

Abstract
A series of novel derivatives of the nonsteroidal anti-inflammatory drug (NSAID) sulindac sulfide were synthesized as potential agonists of the peroxisome proliferator-activated receptor gamma (PPARγ). Nonpolar and aromatic substitutions on the benzylidene ring as well as retention of the carboxylic acid side chain were required for optimal activity. Compound 24 was as potent a compound as any other in the series with an EC50 of 0.1 μM for the induction of peroxisome proliferator response element (PPRE)-luciferase activity. Direct binding of compound 24 to PPARγ was demonstrated by the displacement of [3H]troglitazone, a PPARγ agonist, in a scintillation proximity assay. Compound 24 also stimulated the binding of PPARγ to a PPRE-containing oligonucleotide and induced expression of liver fatty-acid binding protein (L-FABP) and adipocyte fatty acid-binding protein (aP2), two established PPARγ target genes. Taken together, these compounds represent potential leads in the development of novel PPARγ agonists.
Introduction
Peroxisome proliferator-activated receptors (PPARs)a are a group of nuclear hormone receptors that control cellular metabolism and proliferation through the modulation of gene expression. They exist as three distinct subtypes: PPARα, PPARδ, and PPARγ. Upon ligand binding, PPARs, including PPARγ, form a heterodimer with the retinoid X receptor and activate gene expression by binding to PPAR response elements.(1) The binding of various endogenous and exogenous ligands to PPARγ has been shown to regulate cellular differentiation, apoptosis, glucose homeostasis, and anti-inflammatory responses(2) (and references within). Thiazolidinediones, a class of PPARγ agonists that reduce insulin resistance3,4 are used to treat type 2 diabetes.(3) A wealth of crystallographic information for agonist bound to PPAR ligand binding domains (LBDs) as well as extensive SARs have provided structural insight into the factors controlling receptor binding and activation as well as subtype selectivity.5–8 These reports, and others, have described similar binding pockets for all three receptor subtypes that include a large, concave, mainly hydrophobic channel. This binding pocket is significantly larger than other known nuclear receptors, particularly in PPARα and PPARγ (∼1400 Å)(6) and may explain the observed promiscuity of the PPARs for various types of ligands.5,9,10 Typically, PPAR agonists consist of an acidic headgroup, traditionally either a carboxylic acid or a 2,4-thiazolidinedione tethered to an aromatic center that is linked to cyclic tail region. The acidic headgroup is crucial for PPAR activation in that it forms key interactions with residues on the AF-2 helix that anchor it close to the protein, which in turn allows for successful coactivator binding.(8) The aromatic center forms a variety of van der Waals interactions with various hydrophobic residues in the binding site, while the cyclic tail tolerates a more diverse set of substituents that are free to interact with a fairly large, hydrophobic pocket.(6)
Previous studies found that a variety of nonsteroidal anti-inflammatory drugs (NSAIDs) activate PPARs at micromolar concentrations.11–13 However, the effects of NSAIDs were heterogeneous. For example, ibuprofen was demonstrated to be a coactivator of PPARα and γ, while aspirin, acetaminophen, and piroxicam showed no agonistic activity for any PPAR subtype.(13) Indomethacin and diclofenac were shown to be selective for PPARγ although certain reports have claimed that indomethacin can also act as an agonist for PPARα.(12) These compounds are attractive targets for redesign because their pharmacokinetics have already been extensively studied in humans. Chemical removal of cyclooxygenase (COX) inhibition could reduce potential gastrointestinal and cardiovascular side effects and provide excellent tools for evaluating the contribution of COX to their overall biological effects. Previous work in our laboratory has shown that a subtle chemical modification of sulindac sulfide (i.e., removal of the indenyl methyl group) abolishes inhibition of cyclooxygenase while retaining activity at other targets including PPARγ (Figure 1).(14)
Figure 1.

Structures of sulindac sulfide and 2.
Herein we report the design, synthesis, and biological evaluation of a unique class of PPARγ agonists derived from sulindac sulfide, an active metabolite of the NSAID sulindac. Sulindac sulfide has been reported to activate PPARγ(15) although other reports indicate that it does not.(16) Furthermore, sulindac sulfide has been reported to act as an antagonist for PPARδ by preventing PPARδ/RXR heterodimer from binding to their recognition sequences.(17) The stereochemistry of the benzylidene double bond is Z in sulindac and sulindac sulfide, whereas it is E in the 2′-des-methyl derivatives. The altered stereochemistry presents a unique scaffold from which to evolve novel PPARγ agonists. Evaluation of a series of 2′-des-methyl sulindac sulfide analogues identified highly potent compounds that were essentially equipotent to troglitazone, a thiazolidinedione, in the ability to stimulate PPARγ-dependent transcription and trigger adipocyte differentiation in cell culture. shRNA knockdown experiments indicate these compounds are relatively selective PPARγ agonists, with weak activity at PPARα.
Results
Chemistry
A series of modifications was made to 2′-des-methyl sulindac sulfide (2) in an effort to explore the chemical functionalities required for effective activation of PPARγ. Synthesis of indene 1 was performed according to the procedure described previously.(14) Condensation of the indene with the appropriate aldehyde was performed using 1 N NaOH in refluxing methanol (Scheme 1). The aldehydes used were either commercially available or synthesized using established methods. Acidic workup followed by filtration or purification using column chromatography afforded the requisite 2′-des-methyl sulindac sulfide derivative. Amides of the 2′-des-methyl sulindac sulfide derivatives were synthesized using standard EDCI carbodiimide coupling reactions. The final products were characterized using electrospray ionization mass spectrometry and 1H NMR and purity was determined using HPLC.
Scheme 1. Synthesis of Benzylidene Derivatives.

Reagents and conditions: (a) R-CHO, 1 N NaOH, MeOH, reflux, 2 h.
PPRE-Luciferase Reporter Assay
A PPRE-luciferase reporter assay was used to test the ability of the 2′-des-methyl sulindac sulfide derivatives to activate PPARs. HCA-7 colon cancer cells, which express PPARα, PPARδ, and PPARγ, were transiently transfected with an expression vector containing a PPRE-luciferase reporter plasmid.(18) Following transfection, cells were treated with various concentrations of compound for 12 h and assayed for luciferase activity.
SAR
The series of derivatives of 2 were synthesized, and the results of the PPRE-luciferase screening assays are summarized in Table 1. A general descriptor of traditional PPARγ agonists is the requirement of an acidic headgroup that forms essential hydrogen bonds with key residues on the AF-2 helix in the LBD. This was verified in the present series with a number of amide derivatives, none of which activated PPRE-luciferase (data not shown).
Table 1. Induction of PPRE-Luciferase Activity by Derivatives of 2′-des-Methyl Sulindac Sulfide.
