


Abstract
Recently, we synthesized pyrimidine derivatives of the 2′-O,4′-C-methylenoxymethylene-bridged nucleic-acid (2′,4′-BNACOC) monomer, the sugar conformation of which is restricted in N-type conformation by a seven-membered bridged structure. Oligonucleotides (BNACOC) containing this monomer show high affinity with complementary single-stranded RNA and significant resistance to nuclease degradation. Here, BNACOC consisting of 2′,4′-BNACOC monomers bearing all four bases, namely thymine, 5-methylcytosine, adenine and guanine was efficiently synthesized and properties of duplexes containing the 2′,4′-BNACOC monomers were investigated by UV melting experiments and circular dichroism (CD) spectroscopy. The UV melting curve analyses showed that the BNACOC/BNACOC duplex possessed excellent thermal stability and that the BNACOC increased thermal stability with a complementary RNA strand. On the other hand, BNACOC/DNA heteroduplexes showed almost the same thermal stability as RNA/DNA heteroduplexes. Furthermore, mismatched sequence studies showed that BNACOC generally improved the sequence selectivity with Watson–Crick base-pairing compared to the corresponding natural DNA and RNA. A CD spectroscopic analysis indicated that the BNACOC formed duplexes with complementary DNA and RNA in a manner similar to natural RNA.
INTRODUCTION
Antisense oligonucleotides are now attracting interest for their potential to be developed as a new class of drugs for treatment of inveterate diseases such as cancer and viral diseases. For practical application of antisense methodology, it is essential to develop modified oligonucleotides, which strongly interact with single-stranded RNA (ssRNA) in a sequence-specific manner (1–3). The sugar moiety of natural nucleosides and single-stranded oligonucleotides exists in an individual equilibrium mixture between S-type and N-type conformations. However, it is well known that the B-form DNA duplex possesses the S-type sugar conformation, and that a range of N-type sugar conformation (pseudorotation phase angle, 0 ≤ P ≤ 36°) are adapted to the A-form RNA duplex structure (Figure 1) (4–6). Therefore, modified oligonucleotides, which have sugar moiety restricted to the S-type or N-type conformations in advance, are expected to have high binding affinity with complementary ssDNA or ssRNA respectively. We have so far developed various kinds of bridged nucleic acids (BNAs) (7–19), the sugar conformation of which is restricted or locked by introduction of an additional bridged structure to the furanose skeleton. It has been observed that 2′,4′-BNA (9,10)/LNA [The 2′,4′-BNA was independently synthesized by the group of Wengel et al. immediately after our first report (9), and it is called a locked nucleic acid (LNA). See refs (3,20–22)] (Figure 2), which is a BNAs with its sugar moiety fixed to an N-type conformation by five-membered ring, prominently hybridizes to ssRNA targets. An X-ray crystallographic analysis of 2′,4′-BNA showed that the maximum out-of-plane (νmax) value was 57°, and that this value is larger than an adapted value (νmax = 38.6° ± 3°) to natural A-form RNA duplex (4–6), so the N-type nature of 2′,4′-BNA was emphasized due to the restriction of the sugar moiety by the small five-membered ring. Other nucleic-acid analogues with a different type of bridged structure between the 2′- and 4′-positions, which have six- and seven-membered ring, have been reported (16–19,23–29). In one of these studies, we described the synthesis and properties of 2′-O,4′-C-aminomethylene BNA (2′,4′-BNANC) (Figure 2), and showed that oligonucleotides containing 2′,4′-BNANC have high hybridizing affinity with RNA compliments (16–19). X-ray crystallographic analysis revealed that the P and νmax values of 2′,4′-BNANC[NMe] were 23° and 49°, respectively (19). These values indicated that the conformation of 2′,4′-BNANC[NMe] is restricted to the N-type conformation as seen in natural A-form RNA duplex. This suggested that the sugar conformation restricted by the bridged structure between the 2′- and 4′-positions approximated to the adapted sugar conformation of the natural A-form RNA duplex because of increased ring size. Recently, we designed 2′,4′-BNACOC, bearing a seven-membered bridged structure, and successfully synthesized the 2′,4′-BNACOC monomers having pyrimidine nucleobases (Figure 2) (30). The oligonucleotides containing these monomers show high affinity with complementary ssRNA. An X-ray crystallographic analysis of 2′,4′-BNACOC bearing thymine showed that the P and νmax values were 17° and 38°, respectively. This revealed that the sugar conformation of 2′,4′-BNACOC, which is restricted by a large seven-membered ring, is identical with the N-type sugar puckering fit to canonical A-form RNA duplex, and that 2′,4′-BNACOC has the closest sugar conformation among nucleic-acid analogues with a different type of bridged structure between the 2′- and 4′-positions.
Figure 1.

Sugar conformations and helix structures of double-stranded nucleic acids.
Figure 2.

Structures of 2′,4′-BNA/LNA, ENA, 2′,4′-BNANC, PrNA and 2′,4′-BNACOC monomers.
