

Abstract
Synaptotagmin and complexin regulate SNARE-mediated synaptic vesicle exocytosis. It has been proposed that complexin clamps membrane fusion and that Ca2+-synaptotagmin displaces complexin from SNARE complexes to relieve this clamping activity. Using a reconstituted system, we demonstrate that complexin and synaptotagmin simultaneously bind to neuronal SNARE complexes and that both apo-synaptotagmin and complexin inhibit SNARE-mediated membrane fusion. Moreover, the clamping ability of apo-synaptotagmin occluded the clamping activity of complexin until the arrival of a Ca2+ trigger, at which point synaptotagmin accelerated fusion while high concentrations of complexin inhibited fusion. Thus, the inhibitory patterns of synaptotagmin and complexin are different, suggesting that SNAREs assemble into distinct states along the fusion pathway. These data also suggest that during synaptotagmin-regulated vesicle−vesicle fusion, complexin does not function as a fusion clamp that is relieved by Ca2+-synaptotagmin.
Rapid point-to-point communication in the nervous system is mediated by Ca2+-triggered exocytosis of neurotransmitter-filled synaptic vesicles (SVs). The speed of this fusion event suggests that the membrane fusion machinery is poised and ready to respond to Ca2+ with submillisecond kinetics (1). A number of the components of this machine have been identified. It is now established that soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins form the core of the SV fusion apparatus. It is believed that trans interactions between the vesicle SNARE (v-SNARE) VAMP 2/synaptobrevin 2 and target membrane SNAREs (t-SNAREs) syntaxin 1A and SNAP-25B serve to directly catalyze fusion of SVs with the plasma membrane. However, little is known concerning the mechanisms by which accessory proteins control the SNARE complex to regulate fusion.
Considerable attention has been placed on identifying a molecular “clamp” that prevents SNARE-mediated fusion under resting conditions (i.e., in the absence of a Ca2+ signal); Ca2+ would then serve to relieve the clamp, thus allowing exocytosis to proceed. Recent efforts to identify this clamp have focused on the interplay between two proteins that interact with neuronal SNAREs at various stages of SNARE complex assembly: synaptotagmin I (syt) and members of the complexin family of proteins. Syt is a Ca2+ binding protein found on SVs and serves as the Ca2+ sensor for synaptic vesicle exocytosis (2). Complexins are small, soluble proteins expressed in numerous cell types though complexins I and II are predominantly found in neurons where they serve a regulatory role during Ca2+-triggered neurotransmitter release (3).
A handful of recent reports concluded that complexins clamp SNARE-catalyzed membrane fusion (4−8) and that Ca2+-syt displaces complexin from SNARE complexes to alleviate the clamp (4−6,8,13). These results support a model of SV exocytosis in which complexins plays a negative regulatory role under resting conditions (i.e., prior to the Ca2+ trigger) and also suggest that syt acts simply as a positive regulator in response to Ca2+. However, it has also been reported that complexins are not displaced from SNARE complexes by syt (6,9) and that complexins are not clamps, but instead play a positive role in SNARE-mediated fusion (10−12). Hence, a consensus about the physiological and biochemical function of complexin has not been reached. Here, we have used a reconstituted system to analyze directly the functional and biochemical roles of full-length complexin I (cpx-I), and a complexin I fragment [cpx-I(26−83)] that constitutes the proposed inhibitory domain of cpx-I used in recent studies (5,8,13).
We first characterized the interaction of complexin with membrane-embedded SNAREs. We assembled ternary SNARE complexes, the favored binding partner for complexin (14), on liposomes (Figure S1a,b of the Supporting Information). Saturation of membrane-embedded t-SNARE heterodimers with the cytoplasmic domain of synaptobrevin 2 was confirmed using a coflotation assay. Next, we saturated fully assembled ternary SNARE complexes with cpx-I or cpx-I(26−83) (Figure S1c,d of the Supporting Information). Both cpx-I and cpx-I(26−83) preferentially bound ternary SNARE complexes as compared to t-SNARE heterodimers, although some degree of full-length cpx-I binding to t-SNAREs was observed (Figure S1d of the Supporting Information).