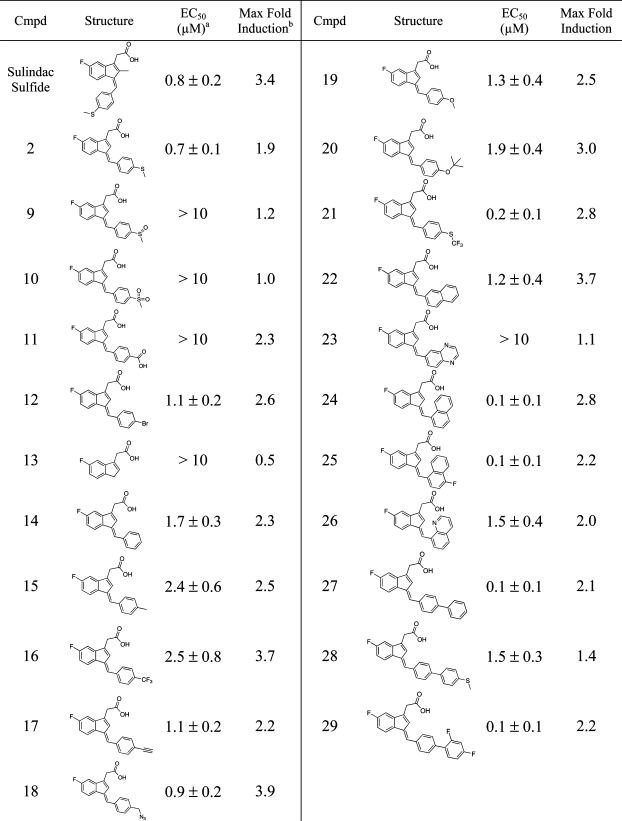 |
EC50 is the concentration of compound required to achieve 50% of the maximum induction of PPRE-luciferase activity. Means ± SD are shown, n = 3.
A 2-fold induction corresponds to a doubling of luciferase activity relative to vehicle-treated control.
Having confirmed the requirement of the acidic acid side chain for PPRE-luciferase activity, a variety of substitutions on the benzylidene ring were explored in an effort to increase potency (Table 1). Polar substitutions, including the sulfoxide (9) and sulfone (10), gave compounds that were unable to induce PPRE-luciferase activity. These results would be expected if the phenyl ring inserted into the large hydrophobic pocket at the top of the active site analogous to the phenyloxazole tail of many traditional PPARγ agonists.(6) Removal of the benzylidene ring to give compound 13 also gave an inactive compound.
Anticipating that the benzylidene ring may be making key hydrophobic interactions in the PPARγ LBD, a series of nonpolar and aromatic substitutions were examined. Various substitutions, including hydrogen (14), methyl (15), trifluoromethyl (16), and t-butoxy (20), gave derivatives that were less potent inducers of PPRE-luciferase activity than 2. Other substitutions, including bromo (12), alkynyl (17), azido (18), and methoxy (19), yielded derivatives with comparable PPRE-luciferase activity to 2.
Large aromatic and heteroaromatic systems comprising the tail end of the pharmacophore are a common feature of other known PPARγ agonists.7,8,19 A series of naphthyl and biphenyl substitutions were introduced, resulting in a series of derivatives that showed a marked increase in potency relative to 2. Although attachment of a naphthyl ring at the 2-position gave a derivative (22), which did not result in a significant increase in potency, attachment at the 1-position yielded highly potent derivatives (i.e., 24 and 25) with EC50′s of 0.1 μM. It is important to note that the EC50 as well as the magnitude of the response for 24 was comparable to troglitazone (see below). Interestingly, incorporation of a nitrogen to give a quinoline derivative decreased potency relative to 2. Biphenyl substitutions also resulted in potent derivatives, 27 and 29, with EC50′s of 0.1 μM, although incorporation of a thiomethyl group at the para postion of the terminal phenyl ring (28) resulted in a significant drop in potency.
A selection of these derivatives was tested for their ability to inhibit the two isoforms of cyclooxygenase. As expected, none of these compounds showed inhibition of either COX-1 or COX-2 (data not shown) up to concentrations of 4 μM. This further confirmed that the lack of a 2′-methyl group combined with the isomeric reversal of configuration around the benzylidene double bond eliminates the ability of this class of compounds to inhibit cyclooxygenase.(14)
These results point to an interesting SAR for the activation of PPRE luciferase activity by 2′-des-methyl sulindac sulfide derivatives. The carboxylic side chain is absolutely required for optimal activity, consistent with previous observations. The benzylidene ring shows a bit more flexibility but an overall preference for nonpolar substituents on the benzylidene ring was observed. Finally, aromatic systems, particularly naphthyl and biphenyl, appear to be highly favored at this position.
Compound 24 Activates PPARγ and Induces Adipogenesis
On the basis of the SAR, 24 was as potent an inducer of PPRE luciferase activity as any other compound in the series. Hence, this compound was investigated in greater detail. Initially, 24 was compared with troglitazone, a prototypic PPARγ agonist. Cells were transfected with an expression vector containing luciferase under the control of a PPRE. Addition of either compound stimulated comparable concentration-dependent increases in PPRE luciferase activity (Figure 2a). To determine whether the increase in PPRE luciferase activity reflected direct binding of 24 to PPARγ, a scintillation proximity assay was performed. Compound 24 caused a dose-dependent displacement of [3H]-troglitazone, indicating that 24 binds directly to PPARγ (Figure 2b). Next, electrophoretic mobility shift assays were performed to probe for nuclear translocation of PPARγ. Cells were treated with 24, nuclear lysates were prepared, and the lysates exposed to radiolabeled oligonucleotides containing a PPRE sequence. Treatment with 24 led to a concentration-dependent increase in binding of nuclear protein to labeled PPRE oligonucleotides (Figure 3, lanes 1−5). The specificity of this binding was also evaluated; binding was abolished when an excess of unlabeled consensus PPRE oligonucleotide was added (Figure 3, lane 6). By contrast, binding was unaffected by the addition of an excess of oligonucleotide in which the PPRE site was scrambled (Figure 3, lane 10). Supershift analysis identified PPARγ in the binding complex (Figure 3, lane 9).
Figure 2.

Compound 24 is a PPARγ agonist. (a) HCA7 cells were transfected with 1.8 μg of PPRE luciferase and 0.2 μg of pSVβgal. Cells were treated with 0−2.5 μM 24 or troglitazone for 12 h. Luciferase activity represents data that have been normalized to β-galactosidase. Columns, means; bars, SD; n = 6. *: p < 0.05, **: p < 0.01. (b) 24 binds to PPARγ. Competitive binding assays were performed by scintillation proximity assay for human PPARγ-ligand binding domain and 50 nM 3H-troglitazone in the presence of increasing concentrations of nonradioactive 24 as a competitor. Means and SD are shown, n = 3.
Figure 3.

Compound 24 stimulates PPARγ binding. Nuclear proteins from HCA7 cells were incubated with a 32P-labeled PPRE containing oligonucleotide. Cells were treated with vehicle (lane 1) or 24 (0.1 μM, lane 2; 0.25 μM, lane 3; 0.5 μM, lane 4; 1 μM, lane 5) for 30 min. Lanes 6 and 10 represent nuclear extract from cells treated with 1 μM 24 incubated with a 32P-labeled PPRE containing oligonucleotide and a 100-fold excess of unlabeled oligonucleotide (lane 6) or mutant PPRE containing oligonucleotide (lane 10). Lanes 7−9 represent nuclear extract from cells treated with 1 μM 24 incubated with a 32P-labeled PPRE containing oligonucleotide incubated with 0 μL (lane 7), 1 μL (lane 8) or 2 μL (lane 9) of antibody to PPARγ. The protein−DNA complex that formed was separated on a 4% polyacrylamide gel.