It is of great interest to synthesize 2′,4′-BNACOC bearing a purine nucleobase for potential use in a wide variety of applications and to investigate the conformations of oligonucleotides containing 2′,4′-BNACOC. Moreover, conformational analysis of duplex, which is formed from fully modified oligonucleotides consisted of 2′,4′-BNACOC, is very fascinating. In general, a purine glycosidic linkage is more acid-labile than a pyrimidine glycosidic linkage (31). Therefore, it is readily expected that the acidic conditions used for the synthesis of 2′,4′-BNACOC with a pyrimidine nucleobase are not suitable for the purine congener (Scheme 1). Here, we report the synthesis of 2′,4′-BNACOC monomers having a purine nucleobase via formation of a COC linkage under mild conditions, and we describe the properties of the corresponding oligonucleotide derivatives.
Scheme 1.

Reagents and conditions: (a) p-TsOH·H2O, (CH2O)n, DCE, reflux, 3 h, 81%; (b) TBAF, THF, rt, 3 h, 91%; (c) 20% Pd(OH)2-C, cyclohexene, EtOH, reflux, 3 h, 89%.
MATERIALS AND METHODS
General aspects and instrumentation
All reactions were performed under an atmosphere of nitrogen. Unless otherwise mentioned, all chemicals from commercial sources were used without further purification. Acetonitrile (MeCN), dichloromethane (CH2Cl2), dichloroethane (DCE), triethylamine and pyridine were distilled from CaH2. Tetrahydrofuran (THF) was distilled from CaH2 followed by LiAlH4 just before use. All melting points were measured with a Yanagimoto micro melting point apparatus and are uncorrected. 1H NMR (270 or 300 MHz), 13C NMR (67.8 or 75.5 MHz) and 31P NMR (202.4 MHz) spectra were recorded on JEOL EX-270, JEOL-AL-300 and JEOL GX-500 spectrometers, respectively. Chemical shifts are reported in parts per million downfield from tetramethylsilane or deuterated solvent as internal standard for 1H and 13C spectra, and 85% H3PO4 as external standard for 31P spectra. IR spectra were recorded on a JASCO FT/IR-200 spectrometer. Optical rotations were recorded on a JASCO DIP-370 instrument. Mass spectra were measured on JEOL JMS-600 or JMS-700 mass spectrometers. Column chromatography was carried out using Fuji Silysia PSQ100B or FL-100D. Matrix-assisted laser desorption ionization-time of flight (MALDI-TOF) mass spectra were recorded on Bruker Daltonics® Autoflex II TOF/TOF instruments.
4′-C-Acetoxymethyl-2′-O-acetyl-6-N-benzoyl-3′-O-benzyl-5′-O-tert-butyldiphenylsilyladenosine (5)
Trimethylsilyl chloride (TMSCl) (0.5 ml) was added to a suspension of N6-benzoyladenine (2.17 g, 9.07 mmol) in anhydrous hexamethyldisilazane (HMDS) (35 ml) at room temperature and the mixture was refluxed for 24 h. After the resulting clear solution was evaporated under reduced pressure, to the residue a solution of compound 4 (30) (4.80 g, 7.56 mmol) in anhydrous DCE (25 ml) and trimethylsilyl trifluorimethanesulfonate (TMSOTf) (0.14 ml, 0.756 mmol) were added and the mixture was refluxed for 11 h. After addition of saturated aqueous NaHCO3 solution at 0°C, the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (3:4→1:2, v/v)] to give compound 5 (4.68 g, 76%). White powder; m.p. 71–73°C. [α]D26 − 6.80 (c 1.03, CHCl3). IR νmax (KBr): 3065, 2939, 2862, 1744, 1703, 1607, 1458, 1368, 1235, 1106 cm−1. 1H NMR (CDCl3): δ 1.06 (9H, s), 1.97 (3H, s), 2.05 (3H, s), 3.81(1H, d, J = 11 Hz), 3.96 (1H, d, J = 11 Hz), 4.30 (1H, d, J = 12 Hz), 4.66 (1H, d, J = 12 Hz), 4.62 (2H, s), 4.91 (1H, d, J = 6 Hz), 6.08 (1H, t, J = 6 Hz), 6.25 (1H, d, J = 5 Hz), 7.31–7.66 (18H, m), 8.01 (2H, d, J = 7 Hz), 8.09 (1H, s), 8.59 (1H, s), 9.04 (1H, brs). 13C NMR (CDCl3): δ 19.3, 20.7, 20.9, 26.9, 26.9, 26.9, 62.7, 64.0, 74.1, 74.7, 78.1, 86.6, 87.0, 123.4, 127.7, 127.7, 127.7, 127.7, 127.7, 127.7, 127.8, 127.8, 128.0, 128.4, 128.4, 128.7, 128.7, 129.8, 129.8, 132.3, 132.4, 132.6, 133.4, 135.4, 135.4, 135.4, 135.4, 136.9, 142.1, 149.4, 151.2, 152.4, 164.4, 169.7, 170.4. Mass (FAB): m/z 814 (MH+). Anal. Calcd for C45H47N5O8Si·1/2H2O: C, 65.67; H, 5.88; N, 8.51. Found: C, 65.44; H, 5.71; N, 8.42.