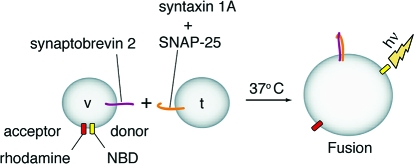
Having established saturable binding of cpx-I and cpx-I(26−83) to assembled SNARE complexes, we carried out in vitro membrane fusion assays to assess the impact of these binding interactions on the function of SNARE proteins (Figure S2a of the Supporting Information). It is important to note that for all fusion experiments in this study, t-SNARE and v-SNARE vesicles were combined in a 9:1 ratio to allow for multiple rounds of fusion (15). Low concentrations of cpx-I stimulated SNARE-mediated fusion to a small but reproducible extent, whereas higher concentrations of cpx-I inhibited fusion in a dose-dependent manner (∼50% inhibition at 60 μM cpx-I) (Figure S2b,c of the Supporting Information). Cpx-I(26−83) failed to show any stimulation of fusion and only weakly inhibited fusion at the highest concentration that was tested (Figure S2d,e of the Supporting Information). We note that saturation occurs at different concentrations of cpx-I for the binding and fusion reactions. This apparent discrepancy is likely due to adding cpx-I to preformed cis-SNARE complexes in the binding assay as compared to trans-SNARE complexes in the fusion assay (see the Supporting Information for more details).
Next, we addressed whether syt and cpx-I simultaneously bind SNAREs and whether cpx-I-mediated inhibition can be overcome by Ca2+-syt (as proposed in refs (4)−(6) and (8)). We titrated syt onto ternary SNARE complexes that had been saturated with cpx-I (Figure 1a,b) in the coflotation assay. For these experiments, we used liposomes that lacked phosphatidylserine (PS). t-SNARE vesicles lacking PS must be used for this experiment, in contrast to ref (6), because Ca2+-syt binds with high affinity to PS (16). With the omission of PS, any coflotation of syt with the vesicles must be solely due to direct interactions between syt and SNARE proteins. Even when present in a 3-fold molar excess of cpx-I, syt had no effect on the extent of cpx-I binding either in the absence or in the presence of Ca2+ (Figure 1b). Similarly, saturation of ternary SNARE complexes with cpx-I did not affect the extent of syt binding (Figure 1b). These data unambiguously demonstrate that syt and complexin simultaneously bind SNARE complexes that have been assembled onto liposomes [similar results were obtained with cpx-I(26−83) (Figure S3 of the Supporting Information)].
Figure 1.

Binding of cpx-I and syt to membrane-embedded ternary SNARE complexes. (a) Diagram of the flotation assay used to monitor binding interactions (see also Figure S1 of the Supporting Information). (b) Cpx-I (10 μM) and increasing concentrations of syt were added to ternary SNARE complexes individually or together and then the complexes subjected to flotation. Vesicles harbored 70% PC and 30% PE. Binding was monitored in the absence or presence of Ca2+ (1 mM), and proteins were visualized by being stained with Coomassie blue. (c) Experiments in panel b were repeated using vesicles harboring PS (55% PC, 30% PE, and 15% PS). All gels are representative from n ≥ 3.
It has been suggested that membranes must contain anionic phospholipids, such as PS, for Ca2+-syt to displace cpx-I from membrane-embedded SNARE complexes (4,13). Therefore, we repeated the competition experiments described above using liposomes that harbored PS (Figure 1c). As expected, Ca2+-syt exhibited even more robust coflotation with these vesicles due to high-affinity binding of Ca2+-syt to PS. Even under these conditions, syt had no effect on the interaction of cpx-I with assembled SNARE complexes. If anything, the presence of Ca2+-syt may have enhanced slightly the ability of cpx-I to bind SNARE complexes [similar results were obtained with cpx-I(26−83) (Figure S3 of the Supporting Information)]. Thus, Ca2+-syt and cpx-I [or cpx-I(26−83) which was used in refs (5), (8), and (13)] interact with SNARE complexes in a noncompetitive manner under the conditions of our binding assays. These data argue against a complexin clamping mechanism that is displaced by Ca2+-syt (4−6,8,13).