To further evaluate the functional significance of 24 as a PPARγ agonist, we investigated its effects on the levels of L-FABP and aP2, two established PPARγ target genes.20,21 Cells were treated with 0−1 μM 24 for 24 h. Following treatment, cell lysates were prepared and Western blotting was performed. Compound 24 induced both L-FABP and aP2 (Figure 4a) over the same concentration range that stimulated PPARγ binding (Figure 3). Induction of L-FABP and aP2 was suppressed by pretreatment with 30 (GW9662)(22) (Figure 5), a PPARγ antagonist (Figure 4b). PPARγ agonists induce adipogenesis;(23) hence, we also investigated whether 24 could stimulate triglyceride accumulation in 3T3-L1 cells, a murine fibroblast cell line. As shown in Figure 6, staining with Oil Red O revealed formation of lipid droplets in cells treated with 24 in a manner similar to the known PPARγ activator, troglitazone.
Figure 4.

Compound 24 modulates the expression of known PPARγ target genes. (a) HCA7 cells were treated with 0−1 μM 24 for 24 h. Cellular lysate protein (100 μg/lane) was loaded onto a 12.5% SDS-polyacrylamide gel, electrophoresed, and subsequently transferred onto nitrocellulose. Immunoblots were probed with antibodies to L-FABP, aP2, and β-actin. (b) 30, a PPARγ antagonist, suppresses 24-mediated induction of L-FABP and aP2. Cells were treated as indicated with vehicle, 24, 30 alone, or 24 and 30 for 24 h. Cellular lysate protein (100 μg/lane) was loaded onto a 12.5% SDS-polyacrylamide gel, electrophoresed, and subsequently transferred onto nitrocellulose. The immunoblot was probed for L-FABP, aP2, and β-actin.
Figure 5.

Structures of 30 and 31.
Figure 6.

Compound 24 activates adipogenesis in 3T3-L1 cells. Images of Oil Red O stained 3T3-L1 cells were taken after treatment with (a) vehicle (b) 1 μM troglitazone or (c) 1 μM 24 for 48 h (200×).
shRNA knockdown of PPARα, PPARδ, and PPARγ
Previous studies have shown that NSAIDs and NSAID derivatives that activate PPARγ also activate PPARα to varying extents. Western blot analysis indicated that HCA-7 cells contain PPARα, PPARγ, and PPARδ (Figure 7a). Overexpressing of shRNAs to the three PPAR isoforms led to selective silencing of each PPAR (Figure 7a). Knocking down PPARγ abrogated troglitazone-mediated activation of PPRE-luciferase (Figure 7b). The inductive effects of 24 were also attenuated when PPARγ was silenced, although induction was not reduced to the level seen in vehicle-treated control (Figure 7b). Compound 31 (GW7647)(24) (Figure 5), a PPARα agonist, caused a marked increase in PPRE-luciferase activity, an effect that was abrogated when PPARα was silenced (Figure 7c). A small but statistically significant reduction in the inductive effects of 24 was found in cells in which PPARα was silenced, indicating that 24 may act as a weak agonist of PPARα. cPGI, an agonist of PPARδ was a potent inducer of PPRE-luciferase, an effect that was abrogated when PPARδ was silenced. In contrast, silencing of PPARδ did not affect the induction of PPRE-luciferase by 24, indicating that 24 does not act as an agonist for PPARδ.
Figure 7.

Compound 24 is a mixed agonist of PPARγ and PPARα. (a) HCA7 cells were transfected with control shRNA or shRNAs to PPARγ, PPARα and PPARδ. Cellular protein was isolated (100 μg/lane) and loaded onto a 12.5% SDS-polyacrylamide gel, electrophoresed, and subsequently transferred onto nitrocellulose sheet. Immunoblots were probed with the indicated antibodies. In each case, the shRNA selectively silenced its receptor. (b−d) shRNA expressing cells were transfected with 1.8 μg PPRE luciferase and 0.2 μg pSVβgal. Cells were treated with vehicle, 1 μM troglitazone, or 1 μM 24 (b); vehicle, 2 μM 31 or 1 μM 24 (c); vehicle, 2.5 μM cPGI or 1 μM 24 (d) for 12 h. Luciferase activity represents data that have been normalized to β-galactosidase. Column, means; bars, S.D; n = 6, *, p < 0.05.
Molecular Docking of 24 in the hPPARγ Active Site
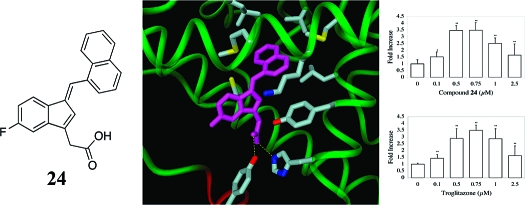
To date, crystal structures of PPARγ in complex with carboxylate-containing full agonists support a model of receptor activation in which the bound agonist hydrogen bonds with H323 and the hydroxyl group of Y473 in the AF-2 helical domain. Stabilized by this hydrogen-bonding network, the AF-2 domain packs closely with the protein core, which closes the binding site and is proposed to activate the receptor.5,7
We generated models for compound 24 bound to PPARγ to determine whether this molecule might be able to stabilize a closed, activated receptor structure comparable to those observed for other full agonists. We used ab initio Hartree−Fock calculations to compute the global minimum energy structure for compound 24 and then docked this geometry-optimized conformation in the receptor using the crystal structure for the PPARγ complex with the full agonist 2-(5-[3-(7-propyl-3-trifluoromethylbenzo[d]isoxazol-6-yloxy)propoxy]indol-1-yl)ethanoic acid (PDB code: 2ATH) as a template. This crystal structure was chosen because it contained similar structural elements of the ligand relative to 24 (indole versus indene core) as well as for sufficient resolution (2.28 Å). Compound 24 was positioned in the binding site so that its carboxylate group could form favorable hydrogen bonds with H323 and Y473 in the AF-2 domain. This ligand orientation placed the large napthalene substituent neatly into a nearby hydrophobic pocket formed by residues C285, Y327, L330, M334, V339, L353, M364, and the aliphatic carbons of K367 (Figure 8). Compound 24 and protein hydrogen atoms were relaxed with 1000 cycles of conjugate gradient energy minimization, followed by 2000 additional cycles of minimization for the full receptor−ligand complex to relieve any residual unfavorable steric interactions. The rmsd for the minimized PPARγ backbone vs crystal structure is 0.27 Å.
Figure 8.