6-N-Benzoyl-3′-O-benzyl-5′-O-tert-butyldiphenylsilyl-4′-C-(hydroxymethyl)adenosine (6)
K2CO3 (98 mg, 0.712 mmol) was added to a solution of compound 5 (193 mg, 0.237 mmol) in MeOH (3.4 ml) at 0°C, and the mixture was stirred at the same temperature for 1.5 h. After neutralization with a 10% aqueous HCl solution (0.26 ml), the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (1:3, v/v)] to give compound 6 (170 mg, 98%). White powder; m.p. 88–90°C. [α]D26 + 4.43 (c 1.43, CHCl3). IR νmax (KBr): 3287, 3062, 2937, 2862, 1702, 1612, 1461, 1328, 1253, 1109 cm−1. 1H NMR (CDCl3): δ 1.00 (9H, s), 2.57 (1H, dd, J = 5, 7 Hz), 3.71 (1H, d, J = 11 Hz), 3.76 (1H, d, J = 11 Hz), 3.86 (1H, dd, J = 7, 12 Hz), 4.02 (1H, dd, J = 5, 12 Hz), 4.52 (1H, d, J = 8 Hz), 4.66 (1H, d, J = 6 Hz), 4.68 (1H, d, J = 11 Hz), 4.85 (1H, d, J = 11 Hz), 4.90 (1H, m), 6.10 (1H, d, J = 4 Hz), 7.22–7.44 (11H, m), 7.50–7.65 (7H, m), 8.02 (2H, d, J = 7 Hz), 8.08 (1H, s), 8.64 (1H, s), 8.99 (1H, brs). 13C NMR (CDCl3): δ 19.3, 26.9, 26.9, 26.9, 63.0, 65.8, 73.8, 74.2, 78.7, 89.3, 91.0, 123.1, 127.6, 127.6, 127.7, 127.7, 127.7, 128.0, 128.0, 128.2, 128.2, 128.5, 128.5, 128.7, 128.7, 129.8, 129.8, 132.2, 132.5, 132.7, 133.4, 135.3, 135.3, 135.4, 135.4, 137.0, 142.0, 149.4, 150.7, 152.3, 164.4. Mass (FAB): m/z 730 (MH+). Anal. Calcd for C41H43N5O6Si·3/2H2O: C, 65.06; H, 6.13; N, 9.25. Found: C, 65.30; H, 5.86; N, 9.19.
4′-C-Acetoxymethyl-2′-O-acetyl-6-N-benzoyl-3′-O-benzyl-6-N-benzyloxymethyl-5′-O-tert-butyldiphenylsilyladenosine (8)
DBU (0.39 ml, 2.58 mmol) was added to a solution of compound 5 (1.05 g, 1.29 mmol) in anhydrous DMF (6.4 ml) at 0°C and the mixture was stirred for 10 min. Benzyloxymethyl chloride (BOMCl) (0.27 ml, 1.93 mmol) was added to the reaction mixture at 0°C and the mixture was stirred at the same temperature for 1 h. After addition of water at 0°C, the mixture was extracted with Et2O. The organic extracts were washed with 0.5 M aqueous KHSO4 solution, water and brine, dried over MgSO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (2:1, v/v)] to give compound 8 (769 mg, 64%). White foam. [α]D26 − 16.4 (c 1.23, CHCl3). IR νmax (KBr): 3064, 2939, 2863, 2251, 1962, 1894, 1745, 1680, 1581, 1456, 1366, 1227, 1105 cm−1. 1H NMR (CDCl3): δ 1.04 (9H, s), 1.96 (3H, s), 2.03 (3H, s), 3.79 (1H, d, J = 11 Hz), 3.92 (1H, d, J = 11 Hz), 4.28 (1H, d, J = 12 Hz), 4.62 (1H, d, J = 12 Hz), 4.59 (2H, s), 4.76 (2H, s), 4.87 (1H, d, J = 6 Hz), 5.87 (2H, s), 5.98 (1H, t, J = 6 Hz), 6.18 (1H, d, J = 5 Hz), 7.14–7.46 (19H, m), 7.50 (2H, d, J = 7 Hz), 7.60-7.64 (4H, m), 7.99 (1H, s), 8.38 (1H, s). 13C NMR (CDCl3): δ 19.3, 20.7, 20.9, 26.9, 26.9, 26.9, 62.8, 63.9, 71.3, 74.0, 74.7, 76.7, 78.0, 86.5, 87.0, 127.1, 127.5, 127.5, 127.7, 127.7, 127.7, 127.8, 127.8, 127.8, 127.9, 127.9, 128.1, 128.1, 128.1, 128.1, 128.4, 128.4, 128.7, 128.7, 129.9, 129.9, 131.0, 132.3, 132.4, 135.3, 135.4, 135.4, 135.5, 135.5, 136.9, 137.6, 142.8, 151.9, 152.1, 153.2, 169.7, 170.4, 172.2. Mass (FAB): m/z 934 (MH+). Anal. Calcd for C53H55N5O9Si·1/3H2O: C, 67.71; H, 5.97; N, 7.45. Found: C, 67.64; H, 5.88; N, 7.40.