In the final series of experiments, cpx-I was titrated into an in vitro fusion assay in the presence of a saturating concentration of syt (30 μM syt; see ref (17)). Fusion was monitored in the absence of Ca2+ (Figure 2a,b), and after addition of 1 mM Ca2+ to reaction mixtures at 20 min (Figure 2c,d) (similar to a method used in ref (6)). Under these conditions, if cpx-I acts as a clamp as reported in refs (4)−(6), (8), and (13), it would be expected to inhibit fusion only in the absence of Ca2+ and not after a Ca2+ trigger since Ca2+-syt would overcome the cpx-I clamp to drive fusion. As shown in Figure 2a,b, in the absence of Ca2+, syt alone inhibited fusion by ∼25%. Addition of cpx-I to reaction mixtures containing apo-syt (Ca2+ free) resulted in a small but reproducible amount of stimulation at low cpx-I concentrations analogous to the findings obtained using SNAREs alone in the fusion assay (Figure 2a,b and Figure S2b,c of the Supporting Information). However, in contrast to what was observed when cpx-I was added to reaction mixtures containing SNAREs alone, addition of higher concentrations of cpx-I to reaction mixtures containing apo-syt only modestly inhibited fusion beyond what was already achieved by apo-syt alone (Figure 2a,b). Thus, cpx-I does not appear to play a major role in clamping SNARE-mediated vesicle−vesicle fusion prior to a Ca2+ signal when syt is also present (see the Supporting Information for more details). In duplicate samples, we triggered syt-stimulated fusion by adding Ca2+ at 20 min (Figure 2c). From these traces, it is evident that Ca2+-syt stimulates fusion but also that high concentrations of cpx-I inhibit Ca2+-syt-triggered fusion (Figure 2c,d).
Figure 2.

In vitro membrane fusion regulated by cpx-I and syt. (a) Increasing concentrations of cpx-I were added to fusion reaction mixtures containing 30 μM syt. t+v denotes fusion reaction mixtures lacking cpx-I and syt. As a control, the cytoplasmic domain of synaptobrevin 2 (cd-syb, 10 μM) was added to t+v to inhibit SNARE-mediated fusion. Fusion was monitored for 120 min at 37 °C, normalized to the maximum donor fluorescence signal (% Max. fluorescence), and plotted as a function of time. Reactions were carried out in 0.2 mM EGTA. (b) The final extent of fusion at each cpx-I concentration tested in panel a was normalized to the final extent of fusion obtained by t+v (% t+v). (c) Experiments were conducted as described for panel a except Ca2+ (1 mM) was added to reaction mixtures at 20 min. (d) Data from panel c were normalized as in panel b. The inset shows the final extent of fusion at each cpx-I concentration tested in panels a and c normalized to the extent of fusion obtained in reaction mixtures containing syt, but lacking cpx-I (% t+v+syt). All fusion traces are representative from n ≥ 3. Data in panels b and d represent the mean ± the standard error of the mean from n ≥ 3.
Therefore, while equal concentrations of apo-syt and cpx-I clamp fusion to similar extents (see 30 μM cpx-I in Figure S2b,c of the Supporting Information as compared to 0 μM cpx-1 in Figure 2a,b), when added to reaction mixtures at the same time, the clamping ability of cpx-I was largely occluded by the clamping ability of apo-syt. Interestingly, after addition of Ca2+ to reaction mixtures containing both syt and cpx-I, the inhibitory ability of cpx-I was unmasked and cpx-I efficiently inhibited Ca2+-syt-driven fusion (Figure 2d, inset). Since syt binds t-SNAREs before, during, and after formation of the ternary SNARE complex, whereas complexin preferentially binds to ternary SNARE complexes (see refs (14) and (17) and Figure S1), it appears that apo-syt inhibits fusion at early stages of trans-SNARE pairing, as suggested in ref (17), whereas cpx-I inhibits fusion after trans-SNARE pairing has progressed further toward complete ternary SNARE complex formation. Thus, our data likely reflect the functional consequences of concurrent binding of syt and cpx-I to SNAREs in the reconstituted system.