PPARγ complex with compound 24. The carboxylate of compound 24 forms hydrogen bonds (shown as dashed yellow lines) with H323 and Y473 of the AF-2 helical domain, displayed in red. The naphthalene moiety of 24 binds in a hydrophobic pocket formed by residues C285, Y327, L330, M334, V339, L353, M364, and the aliphatic carbons of K367, each colored by atom type. Figure 8 was created with the DINO program.(35)
Discussion
The results from these experiments indicate that sulindac sulfide as well as its 2′-des-methyl derivatives are potent inducers of PPARγ. This contradicts an earlier report indicating that sulindac sulfide does not activate PPARγ.(16) The SAR confirms that the carboxylic side chain is required for activity; the neutral amide derivatives showed a complete loss of activity. We also found that nonpolar and aromatic substituents on the benzylidene ring lead to potent PPARγ agonists. Specifically, a naphthalene substitution at the 1-position led to compound 24, which had an EC50 of 0.1 μM in a PPRE-luciferase activation assay. Compound 24 was able to displace [3H]-troglitazone in a scintillation proximity assay demonstrating that activation of PPRE-luciferase reflects direct binding to PPARγ. Nuclear translocation of PPARγ by 24 was confirmed using an electrophoretic mobility shift assay in which a concentration-dependent increase in binding of nuclear protein to labeled PPRE oligonucleotides was observed. Moreover, 24 induced known PPARγ target genes, L-FABP and aP2, and 31, a PPARγ antagonist, suppressed 24-mediated induction of both L-FABP and aP2. Compound 24 induced the accumulation of lipid in cells, a finding that is characteristic of PPARγ agonists. shRNA knockdown of PPARα and PPARγ indicated that 24 is a potent agonist of PPARγ with only weak activity at PPARα. This finding contrasts with previous work, demonstrating that certain NSAIDs are mixed agonists of PPARγ and α with similar potencies of activation.(11) shRNA knockdown of PPARδ indicated that 24 had no effect on PPARδ.(25)
Our model for the PPARγ-compound 24 complex accommodates the ligand well in its global minimum energy conformation and explains the experimental observations. In the model, the ligand carboxylate forms a hydrogen bond network with H323 and Y473, comparable to that observed in crystal structures for other full agonist complexes. This hydrogen-bonding network recruits the AF-2 helix domain to close the binding site and activate the receptor. The naphthalene substituent fits nicely in the large hydrophobic pocket described above and thus increases favorable protein/ligand contacts compared to other ligands in the series.
Traditional NSAIDs inhibit COX activity in addition to having collateral targets such as PPARγ. The derivatives presented in this report represent structurally distinct derivations of sulindac sulfide that could not necessarily have been predicted to retain similar activity. Furthermore, this report also represents the first published exploration of the structural requirements necessary for potent PPARγ activation specific to this class of PPAR agonists. The compounds reported herein represent a new class of PPARγ activators that lack the COX inhibitory activity of sulindac sulfide, the parent NSAID. Derivation of the core structure led to compounds with potency that was similar to troglitazone, a known PPARγ agonist. Hence, compounds such as 24 provide potentially useful tools for understanding the contribution of the PPARγ response to the overall biological responses elicited by sulindac sulfide, a compound that can both activate PPARγ and inhibit COX. Recently, rosiglitazone, a PPARγ agonist that is used to treat diabetes mellitus, was found to be efficacious in the treatment of mild to moderately active ulcerative colitis.(26) Future studies will be needed to determine whether the derivatives of sulindac sulfide such as 24 possess properties that make them attractive leads as drug candidates.
Experimental Section
Materials
BCA protein assay reagent kit and NHC-biotin were purchased from Pierce Biotechnology, Inc. (Rockford, IL). Nitrocellulose membranes were from Whatman, Inc. (Sanford, ME). Antibody to β-actin, secondary antibodies conjugated to horseradish peroxidase (HRP), and 2-nitrophenyl β-d-galactopyranoside were from Sigma-Aldrich Co. (St. Louis, MO). 30 was from EMD Biosciences, Inc. (San Diego, CA). Polyclonal chicken antiadipocyte fatty acid-binding protein (aP2) and adipogenesis assay kits were from Chemicon International Inc. (Temecula, CA). Polyclonal antisera to liver fatty acid binding protein (L-FABP) was from Abcam Inc. (Cambridge, MA). Bovine antichicken IgY-HRP secondary antibody and isoform specific PPAR antibodies were purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Western blotting detection reagents (ECL) and [32P]-ATP were from Perkin-Elmer LAS, Inc. (Boston, MA). Plasmid DNA preparation kits were from Qiagen (Valencia, CA). Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS), OPTI-MEM, and Lipofectamine 2000 were from Invitrogen Corp. (Carlsbad, CA). Calf bovine serum (CBS) and 3T3-L1 cells were from the American Type Culture Collection (Manassas, VA). Luciferase assay substrates A and B and cell lysis buffer were from BD Biosciences & Co. (Franklin Lakes, NJ). pSVβgal was obtained from Promega Corp. (Madison, WI). The PPRE luciferase and PPAR-γligand binding domain (PPARγ-LBD) plasmids were gifts from Dr. Ron Evans (Salk Institute for Biological Sciences, La Jolla, CA). Streptavidin SPA beads were from Amersham Biosciences (Piscataway, NJ). cPGI and 31 were from Cayman Chemical Co. (Ann Arbor, MI). Control shRNA and shRNAs to PPARs were obtained from Superarray Bioscience Corp. (Frederick, MD).
Chemistry
HPLC grade solvents obtained from Fischer (Pittsburgh, PA) were used for column chromatography. Reagent grade chemicals were obtained from Aldrich (Milwaukee, WI), Matrix Scientific (Columbia, SC), Alfa Aesar (Ward Hill, MA), and Oakwood Products (West Columbia, SC). All other chemicals were used without further purification. Thin layer chromatography was performed on silica plates obtained from Analtech (Silica Gel 60 F254 precoated). The plates were read by UV fluorescence (254 nm) or by staining with phosphomolybdic acid (PMA) followed by heating. Column chromatography was performed using a Biotage SP1 purification system.
Tissue Culture
The HCA7 human colon cancer cell line was established from moderately differentiated adenocarcinoma of the colon.(27) The cells were maintained in DMEM supplemented with 100 IU/mL penicillin, 100 mg/mL streptomycin, and 10% FBS. 3T3-L1 cells were grown in DMEM supplemented with 10% CBS.
Creation of Stable Cell Lines
HCA7 cells were transfected with control shRNA or shRNAs to different PPARs. The Amaxa nucleofection system (Gaithersburg, MD) was used and the manufacturer’s instructions employed. Puromycin B resistant cells were selected.
Transient Transfection Assays
Cells were grown to 40% confluence in 6-well dishes. For each well, 2 μg of plasmid DNA were introduced into cells using 6 μg of Lipofectamine 2000 as per the manufacturer’s instructions. After 6 h of incubation, the medium was replaced with growth medium for 16 h, followed by serum-free media containing the compounds for 12 h. The activities of luciferase and β-galactosidase were measured.
Scintillation Proximity Assay
The assay was performed as described previously.(28) The PPARγ-LBD was isolated from Escherichiacoli as a polyhistidine-tagged fusion protein. Radiolabeled troglitazone was synthesized as described previously.(29) The buffer for all assays was 50 mM HEPES (pH 7), 50 mM KCl, 5 mM CHAPS, 0.1 mg/mg BSA. The protein was biotinylated and immobilized on streptavidin-modified SPA beads. Nonradioactive 24 was used to compete for binding to the PPARγ-LBD using 3H-troglitazone as the ligand. The assays were performed in the absence of dithiothreitol.
Electrophoretic Mobility Shift Assay
Nuclear extracts were prepared from cells. For binding studies, oligonucleotides containing PPRE sites were obtained from Active Motif (Carlsbad, CA). The complementary oligonucleotides were annealed in 20 mM Tris (pH7.6), 50 mM NaCl, 10 mM MgCl2, and 1 mM dithiothreitol. The annealed oligonucleotide was phosphorylated at the 5′ end with [γ-32P]ATP and T4 polynucleotide kinase. The binding reaction was performed by incubating 5 μg of nuclear protein in 20 mM HEPES (pH 7.9), 10% glycerol, 300 μg of bovine serum albumin, and 1 μg of poly (dI·dC) in a final volume of 10 μL for 10 min at 25 °C. The labeled oligonucleotide was added to the reaction mixture and allowed to incubate for an additional 20 min at 25 °C. The samples were electrophoresed on a 4% nondenaturing polyacrylamide gel. The gel was then dried and subjected to autoradiography at −80 °C.