6-N-Benzoyl-3′-O-benzyl-6-N-benzyloxymethyl-5′-O-tert-butyldiphenylsilyl-4′-C-(hydroxymethyl)adenosine (9)
K2CO3 (1.61 g, 11.6 mmol) was added to a solution of compound 8 (3.62 g, 3.88 mmol) in MeOH (80 ml) at 0°C and the mixture was stirred for 50 min. After neutralization with a 10% aqueous HCl solution (4.3 ml), the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (3:2 to 1:1, v/v)] to give compound 9 (3.21 g, 97%). White powder; m.p. 54–57°C. [α]D26 − 1.94 (c 1.23, CHCl3). IR νmax (KBr): 3326, 3064, 2940, 2864, 2247, 1962, 1894, 1820, 1677, 1581, 1458, 1281, 1210, 1070 cm−1. 1H NMR (CDCl3): δ 0.99 (9H, s), 2.51 (1H, dd, J = 5, 7 Hz), 3.70 (2H, s), 3.80 (1H, dd, J = 7, 12 Hz), 3.97 (1H, dd, J = 5, 12 Hz), 4.47 (1H, d, J = 9 Hz), 4.58 (1H, d, J = 6 Hz), 4.63 (1H, d, J = 11 Hz), 4.80 (1H, d, J = 11 Hz), 4.77 (1H, m), 4.78 (2H, s), 5.88 (2H, s), 6.04 (1H, d, J = 4 Hz), 7.10-7.44 (19H, m), 7.48–7.56 (6H, m), 8.00 (1H, s), 8.43 (1H, s). 13C NMR (CDCl3): δ 19.3, 26.9, 26.9, 26.9, 63.2, 65.7, 71.3, 73.9, 74.2, 76.7, 78.7, 89.1, 90.9, 127.1, 127.5, 127.5, 127.7, 127.7, 127.7, 127.7, 127.9, 127.9, 128.0, 128.0, 128.1, 128.1, 128.3, 128.3, 128.6, 128.6, 128.7, 128.7, 129.9, 129.9, 131.1, 132.2, 132.4, 135.3, 135.3, 135.3, 135.4, 135.4, 136.9, 137.6, 142.6, 151.6, 151.8, 153.2, 172.2. Mass (FAB): m/z 850 (MH+). Anal. Calcd for C49H51N5O7Si·3/4H2O: C, 68.15; H, 6.13; N, 8.11. Found: C, 68.30; H, 6.06; N, 8.07.
6-N-Benzoyl-3′-O-benzyl-6-N-benzyloxymethyl-5′-O-tert-butyldiphenylsilyl-2′-O,4′-C- (methylenoxymethylene)adenosine (10)
N-bromosuccinimide (NBS) (312 mg, 1.75 mmol) was added to a solution of compound 9 (373 mg, 0.438 mmol) in anhydrous dimethylsulfoxide (DMSO) (5.5 ml) at room temperature and the mixture was stirred at 60°C for 1.5 h. After addition of saturated aqueous NaHCO3 solution at 0°C, the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (3:1, v/v)] to give compound 10 (281 mg, 74%). White foam. [α]D27 − 3.97 (c 1.01, CHCl3). IR νmax (KBr): 3064, 2955, 2865, 1679, 1580, 1456, 1362, 1281, 1209, 1176, 1117, 1074 cm−1. 1H NMR (CDCl3): δ 0.97 (9H, s), 3.68 (1H, d, J = 12 Hz), 3.81 (1H, d, J = 12 Hz), 3.72 (1H, d, J = 12 Hz), 3.88 (1H, d, J = 12 Hz), 4.62 (1H, d, J = 11 Hz), 4.81 (1H, d, J = 11 Hz), 4.79 (2H, s), 4.83 (1H, d, J = 6 Hz), 4.99 (1H, d, J = 6 Hz), 5.25 (1H, d, J = 6 Hz), 5.40 (1H, d, J = 6 Hz), 5.91 (2H, s), 6.42 (1H, s), 7.10-7.42 (19H, m), 7.50 (2H, d, J = 7 Hz), 7.54–7.59 (4H, m), 8.14 (1H, s), 8.50 (1H, s). 13C NMR (CDCl3): δ 19.2, 26.8, 26.8, 26.8, 62.7, 71.3, 71.7, 72.8, 75.8, 76.0, 76.7, 89.0, 91.8, 96.0, 127.3, 127.5, 127.5, 127.6, 127.6, 127.7, 127.7, 127.8, 127.8, 127.9, 127.9, 127.9, 127.9, 128.1, 128.1, 128.4, 128.4, 128.7, 128.7, 129.8, 129.8, 130.9, 132.2, 132.3, 135.3, 135.3, 135.4, 135.4, 135.4, 136.8, 137.7, 142.6, 151.5, 151.9, 153.1, 172.2. Mass (FAB): m/z 862 (MH+). Anal. Calcd for C50H51N5O7Si·1/10H2O: C, 69.52; H, 5.97; N, 8.11. Found: C, 69.29; H, 5.86; N, 7.96.