In summary, we have analyzed the putative clamping role of cpx-I (4−6,8) and addressed the controversy regarding whether syt and cpx-I compete for binding to SNARE complexes (6,8,9,13). We demonstrate that Ca2+-syt and cpx-I bind to fully assembled membrane-embedded SNARE complexes (formed on liposomes) at the same time, that cpx-I(26−83) does not substitute for full-length cpx-I in our fusion experiments, and that both apo-syt and cpx-I can inhibit SNARE-mediated fusion. Importantly, inhibition by cpx-I was not overcome by Ca2+-syt. Therefore, in our system, it does not appear that cpx-I acts as a fusion clamp that is relieved by Ca2+-syt but rather that apo-syt and cpx-I inhibit SNAREs at distinct stages of SNARE complex assembly.
We note that key differences in the experimental conditions used by various groups to study the function of complexins could potentially explain why some groups have observed competition between Ca2+-syt and cpx-I for SNARE binding while others have not. For example, refs (8) and (13) report that Ca2+-syt can displace cpx-I(26−83) from cis-SNARE complexes embedded in planar lipid bilayers, whereas our experiments suggest that Ca2+-syt cannot displace cpx-I from cis-SNARE complexes formed on liposomes (diameter of ∼50 nm). The differences in these reports suggest that interactions between regulatory proteins and the SNARE complex might be profoundly affected by membrane curvature. This is an important consideration since highly curved membranes form critical intermediates during the fusion reaction (18).
Supporting Information Available
Detailed methods, Figures S1−S3, and supporting text and references. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Llinas R.; Steinberg I. Z.; Walton K. (1981) Biophys. J. 33, 323–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E. R. (2008) Annu. Rev. Biochem. 77, 615–641. [DOI] [PubMed] [Google Scholar]
- Melia T. J. Jr. (2007) FEBS Lett. 581, 2131–2139. [DOI] [PubMed] [Google Scholar]
- Giraudo C. G.; Eng W. S.; Melia T. J.; Rothman J. E. (2006) Science 313, 676–680. [DOI] [PubMed] [Google Scholar]
- Giraudo C. G.; Garcia-Diaz A.; Eng W. S.; Yamamoto A.; Melia T. J.; Rothman J. E. (2008) J. Biol. Chem. 283, 21211–21219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaub J. R.; Lu X.; Doneske B.; Shin Y. K.; McNew J. A. (2006) Nat. Struct. Mol. Biol. 13, 748–750. [DOI] [PubMed] [Google Scholar]
- Huntwork S.; Littleton J. T. (2007) Nat. Neurosci. 10, 1235–1237. [DOI] [PubMed] [Google Scholar]
- Tang J.; Maximov A.; Shin O. H.; Dai H.; Rizo J.; Sudhof T. C. (2006) Cell 126, 1175–1187. [DOI] [PubMed] [Google Scholar]
- McMahon H. T.; Missler M.; Li C.; Sudhof T. C. (1995) Cell 83, 111–119. [DOI] [PubMed] [Google Scholar]
- Reim K.; Mansour M.; Varoqueaux F.; McMahon H. T.; Sudhof T. C.; Brose N.; Rosenmund C. (2001) Cell 104, 71–81. [DOI] [PubMed] [Google Scholar]
- Xue M.; Reim K.; Chen X.; Chao H. T.; Deng H.; Rizo J.; Brose N.; Rosenmund C. (2007) Nat. Struct. Mol. Biol. 14, 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M.; Stradomska A.; Chen H.; Brose N.; Zhang W.; Rosenmund C.; Reim K. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 7875–7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H.; Shen N.; Arac D.; Rizo J. (2007) J. Mol. Biol. 367, 848–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Tomchick D. R.; Kovrigin E.; Arac D.; Machius M.; Sudhof T. C.; Rizo J. (2002) Neuron 33, 397–409. [DOI] [PubMed] [Google Scholar]
- Weber T.; Zemelman B. V.; McNew J. A.; Westermann B.; Gmachl M.; Parlati F.; Sollner T. H.; Rothman J. E. (1998) Cell 92, 759–772. [DOI] [PubMed] [Google Scholar]
- Bai J.; Tucker W. C.; Chapman E. R. (2004) Nat. Struct. Mol. Biol. 11, 36–44. [DOI] [PubMed] [Google Scholar]
- Chicka M. C.; Hui E.; Liu H.; Chapman E. R. (2008) Nat. Struct. Mol. Biol. 15, 827–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik L. V.; Kozlov M. M. (2008) Nat. Struct. Mol. Biol. 15, 675–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.