Western Blotting
Cell lysates were prepared by treating cells with lysis buffer (150 mM NaCl, 100 mM Tris (pH 8.0), 1% Tween 20, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10 μg/mL trypsin inhibitor, 10 μg/mL aprotinin, and 10 μg/mL leupeptin). Following sonication, lysates were centrifuged at 4 °C for 10 min at 10000g to sediment the particulate material. The protein concentration of the supernatant was measured using a BCA protein assay kit. SDS-polyacrylamide gel electrophoresis was performed using 12.5% polyacrylamide gels as described by Laemmli.(30) The resolved proteins (100 mg/lane) were transferred overnight onto a nitrocellulose membrane and then probed with primary antisera. Secondary antibody conjugated to HRP was then used. The blots were then developed with the ECL Western blot detection system according to the manufacturer’s instructions.
Adipogenesis Assay
The adipogenesis assay was performed using a kit from Chemicon International. Briefly, 3T3-L1 cells were grown to 60% confluence. The cells were then placed in growth medium supplemented with 0.1% 3-isobutyl-1-methylxanthine and 1.0% dexamethasone to initiate adipogenesis for 48 h. The cells were then treated in growth medium containing 10 μg/mL of recombinant human insulin supplemented with the test compound for an additional 2 days. The medium was then replaced with fresh growth medium for an additional 48 h prior to staining with Oil Red O. Images were taken using Q-Imaging Retina EX camera and Olympus IX51 bright field microscope (200x).
Statistics
Comparisons between groups were made with Student’s t test. A difference between groups of p < 0.05 was considered significant.
Molecular Modeling
Geometry optimization and conformation analysis calculations were performed with restricted Hartree−Fock (RHF) calculations using a 3-21G* basis set with the Gaussian03 package.(31) The global minimum energy structure for compound 24 was docking manually in the binding site of the 2ATH crystal structure(7) using the interactive molecular graphics program PSSHOW.(32) The protein−ligand complex was refined using conjugate gradient energy minimization and a generalized Born solvent model with the AMBER9 package.(33) A molecular electrostatic potential for the geometry-optimized conformation of compound 24 was computed using RHF calculations with a 6-31G* basis set, and the RESP program(34) was then used to fit a set of atom-centered partial charges that reproduced the electrostatic potential.
Instrumental Analysis
Mass spectra were obtained by electrospray ionization (ESI-MS) on a Finnigan TSQ 7000 triple-quadrupole spectrometer. 1H NMR spectra were obtained on a Bruker AC 300 NMR spectrometer using CDCl3 or DMSO-d6 as the solvent and TMS as an internal standard. All chemical shifts are reported in ppm downfield from TMS and coupling constants are reported in hertz.
General Procedure for the Synthesis of Benzylidene Derivatives of Ethyl 2-(5-fluoro-1H-inden-3-yl)ethanoate
To a solution of the ethyl 2-(5-fluoro-1H-inden-3-yl)ethanoate (1 equiv) and the appropriate aldehyde (1.2 equiv) was added 1 N NaOH and methanol. The mixture was stirred at reflux for 2 h. The solution was cooled, neutralized with 15% HCl, diluted with water, and extracted with CH2Cl2 (3×). The combined organics were washed with water, NaHCO3, water, dried (MgSO4), filtered, and concentrated in vacuo. All purifications were performed using flash chromatography.
(E)-2-(5-Fluoro-1-(4-(methylsulfinyl)benzylidene)-1H-inden-3-yl)ethanoic acid (9)
1H NMR (DMSO) δ 7.87−7.82 (m, 1H), 7.84 (d, J = 8.4 Hz, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.69 (s, 1H), 7.17 (dd, J = 2.4, 9.2 Hz, 1H), 7.09 (s, 1H), 7.06 (td, J = 2.4, 8.7 Hz, 1H), 3.68 (s, 2H), 2.79 (s, 3H). ESI 341 (M − H+).
(E)-2-(5-Fluoro-1-(4-(methylsulfonyl)benzylidene)-1H-inden-3-yl)ethanoic acid (10)
1H NMR (CDCl3) δ 7.99 (d, J = 7.8 Hz, 2H), 7.72 (d, J = 7.8 Hz, 2H), 7.62 (dd, J = 5.1, 7.8 Hz, 1H), 7.38 (s, 1H), 7.04−6.95 (m, 3H), 3.69 (s, 2H), 3.05 (s, 3H). ESI 357 (M − H+).
(E)-4-((3-(Carboxymethyl)-5-fluoro-1H-inden-1-ylidene)methyl)benzoic acid (11)
1H NMR (DMSO) δ 8.01 (d, J = 8.3 Hz, 2H), 7.86 (dd, J = 5.1, 8.3 Hz, 1H), 7.76 (d, J = 8.3 Hz, 2H), 7.69 (s, 1H), 7.16 (dd, J = 2.4, 9.2 Hz, 1H), 7.10−7.04 (m, 2H), 3.68 (s, 2H). ESI 279 (M − CO2H+).
(E)-2-(1-(4-Bromobenzylidene)-5-fluoro-1H-inden-3-yl)ethanoic acid (12)
1H NMR (CDCl3) δ 7.60 (dd, J = 4.9, 8.2 Hz, 1H), 7.56 (d, J = 8.5 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 7.32 (s, 1H), 7.03 (dd, J = 2.3, 8.8 Hz, 1H), 6.99 (s, 1H), 6.95 (td, J = 2.3, 8.4 Hz, 1H), 3.68 (s, 2H). ESI 359 (M − H+).
2-(5-Fluoro-1H-inden-3-yl)ethanoic acid (13)
1H NMR (DMSO) δ 7.44 (dd, J = 5.2, 8.2 Hz, 1H), 7.15 (dd, J = 2.5, 9.5 Hz, 1H), 6.99 (dtd, J = 1.7, 2.5, 10.0 Hz, 1H), 6.53 (s, 1H), 3.54 (s, 2H), 3.34 (s, 2H). ESI 191 (M − H+).
(E)-2-(1-Benzylidene-5-fluoro-1H-inden-3-yl)ethanoic acid (14)
1H NMR (MeOD) δ 7.70 (dd, J = 5.0, 8.3 Hz, 1H), 7.62 (d, J = 7.3 Hz, 2H), 7.48 (s, 1), 7.45−7.40 (m, 2H), 7.37−7.31 (m, 1H), 7.07−7.03 (m, 2H), 6.93 (td, J = 2.4, 8.4 Hz, 1H), 3.65 (s, 2H). ESI 279 (M − H+).
(E)-2-(5-Fluoro-1-(4-methylbenzylidene)-1H-inden-3-yl)ethanoic acid (15)
1H NMR (CDCl3) δ 7.60 (dd, J = 4.9, 8.3 Hz, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.38 (s, 1H), 7.24 (d, J = 8.0 Hz, 2H), 7.07 (s, 1H), 7.02 (dd, J = 2.3, 8.9 Hz, 1H), 6.93 (td, J = 2.3, 9.2 Hz, 1H), 3.68 (s, 2H), 2.39 (s, 3H). ESI 293 (M − H+).