3′-O-Benzyl-6-N-benzyloxymethyl-5′-O-tert-butyldiphenylsilyl-2′-O,4′-C-(methylenoxymethylene)adenosine (11)
Twenty-eight percent aqueous NH3 (1.2 ml) was added to a solution of compound 10 (209 mg, 0.242 mmol) in THF (2.4 ml) at room temperature. The flask was sealed and the mixture was stirred at 50°C for 48 h. The mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (2:1, v/v)] to give compound 11 (186 mg, quant.). White foam. [α]D27 + 2.62 (c 1.33, CHCl3). IR νmax (KBr): 3266, 3037, 2938, 2246, 1959, 1897, 1615, 1475, 1418, 1357, 1292, 1176, 1116 cm−1. 1H NMR (CDCl3): δ 0.99 (9H, s), 3.69 (1H, d, J = 11 Hz), 3.73 (1H, d, J = 12 Hz), 3.83 (1H, d, J = 11 Hz), 3.90 (1H, d, J = 12 Hz), 4.62 (1H, d, J = 11 Hz), 4.66 (2H, s), 4.82 (1H, d, J = 11 Hz), 4.92 (1H, d, J = 6 Hz), 5.04 (1H, d, J = 6 Hz), 5.27 (1H, d, J = 6 Hz), 5.28 (2H, brd, J = 7 Hz), 5.41 (1H, d, J = 6 Hz), 6.43 (1H, s), 6.52 (1H, brs), 7.21–7.43 (16H, m), 7.57–7.60 (4H, m), 8.03 (1H, s), 8.38 (1H, s). 13C NMR (CDCl3): δ 19.2, 26.7, 26.7, 26.7, 62.9, 70.0, 71.6, 72.7, 75.9, 76.0, 88.7, 91.8, 95.9, 120.5, 127.5, 127.5, 127.6, 127.6, 127.6, 127.6, 127.7, 127.7, 127.8, 127.8, 128.0, 128.0, 128.2, 128.2, 128.4, 128.4, 129.7, 129.7, 132.2, 132.4, 135.3, 135.3, 135.4, 135.4, 136.9, 137.8, 139.5, 152.7, 154.3. Mass (FAB): m/z 758 (MH+). Anal. Calcd for C43H47N5O6Si: C, 68.14; H, 6.25; N, 9.24. Found: C, 68.11; H, 6.29; N, 9.10.
3′-O-Benzyl-6-N-benzyloxymethyl-2′-O,4′-C-(methylenoxymethylene)adenosine (12)
Tetrabutylammonium fluoride (TBAF) (1.0 M solution in THF, 0.19 ml, 0.19 mmol) was added to a solution of compound 11 (128 mg, 0.169 mmol) in THF (1.7 ml) at room temperature and the mixture was stirred at room temperature for 3 h. After addition of hexane/AcOEt (1:1, v/v, 2.0 ml) at room temperature, the mixture was evaporated under reduced pressure to one-tenth of its volume. The residue was purified by silica gel column chromatography [hexane/AcOEt (2:5, v/v)] to give compound 12 (80 mg, 91%). White foam. [α]D27 − 8.40 (c 1.17, CHCl3). IR νmax (KBr): 3286, 2919, 2245, 1959, 1616, 1484, 1358, 1293, 1213, 1176, 1116, 1056 cm−1. 1H NMR (CDCl3): δ 2.21 (1H, brs), 3.69 (1H, d, J = 12 Hz), 3.82 (1H, d, J = 12 Hz), 3.79 (2H, s), 4.52 (1H, d, J = 6 Hz), 4.65 (2H, s), 4.70 (1H, d, J = 11 Hz), 4.78 (1H, d, J = 11 Hz), 5.25 (1H, d, J = 6 Hz), 5.26 (2H, brs), 5.26 (1H, d, J = 6 Hz), 5.43 (1H, d, J = 6 Hz), 6.35 (1H, s), 7.14 (1H, brs), 7.22–7.38 (10H, m), 7.86 (1H, s), 8.35 (1H, s). 13C NMR (CDCl3): δ 61.1, 70.1, 71.7, 73.0, 74.9, 77.5, 89.7, 93.1, 96.0, 120.5, 127.6, 127.7, 127.7, 127.8, 127.8, 127.8, 128.0, 128.3, 128.3, 128.3, 128.3, 128.3, 136.9, 137.7, 139.5, 152.5, 154.5. Mass (FAB): m/z 520 (MH+). Anal. Calcd for C27H29N5O6·1/6H2O: C, 62.06; H, 5.66; N, 13.40. Found: C, 61.79; H, 5.63; N, 13.29.
