and EEC 444771 and NIH Grant DE10959.
Supporting Information Available
Experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Woods J. G. In Radiation Curing: Science and Technology; Pappas S. P., Ed.; Plenum Press: New York, 1992; pp 333−398. [Google Scholar]
- Jacobine A. F. In Radiation Curing in Polymer Science and Technology; Fouassier J. P., Rabek J. F., Eds.; Elsevier Applied Science: London, 1993; Vol. III, pp 219−268. [Google Scholar]
- Natarajan L. V.; Shepherd C. K.; Brandelik D. M.; Sutherland R. L.; Chandra S.; Tondiglia V. P.; Tomlin D.; Bunning T. J. Chem. Mater. 2003, 15, 2477–2484. [Google Scholar]
- Lu H.; Carioscia J. A.; Stansbury J. W.; Bowman C. N. Dent. Mater. 2005, 21, 1129–1136. [DOI] [PubMed] [Google Scholar]
- Carioscia J. A.; Lu H.; Stansbury J. W.; Bowman C. N. Dent. Mater. 2005, 21, 1137–1143. [DOI] [PubMed] [Google Scholar]
- Hagberg E. C.; Malkoch M.; Ling Y.; Hawker C. J.; Carter K. R. Nano Lett. 2007, 7, 233–237. [DOI] [PubMed] [Google Scholar]
- Hoyle C. E.; Lee T. Y.; Roper T. J. Polym. Sci., Part A: Polym. Chem. 2004, 42, 5301–5338. [Google Scholar]
- Szmant H. H.; Mata A. J.; Namis A. J.; Panthananickal A. M. Tetrahedron 1976, 32, 2665–2680. [Google Scholar]
- Kloosterboer J. G. Adv. Polym. Sci. 1988, 84, 1–61. [Google Scholar]
- Flory P. J.Principles of Polymer Chemistry; Cornell University Press: Ithaca, NY, 1953. [Google Scholar]
- Reddy S. K.; Cramer N. B.; Kalvaitas M.; Lee T. Y.; Bowman C. N. Aust. J. Chem. 2006, 59, 586–593. [Google Scholar]
- Lee T. Y.; Carioscia J.; Smith Z.; Bowman C. N. Macromolecules 2007, 40, 1473–1479. [Google Scholar]
- Senyurt A. F.; Wei H. Y.; Hoyle C. E.; Piland S. G.; Gould T. E. Macromolecules 2007, 40, 4901–4909. [Google Scholar]
- Finzi C. Gazz. Chim. Ital. 1930, 60, 798–811. [Google Scholar]
- Kohler E. P.; Potter H. J. Am. Chem. Soc. 1935, 57, 1316–1321. [Google Scholar]
- Bader H.; Cross L. C.; Heilbron I.; Jones E. R. H. J. Chem. Soc. 1949, 619–623. [Google Scholar]
- Bader H. J. Chem. Soc. 1956, 116–121. [Google Scholar]
- Kharasch N.; Meyers C. Y.. The Chemistry of Organic Sulfur Compounds, 1st ed.; Pergamon Press: Norwich, 1966; Vol. 2. [Google Scholar]
- Graham D. M.; Soltys J. F. Can. J. Chem. 1969, 47, 2529–2533. [Google Scholar]
- Ichinose Y.; Wakamatsu K.; Nozaki K.; Birbaum J. L.; Oshima K.; Utimoto K. Chem. Lett. 1987, 16, 1647–1650. [Google Scholar]
- Benati L.; Montevecchi P. C.; Spagnolo P. J. Chem. Soc., Perkin Trans. 1 1991, 2103–2109. [Google Scholar]
- Benati L.; Capella L.; Montevecchi P. C.; Spagnolo P. J. Org. Chem. 1995, 60, 7941–7946. [Google Scholar]
- Montevecchi P. C.; Navacchia M. L. Tetahedron Lett. 1998, 39, 9077–9080. [Google Scholar]
- Yadav J. S.; Reddy B. V. S.; Raju A.; Ravindar K.; Baishya G. Chem. Lett. 2007, 36, 1474–1475. [Google Scholar]
- Ohashi T.; Kobayashi E.; Jinbo T.; Furukawa J. J. Polym. Sci., Part A: Polym. Chem. 1997, 35, 1621–1625. [Google Scholar]
- Kobayashi E.; Yoshino T.; Aoshima S.; Furukawa J. J. Polym. Sci., Part A: Polym. Chem. 1995, 33, 2403–2414. [Google Scholar]
- Ochiai B.; Tomita I.; Endo T. Polym. Bull. 2004, 51, 263–269. [Google Scholar]
- Skvortsova G. G.; Trzhtsinskaya B. V.; Teterina L. F.; Voronov V. K. Chem. Heterocycl. Compd. 1976, 12, 1278–1280. [Google Scholar]
- Flory P. J. J. Am. Chem. Soc. 1941, 63, 3083–3090. [Google Scholar]
- Stockmayer W. H. J. Chem. Phys. 1943, 11, 45–55. [Google Scholar]
- Dusek K.; Ilavsky M.; Lunak S. J. Polym. Sci., Part C: Polym. Symp. 1975, 53, 29–44. [Google Scholar]
- Walling C. J. Am. Chem. Soc. 1945, 67, 441–447. [Google Scholar]
- Rey L.; Galy J.; Sautereau H. Macromolecules 2000, 33, 6780–6786. [Google Scholar]
- Cramer N. B.; Reddy S. K.; O’Brien A. K.; Bowman C. N. Macromolecules 2003, 36, 7964–7969. [Google Scholar]
- Price C. C.; Zomlefer J. J. Am. Chem. Soc. 1950, 72, 14–17. [Google Scholar]
- Viehe H. G.; Janousek Z.; Merenyi R.. Substituent Effects in Radical Chemistry: Proceedings of the NATO Advanced Research Workshop on Substituent Effects in Radical Chemistry; Reidel: Louvain-la-Neuve, Belgium, 1986. [Google Scholar]
- Selli E.; Bellobono I. R. In Radiation Curing in Polymer Science and Technology; Fouassier J. P., Rabek J. F., Eds.; Elsevier Applied Science: London, 1993; Vol. III, pp 1−32. [Google Scholar]
- Pavlinec J.; Moszner N. J. Appl. Polym. Sci. 2003, 89, 579–588. [Google Scholar]
- Scott T. F.; Kloxin C. J.; Draughon R. B.; Bowman C. N. Macromolecules 2008, 41, 2987–2989. [Google Scholar]
- Halary J. L.; Cukierman S.; Monnerie L. Bull. Soc. Chim. Belg. 1989, 98, 623–634. [Google Scholar]
- Scott T. F.; Cook W. D.; Forsythe J. S. Eur. Polym. J. 2002, 38, 705–716. [Google Scholar]
- Tobita H. Polymer 1992, 33, 3647–3657. [Google Scholar]
- Anseth K. S.; Anderson K. J.; Bowman C. N. Macromol. Chem. Phys. 1995, 197, 833–848. [Google Scholar]
- Pavlinec J.; Moszner N. J. Appl. Polym. Sci. 1997, 65, 165–178. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.