(E)-2-(5-Fluoro-1-(4-(trifluoromethyl)benzylidene)-1H-inden-3-yl)ethanoic acid (16)
1H NMR (CDCl3) δ 7.69−7.59 (m, 5H), 7.38 (s, 1H), 7.02 (dd, J = 2.3, 8.8 Hz, 1H), 6.99−6.92 (m, 2H), 3.68 (s, 2H). ESI 347 (M − H+).
(E)-2-(1-(4-Ethynylbenzylidene)-5-fluoro-1H-inden-3-yl)ethanoic acid (17)
1H NMR (DMSO) δ 7.84 (dd, J = 5.2, 8.3 Hz, 1H), 7.68 (d, J = 8.3 Hz, 2H), 7.63 (s, 1H), 7.56 (d, J = 8.3 Hz, 2H), 7.16 (dd, J = 2.1, 9.2 Hz, 1H), 7.09 (s, 1H), 7.06 (td, J = 2.3, 9.1 Hz, 1H), 4.35 (s, 1H), 3.67 (s, 2H). ESI 303 (M − H+).
(E)-2-(1-(4-(Azidomethyl)benzylidene)-5-fluoro-1H-inden-3-yl)ethanoic acid (18)
1H NMR (CDCl3) δ 7.62−7.56 (m, 3H), 7.38 (s, 1H), 7.37 (d, J = 8.0 Hz, 2H), 7.03−7.00 (m, 2H), 6.94 (td, J = 2.25, 9.1 Hz, 1H), 4.38 (s, 1H), 3.67 (s, 1H). ESI 334 (M − H+).
(E)-2-(5-Fluoro-1-(4-methoxybenzylidene)-1H-inden-3-yl)ethanoic acid (19)
1H NMR (CDCl3) δ 7.60 (dd, J = 4.9, 8.3 Hz, 1H), 7.55 (d, J = 8.7 Hz, 2H), 7.35 (s, 1H), 7.08 (s, 1H), 7.02 (dd, J = 2.3, 8.9 Hz, 1H), 6.96 (d, J = 8.8 Hz, 2H), 6.97−6.90 (m, 1H), 3.86 (s, 3H), 3.69 (s, 2H). ESI 309 (M − H+).
(E)-2-(1-(4-tert-Butoxybenzylidene)-5-fluoro-1H-inden-3-yl)ethanoic acid (20)
1H NMR (CDCl3) δ 7.60 (dd, J = 4.9, 8.3 Hz, 1H), 7.51 (d, J = 8.5 Hz, 2H), 7.36 (s, 1H), 7.08 (s, 1H), 7.07−7.01 (m, 3H), 6.93 (td, J = 2.3, 8.3 Hz, 1H), 3.69 (s, 2H). ESI 351 (M − H+).
(E)-2-(5-Fluoro-1-(4-(trifluoromethylthio)benzylidene)-1H-inden-3-yl)ethanoic acid (21)
1H NMR (CDCl3) δ 7.70 (d, J = 8.3 Hz, 2H), 7.58 (d, J = 8.3 Hz, 2H), 7.63−7.57 (m, 1H), 7.35 (s, 1H), 7.02 (dd, J = 2.3, 8.7 Hz, 1H), 6.98 (s, 1H), 6.95 (td, J = 2.3, 8.4 Hz, 1H), 3.68 (s, 1H). ESI 379 (M − H+).
(E)-2-(5-Fluoro-1-(naphthalen-2-ylmethylene)-1H-inden-3-yl)ethanoic acid (22)
1H NMR (CDCl3) δ 8.01 (s, 1H), 7.90−7.81 (m, 3H), 7.72 (dd, J = 1.6, 8.6 Hz, 1H), 7.65 (dd, J = 4.9, 8.3 Hz, 1H), 7.55−7.48 (m, 3H), 7.15 (s, 1H), 7.04 (dd, J = 2.3, 8.8 Hz, 1H), 6.96 (td, J = 2.3, 9.1 Hz, 1H), 3.71 (s, 1H). ESI 329 (M − H+).
(E)-2-(5-Fluoro-1-(quinoxalin-6-ylmethylene)-1H-inden-3-yl)ethanoic acid (23)
1H NMR (DMF) δ 8.97 (d, J = 10.2 Hz, 2H), 8.32 (s, 1H), 8.23 (d, J = 8.7 Hz, 1H), 8.14 (d, J = 9.0 Hz, 1H), 7.92−7.87 (m, 1H), 7.84 (s, 1H), 7.25 (m, 2H), 7.04 (t, J = 8.1 Hz, 1H), 3.72, (s, 2H). ESI 331 (M − H+).
(E)-2-(5-Fluoro-1-(naphthalen-1-ylmethylene)-1H-inden-3-yl)ethanoic acid (24)
1H NMR (CDCl3) δ 8.12−8.07 (m, 1H), 8.04 (s, 1H), 7.93−7.86 (m, 2H), 7.74 (dd, J = 4.9, 8.2 Hz, 1H), 7.60−7.50 (m, 4H), 7.04 (dd, J = 2.3, 8.9 Hz, 1H), 6.99 (td, J = 2.3, 8.3 Hz, 1H), 6.85 (s, 1H), 3.64 (s, 2H). ESI 329 (M − H+).
(E)-2-(5-Fluoro-1-((4-fluoronaphthalen-1-yl)methylene)-1H-inden-3-yl)ethanoic acid (25)
1H NMR (DMSO) δ 8.32−8.29 (m, 2H), 8.15−8.06 (m, 2H), 7.75−7.69 (m, 2H), 7.62 (dd, J = 5.7, 7.9 Hz, 1H), 7.46 (dd, J = 8.0, 10.6 Hz, 1H), 7.18 (dd, J = 2.3, 9.3 Hz, 1H), 7.11 (dtd, J = 2.2, 2.4, 9.7 Hz, 1H), 6.82 (s, 1H), 3.65 (s, 2H). ESI 347 (M − H+).
(E)-2-(5-Fluoro-1-(quinolin-8-ylmethylene)-1H-inden-3-yl)ethanoic acid (26)
1H NMR (DMSO) δ 9.00 (dd, J = 1.8, 4.2 Hz, 1H), 8.61 (s, 1H), 8.44 (dd, J = 1.5, 8.1 Hz, 1H), 8.06−8.00 (m, 2H), 7.93 (dd, J = 5.1, 8.4 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.63 (dd, J = 4.2, 8.4 Hz, 1H), 7.19 (dd, J = 2.4, 9.3 Hz, 1H), 7.11−7.04 (m, 2H), 3.68 (s, 2H). ESI 330 (M − H+).
(E)-2-(1-(Biphenyl-4-ylmethylene)-5-fluoro-1H-inden-3-yl)ethanoic acid (27)
1H NMR (DMSO) δ 7.86 (dd, J = 5.1, 8.3 Hz, 1H), 7.78 (m, 4H), 7.74 (d, J = 7.2 Hz, 2H), 7.67 (s, 1H), 7.49 (td, J = 1.7, 7.1 Hz, 2H), 7.39 (tt, J = 2.1, 7.3 Hz, 1H), 7.19−7.15 (m, 2H), 7.07 (td, J = 2.4, 8.4 Hz, 1H), 3.69 (s, 2H). ESI 355 (M − H+).