2′-O,4′-C-(Methylenoxymethylene)adenosine (13)
Twenty percent Pd(OH)2-C (750 mg) and ammonium formate (3.00 g, 47.43 mmol) were added to a solution of compound 12 (493 mg, 0.949 mmol) in EtOH/AcOH (100:3, v/v, 24 ml) at room temperature and the mixture was refluxed for 3.5 h. The hot solution was filtrated through a Celite pad and Celite was washed with boiling MeOH (200 ml). After addition of SiO2 (3.0 g) to the filtrate, the mixture was concentrated under reduced pressure. The residue was purified by silica gel column chromatography [CHCl3/MeOH (15:1→12:1, v/v)] to give compound 13 (162 mg, 55%). Colorless crystals; m.p. 255–257°C (MeOH). [α]D23 − 26.8 (c 0.86, DMSO). IR νmax (KBr): 3132, 1683, 1611, 1477, 1418, 1330, 1175, 1119, 1080 cm−1. 1H NMR (DMSO-d6): δ 3.53 (1H, dd, J = 6, 12 Hz), 3.61 (1H, dd, J = 6, 12 Hz), 3.65 (1H, d, J = 12 Hz), 3.80 (1H, d, J = 12 Hz), 4.41 (1H, d, J = 6 Hz), 4.85 (1H, t, J = 6 Hz), 5.09 (1H, d, J = 6 Hz), 5.30 (1H, d, J = 6 Hz), 5.14 (1H, t, J = 6 Hz), 6.09 (1H, brd, J = 4 Hz), 6.23 (1H, s), 7.27 (2H, s), 8.13 (1H, s), 8.32 (1H, s). 13C NMR (DMSO-d6): δ 60.9, 68.9, 70.7, 78.7, 88.8, 90.0, 94.9, 119.2, 139.0, 148.5, 152.5, 156.0. Mass (FAB): m/z 310 (MH+). Anal. Calcd for C12H15N5O5: C, 46.60; H, 4.89; N, 22.64. Found: C, 46.39; H, 4.99; N, 22.38.
6-N-Benzoyl-2′-O,4′-C-(methylenoxymethylene)adenosine (14)
Benzoyl chloride (0.016 μl, 0.136 mmol) was added to a solution of compound 13 (8.4 mg, 0.027 mmol) in anhydrous pyridine (0.3 ml) at room temperature and the mixture was stirred at room temperature for 12 h. After addition of saturated aqueous NaHCO3 solution at 0°C, the mixture was extracted with AcOEt. The organic extracts were washed with saturated aqueous NaHCO3 solution twice, followed by washes with water and brine, then dried over Na2SO4, and concentrated under reduced pressure. The residue was dissolved in MeOH/1,4-dioxane (2:1, v/v, 0.81 ml) and NaOMe (28% solution in MeOH, 0.052 ml) was added. After stirring for 5 min at room temperature, the mixture was neutralized with DOWEX 50W-X2 and filtrated. After addition of SiO2 (1.0 g) to the filtrate, the mixture was concentrated under reduced pressure. The residue was purified by silica gel column chromatography [CHCl3/MeOH (40:1→20:1, v/v)] to give compound 14 (7.7 mg, 68% over two steps). White powder; m.p. 133–136°C. [α]D24 − 17.7 (c 0.88, DMSO). IR νmax (KBr): 3289, 2924, 1692, 1610, 1517, 1457, 1400, 1255, 1176, 1110 cm−1. 1H NMR (DMSO-d6): δ 3.55 (1H, dd, J = 6, 12 Hz), 3.63 (1H, dd, J = 6, 12 Hz), 3.66 (1H, d, J = 12 Hz), 3.81 (1H, d, J = 12 Hz), 4.55 (1H, d, J = 6 Hz), 4.87 (1H, t, J = 6 Hz), 5.12 (1H, d, J = 6 Hz), 5.32 (1H, d, J = 6 Hz), 5.20 (1H, t, J = 6 Hz), 6.19 (1H, brd, J = 5 Hz), 6.36 (1H, s), 7.52–7.67 (3H, m), 8.04 (2H, d, J = 7 Hz), 8.66 (1H, s), 8.74 (1H, s), 11.24 (1H, brs). 13C NMR (DMSO-d6): δ 60.6, 68.8, 70.6, 78.5, 89.1, 90.2, 95.0, 126.0, 128.5, 128.5, 128.5, 128.5, 132.4, 133.6, 142.5, 150.7, 151.3, 151.5, 165.7. Mass (FAB): m/z 414 (MH+). High-resolution MS (FAB): Calcd for C19H20N5O6 (MH+): 414.1414. Found: 414.1397.
6-N-Benzoyl-5′-O-(4,4′-dimethoxytrityl)-2′-O,4′-C-(methylenoxymethylene)adenosine (15)
Synthesis of compound 15 from compound 14 (path A)
To a suspension of AgOTf (622 mg, 2.42 mmol) in CH2Cl2 (2.0 ml) was added dropwise DMTrCl (820 mg, 2.42 mmol) in CH2Cl2 (8.0 ml) at room temperature, and the mixture was stirred for 1 h at room temperature. The supernatant fluid (2.5 ml, 2.5 eq.) was added to a solution of compound 14 (101 mg, 0.244 mmol) in CH2Cl2/pyridine (1:1, v/v, 4.8 ml) at room temperature and the mixture was stirred for 1 h. After addition of saturated aqueous NaHCO3 solution at 0°C, the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (1:2, v/v)] to give compound 15 (183 mg, quant.). White powder; m.p. 125–127°C. [α]D24 − 27.4 (c 0.71, CHCl3). IR νmax (KBr): 3325, 3062, 2922, 2245, 2050, 1901, 1703, 1609, 1509, 1456, 1251, 1178, 1073 cm−1. 1H NMR (CDCl3): δ 3.37 (1H, d, J = 10 Hz), 3.42 (1H, d, J = 10 Hz), 3.39 (1H, d, J = 9 Hz), 3.77 (6H, s), 4.01 (1H, d, J = 13 Hz), 4.06 (1H, d, J = 13 Hz), 4.92 (1H, d, J = 6 Hz), 4.99 (1H, dd, J = 6, 9 Hz), 5.23 (1H, d, J = 7 Hz), 5.33 (1H, d, J = 7 Hz), 6.39 (1H, s), 6.78 (2H, d, J = 9 Hz), 6.79 (2H, d, J = 9 Hz), 7.19–7.27 (7H, m), 7.34–7.38 (2H, m), 7.48–7.64 (3H, m), 8.01 (2H, d, J = 7 Hz), 8.10 (1H, s), 8.74 (1H, s), 9.07 (1H, s). 13C NMR (CDCl3): δ 55.2, 55.2, 64.5, 72.8, 73.4, 78.9, 86.6, 88.6, 88.9, 93.5, 113.1, 113.1, 113.1, 113.1, 123.3, 126.9, 127.7, 127.7, 127.8, 127.8, 127.8, 127.8, 128.7, 128.7, 129.8, 129.8, 129.8, 129.8, 132.7, 133.4, 135.0, 135.1, 141.3, 143.9, 149.4, 150.5, 152.4, 158.4, 158.4, 164.5. Mass (FAB): m/z 716 (MH+). High-resolution MS (FAB): Calcd for C40H38N5O8 (MH+): 716.2720. Found: 716.2716.
Synthesis of compound 15 from compound 13 (path B)
To a suspension of AgOTf (1.06 g, 4.13 mmol) in CH2Cl2 (3.0 ml) was added dropwise DMTrCl (1.40 g, 4.13 mmol) in CH2Cl2 (7.0 ml) at room temperature and the mixture was stirred for 1 h. The supernatant fluid (3.0 ml, 3.0 eq.) was added to a solution of compound 13 (128 mg, 0.413 mmol) in CH2Cl2/pyridine (1:2, v/v, 8.4 ml) at room temperature and the mixture was stirred for 1.5 h. Benzoyl chloride (0.24 ml, 2.07 mmol) was added to the mixture at room temperature and the mixture was stirred for 24 h. After addition of saturated aqueous NaHCO3 solution at 0°C, the mixture was extracted with AcOEt. The organic extracts were washed with saturated aqueous NaHCO3 solution twice, followed by washes with water and brine, then dried over Na2SO4, and concentrated under reduced pressure. The residue was dissolved in THF (5.6 ml) and 1 M aqueous LiOH (2.8 ml, 2.8 mmol) was added. After stirring for 5 h at room temperature, the mixture was extracted with AcOEt. The organic extracts were washed with saturated aqueous NaHCO3 solution twice, followed by washes with water and brine, then dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (1:2, v/v)] to give compound 15 (272 mg, 92% over two steps).
6-N-Benzoyl-3′-O-[2-cyanoethoxy(diisopropylamino)phosphino]-5′-O-(4,4′-dimethoxytrityl)-2′-O,4′-C-(methylenoxymethylene)adenosine (17)
To a solution of compound 15 (283 mg, 0.396 mmol) in MeCN (3.0 ml) were added 4,5-dicyanoimidazole (0.25 M in MeCN, 1.9 ml, 0.48 mmol) and (i-Pr2N)2POCH2CH2CN (0.19 ml, 0.59 mmol) at room temperature and the mixture was stirred at room temperature for 16 h. After removal of the solvent under reduced pressure, the residue was diluted with AcOEt, washed with saturated aqueous NaHCO3 solution, water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (2:3, v/v)] followed by the precipitation from hexane/AcOEt to give compound 17 (304 mg, 84%). White powder; m.p. 99–102°C. 31P NMR (CDCl3): δ 150.7, 151.2. MS (FAB): m/z 916 (MH+). High-resolution MS (FAB): Calcd for C49H55N7O9P (MH+): 916.3799. Found: 916.3808.