(E)-2-(5-Fluoro-1-((4′-(methylthio)biphenyl-4-yl)methylene)-1H-inden-3-yl)ethanoic acid (28)
1H NMR (DMSO) δ 7.86 (dd, J = 5.1, 8.3 Hz, 1H), 7.80−7.55 (m, 7H), 7.36 (d, J = 7.2 Hz, 2H), 7.18−7.16 (m, 2H), 7.06 (td, J = 2.5, 8.4 Hz, 1H), 3.67 (s, 2H), 2.52 (s, 3H). ESI 401 (M − H+).
(E)-2-(1-((2′,4′-Difluorobiphenyl-4-yl)methylene)-5-fluoro-1H-inden-3-yl)ethanoic acid (29)
1H NMR (DMSO) δ 7.85 (dd, J = 5.1, 8.3 Hz, 1H), 7.77 (d, J = 8.4 Hz, 2H), 7.68−7.60 (m, 4H), 7.38 (dtd, J = 2.1, 2.6, 9.3 Hz, 1H), 7.24−7.15 (m, 3H), 7.06 (dtd, J = 1.3, 2.4, 8.4 Hz, 1H), 3.69 (s, 2H). ESI 391 (M − H+).
Acknowledgments
This research was supported by research grants from the National Institutes of Health (CA89450, CA111469), the New York Crohn’s Foundation, and XL Tech Group.
Supporting Information Available
HPLC traces for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: NSAID, nonsteroidal anti-inflammatory drug; PPAR, peroxisome proliferator-activated receptor; PPRE, peroxisome proliferator response element; L-FABP, liver fatty-acid binding protein; aP2, adipocyte fatty acid-binding protein; COX, cyclooxygenase; DMEM, Dulbecco’s modified eagle’s medium; FBS, fetal bovine serum; LBD, ligand binding domain; HRP, horseradish peroxidase.
Supplementary Material
References
- Kersten S.; Desvergne B.; Wahli W. Roles of PPARs in health and disease. Nature 2000, 405, 421–424. [DOI] [PubMed] [Google Scholar]
- Subbaramaiah K.; Lin D. T.; Hart J. C.; Dannenberg A. J. Peroxisome proliferator-activated receptor gamma ligands suppress the transcriptional activation of cyclooxygenase-2. Evidence for involvement of activator protein-1 and CREB-binding protein/p300. J. Biol. Chem. 2001, 276, 12440–12448. [DOI] [PubMed] [Google Scholar]
- Willson T. M.; Brown P. J.; Sternbach D. D.; Henke B. R. The PPARs: from orphan receptors to drug discovery. J. Med. Chem. 2000, 43, 527–550. [DOI] [PubMed] [Google Scholar]
- Basu R.; Shah P.; Basu A.; Norby B.; Dicke B.; Chandramouli V.; Cohen O.; Landau B. R.; Rizza R. A. Comparison of the effects of pioglitazone and metformin on hepatic and extrahepatic insulin action in people with type 2 diabetes. Diabetes 2008, 57, 24–31. [DOI] [PubMed] [Google Scholar]
- Nolte R. T.; Wisely G. B.; Westin S.; Cobb J. E.; Lambert M. H.; Kurokawa R.; Rosenfeld M. G.; Willson T. M.; Glass C. K.; Milburn M. V. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature 1998, 395, 137–143. [DOI] [PubMed] [Google Scholar]
- Xu H. E.; Lambert M. H.; Montana V. G.; Plunket K. D.; Moore L. B.; Collins J. L.; Oplinger J. A.; Kliewer S. A.; Gampe R. T. Jr.; McKee D. D.; Moore J. T.; Willson T. M. Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. U.S.A. 2001, 98, 13919–13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahindroo N.; Huang C. F.; Peng Y. H.; Wang C. C.; Liao C. C.; Lien T. W.; Chittimalla S. K.; Huang W. J.; Chai C. H.; Prakash E.; Chen C. P.; Hsu T. A.; Peng C. H.; Lu I. L.; Lee L. H.; Chang Y. W.; Chen W. C.; Chou Y. C.; Chen C. T.; Goparaju C. M. V.; Chen Y. S.; Lan S. J.; Yu M. C.; Chen X.; Chao Y. S.; Wu S. Y.; Hsieh H. P. Novel indole-based peroxisome proliferator-activated receptor agonists: design, SAR, structural biology, and biological activities. J. Med. Chem. 2005, 48, 8194–8208. [DOI] [PubMed] [Google Scholar]
- Kuhn B.; Hilpert H.; Benz J.; Binggeli A.; Grether U.; Humm R.; Marki H. P.; Meyer M.; Mohr P. Structure-based design of indole propionic acids as novel PPARalpha/gamma co-agonists. Bioorg. Med. Chem. Lett. 2006, 16, 4016–4020. [DOI] [PubMed] [Google Scholar]
- Xu H. E.; Lambert M. H.; Montana V. G.; Parks D. J.; Blanchard S. G.; Brown P. J.; Sternbach D. D.; Lehmann J. M.; Wisely G. B.; Willson T. M.; Kliewer S. A.; Milburn M. V. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [DOI] [PubMed] [Google Scholar]
- Uppenberg J.; Svensson C.; Jaki M.; Bertilsson G.; Jendeberg L.; Berkenstam A. Crystal structure of the ligand binding domain of the human nuclear receptor PPARgamma. J. Biol. Chem. 1998, 273, 31108–31112. [DOI] [PubMed] [Google Scholar]
- Lehmann J. M.; Lenhard J. M.; Oliver B. B.; Ringold G. M.; Kliewer S. A. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other nonsteroidal anti-inflammatory drugs. J. Biol. Chem. 1997, 272, 3406–3410. [DOI] [PubMed] [Google Scholar]
- Jaradat M. S.; Wongsud B.; Phornchirasilp S.; Rangwala S. M.; Shams G.; Sutton M.; Romstedt K. J.; Noonan D. J.; Feller D. R. Activation of peroxisome proliferator-activated receptor isoforms and inhibition of prostaglandin H(2) synthases by ibuprofen, naproxen, and indomethacin. Biochem. Pharmacol. 2001, 62, 1587–1595. [DOI] [PubMed] [Google Scholar]
- Kojo H.; Fukagawa M.; Tajima K.; Suzuki A.; Fujimura T.; Aramori I.; Hayashi K.; Nishimura S. Evaluation of human peroxisome proliferator-activated receptor (PPAR) subtype selectivity of a variety of anti-inflammatory drugs based on a novel assay for PPAR delta(beta). J. Pharmacol. Sci. 2003, 93, 347–355. [DOI] [PubMed] [Google Scholar]
- Felts A. S.; Ji C.; Stafford J. B.; Crews B. C.; Kingsley P. J.; Rouzer C. A.; Washington M. K.; Subbaramaiah K.; Siegel B. S.; Young S. M.; Dannenberg A. J.; Marnett L. J. Desmethyl derivatives of indomethacin and sulindac as probes for cyclooxygenase-dependent biology. ACS Chem. Biol. 2007, 2, 479–483. [DOI] [PubMed] [Google Scholar]
- Wick M.; Hurteau G.; Dessev C.; Chan D.; Geraci M. W.; Winn R. A.; Heasley L. E.; Nemenoff R. A. Peroxisome proliferator-activated receptor-gamma is a target of nonsteroidal anti-inflammatory drugs mediating cyclooxygenase-independent inhibition of lung cancer cell growth. Mol. Pharmacol. 2002, 62, 1207–1214. [DOI] [PubMed] [Google Scholar]
- Sastre M.; Dewachter I.; Rossner S.; Bogdanovic N.; Rosen E.; Borghgraef P.; Evert B. O.; Dumitrescu-Ozimek L.; Thal D. R.; Landreth G.; Walter J.; Klockgether T.; van Leuven F.; Heneka M. T. Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T. C.; Chan T. A.; Vogelstein B.; Kinzler K. W. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell 1999, 99, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R. A.; Sarraf P.; Mueller E.; Brockman J. A.; Prusakiewicz J. J.; Eng C.; Willson T. M.; DuBois R. N. Peroxisome proliferator-activated receptor gamma-mediated differentiation: a mutation in colon cancer cells reveals divergent and cell type-specific mechanisms. J. Biol. Chem. 2003, 278, 22669–22677. [DOI] [PubMed] [Google Scholar]
- Sauerberg P.; Pettersson I.; Jeppesen L.; Bury P. S.; Mogensen J. P.; Wassermann K.; Brand C. L.; Sturis J.; Woldike H. F.; Fleckner J.; Andersen A. S.; Mortensen S. B.; Svensson L. A.; Rasmussen H. B.; Lehmann S. V.; Polivka Z.; Sindelar K.; Panajotova V.; Ynddal L.; Wulff E. M. Novel tricyclic-alpha-alkyloxyphenylpropionic acids: dual PPARalpha/gamma agonists with hypolipidemic and antidiabetic activity. J. Med. Chem. 2002, 45, 789–804. [DOI] [PubMed] [Google Scholar]
- Gupta R. A.; Brockman J. A.; Sarraf P.; Willson T. M.; DuBois R. N. Target genes of peroxisome proliferator-activated receptor gamma in colorectal cancer cells. J. Biol. Chem. 2001, 276, 29681–29687. [DOI] [PubMed] [Google Scholar]
- Fukuen S.; Iwaki M.; Yasui A.; Makishima M.; Matsuda M.; Shimomura I. Sulfonylurea agents exhibit peroxisome proliferator-activated receptor gamma agonistic activity. J. Biol. Chem. 2005, 280, 23653–23659. [DOI] [PubMed] [Google Scholar]
- Lehmann J. M.; Moore L. B.; Smith-Oliver T. A.; Wilkison W. O.; Willson T. M.; Kliewer S. A. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 1995, 270, 12953–12956. [DOI] [PubMed] [Google Scholar]
- Schoonjans K.; Auwerx J. Thiazolidinediones: an update. Lancet 2000, 355, 1008–1010. [DOI] [PubMed] [Google Scholar]
- Brown P. J.; Stuart L. W.; Hurley K. P.; Lewis M. C.; Winegar D. A.; Wilson J. G.; Wilkison W. O.; Ittoop O. R.; Willson T. M. Identification of a subtype selective human PPARalpha agonist through parallel-array synthesis. Bioorg. Med. Chem. Lett. 2001, 11, 1225–1227. [DOI] [PubMed] [Google Scholar]
- Jarvis M. C.; Gray T. J.; Palmer C. N. Both PPARgamma and PPARdelta influence sulindac sulfide-mediated p21WAF1/CIP1 upregulation in a human prostate epithelial cell line. Oncogene 2005, 24, 8211–8215. [DOI] [PubMed] [Google Scholar]
- Lewis J. D.; Lichtenstein G. R.; Deren J. J.; Sands B. E.; Hanauer S. B.; Katz J. A.; Lashner B.; Present D. H.; Chuai S.; Ellenberg J. H.; Nessel L.; Wu G. D. Rosiglitazone for active ulcerative colitis: a randomized placebo-controlled trial. Gastroenterology 2008, 134, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh K. A.; Stamp G. W.; Kirkland S. C. Isolation and characterization of multiple cell types from a single human colonic carcinoma: tumourigenicity of these cell types in a xenograft system. J. Pathol. 1993, 170, 441–450. [DOI] [PubMed] [Google Scholar]
- Nichols J. S.; Parks D. J.; Consler T. G.; Blanchard S. G. Development of a scintillation proximity assay for peroxisome proliferator-activated receptor gamma ligand binding domain. Anal. Biochem. 1998, 257, 112–119. [DOI] [PubMed] [Google Scholar]
- Mais D. E.; Hamann L. G.; Klausing K. U.; Paterniti J. R.; Mukherjee R. Characterization of a synthetic tritium-labeled peroxisome proliferator activated receptor (hPPAR-gamma) ligand. Med. Chem. Res. 1997, 7, 325–334. [Google Scholar]
- Laemmli U. K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970, 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Frisch M. J..; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Montgomery J., J. A.; Vreven T.; Kudin K. N. ; Burant J. C.; Millam J. M.; Iyengar S. S.; Tomasi J.; Barone V.; Mennucci B.; Cossi M.; Scalmani G.; Rega N.; Petersson G. A.; Nakatsuji H.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y. ; Kitao O.; Nakai H.; Klene M.; Li X.; Knox J. E.; Hratchian H. P.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O. ; Austin A. J.; Cammi R. ; Pomelli C. ; Ochterski J. W.; Ayala P. Y.; Morokuma K. ; Voth G. A.; Salvador P. ; Dannenberg J. J.; Zakrzewski V. G.; Dapprich S. ; Daniels A. D.; Strain M. C.; Farkas O. ; Malick D. K.; Rabuck A. D.; Raghavachari K. ; Foresman J. B.; Ortiz J. V.; Cui Q. ; Baboul A. G.; Clifford S. ; Cioslowski J. ; Stefanov B. B.; Liu G. ; Liashenko A. ; Piskorz P. ; Komaromi I. ; Martin R. L.; Fox D. J.; Keith T. ; Al.Laham M. A.; Peng C. Y.; Nanayakkara A. ; Challacombe M. ; Gill P. M. W.; Johnson B. ; Chen W. ; Wong M. W.; Gonzalez C. ; Pople J. A.. Gaussian 03, revision D.01; Gaussian, Inc.: Wallingford, CT, 2004.
- Swanson E.PSSHOW, version 2.4; Seattle, WA, 1995.
- Case D. A.; Darden T. A.; Cheatham T. E. III; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Merz K. M.; Pearlman D. A.; Crowley M.; Walker R. C.; Zhang W.; Wang B.; Hayik S.; Roitberg A.; Seabra G.; Wong K. F.; Paesani F.; Wu X. ; Brozell S.; Tsui V.; Gohlke H.; Yang L.; Tan C.; Mongan J.; Hornak V.; Cui G.; Beroza P.; Mathews D. H.; Schafmeister C.; Ross W. S.; Kollman P. A.. AMBER 9; University of California: San Francisco, 2006. [Google Scholar]
- Bayly C. I.; Cieplak P.; Cornell W. D.; Kollman P. A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar]
- DINO, Visualizing Structural Biology; 2002; http://www.dino3d.org.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.