4′-C-Acetoxymethyl-2′-O-acetyl-3′-O-benzyl-5′-O-tert-butyldiphenylsilyl-6-O-diphenylcarbamoyl-2-N-isobutyrylguanosine (18)
To a suspension of O6-(diphenylcarbamoyl)-N2-isobutyrylguanine (121 mg, 0.291 mmol) in DCE (1.9 ml) was added N,O-bis(trimethylsily)acetamide (0.17 ml, 0.678 mmol) at room temperature, and the mixture was stirred at 80°C for 30 min. After the resulting clear solution was evaporated under reduced pressure, to the residue a solution of compound 4 (123 mg, 0.194 mmol) in anhydrous toluene (1.9 ml) and TMSOTf (0.04 ml, 0.233 mmol) were added and the mixture was stirred at 80°C for 2 h. After addition of saturated aqueous NaHCO3 solution at 0°C, the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (2:1 to 3:2, v/v)] to give compound 18 (161 mg, 84%). White powder; m.p. 86–88°C. [α]D25 + 15.1 (c 0.97 CHCl3). IR νmax (KBr): 3327, 3063, 2961, 2866, 2463, 2251, 1959, 1744, 1588, 1500, 1453, 1408, 1227, 1106 cm−1. 1H NMR (CDCl3): δ 1.01 (9H, s), 1.15 (3H, d, J = 7 Hz), 1.19 (3H, d, J = 7 Hz), 1.94 (3H, s), 2.06 (3H, s), 2.77 (1H, m), 3.85 (2H, s), 4.31 (1H, d, J = 12 Hz), 4.66 (1H, d, J = 12 Hz), 4.65 (1H, d, J = 11 Hz), 4.74 (1H, d, J = 11 Hz), 5.23 (1H, d, J = 6 Hz), 5.88 (1H, dd, J = 4, 6 Hz), 6.09 (1H, d, J = 4 Hz), 7.18–7.45 (21H, m), 7.54–7.61 (4H, m), 7.82 (1H, brs), 7.98 (1H, s). 13C NMR (CDCl3): δ 19.3, 19.3, 19.3, 20.8, 20.9, 26.9, 26.9, 26.9, 36.0, 62.5, 64.9, 74.3, 74.6, 78.3, 87.0, 87.2, 121.3, 127.4, 127.4, 127.6, 127.6, 127.6, 127.6, 127.7, 127.7, 127.7, 127.7, 127.7, 128.2, 128.2, 128.2, 128.2, 129.0, 129.0, 129.0, 129.0, 129.7, 129.7, 132.5, 132.6, 135.4, 135.4, 135.4, 135.4, 137.7, 137.7, 141.6, 142.9, 150.1, 151.7, 154.0, 156.0, 169.7, 170.5, 174.3. Mass (FAB): m/z 991 (MH+). Anal. Calcd for C55H58N6O10Si·1/10H2O: C, 66.53; H, 5.91; N, 8.46. Found: C, 66.26; H, 5.96; N, 8.43.
3′-O-Benzyl-5′-O-tert-butyldiphenylsilyl-6-O-diphenylcarbamoyl-4′-C-hydroxymethyl-2-N-isobutyrylguanosine (19)
A mixture of 18 (100 mg, 0.10 mmol) and K2CO3 (42 mg, 0.30 mmol) in MeOH (1.4 ml) was stirred at 0°C for 40 min. After neutralization with a 10% aqueous HCl solution (0.11 ml), the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (3:2 to 1:1, v/v)] to give compound 19 (88 mg, 96%). White powder; m.p. 94–96°C. [α]D24 + 58.0 (c 0.91, CHCl3). IR νmax (KBr): 3311, 3063, 2958, 2865, 2248, 1958, 1890, 1744, 1588, 1499, 1452, 1407, 1330, 1180 cm−1. 1H NMR (CDCl3): δ 0.91 (9H, s), 1.24 (3H, d, J = 7 Hz), 1.25 (3H, d, J = 7 Hz), 2.57 (1H, m), 3.55 (1H, d, J = 11 Hz), 3.67 (1H, d, J = 11 Hz), 3.80 (1H, d, J = 12 Hz), 3.96 (1H, d, J = 12 Hz), 4.66 (1H, d, J = 5 Hz), 4.74 (1H, d, J = 12 Hz), 5.16 (1H, d, J = 12 Hz), 4.80 (1H, t, J = 5 Hz), 6.02 (1H, d, J = 5 Hz), 7.18–7.49 (25H, m), 8.05 (1H, brs), 8.11 (1H, s). 13C NMR (CDCl3): δ 19.2, 19.3, 19.4, 26.8, 26.8, 26.8, 36.9, 63.7, 66.1, 74.3, 76.2, 80.2, 89.6, 91.5, 121.2, 127.5, 127.5, 127.6, 127.6, 127.6, 127.6, 127.9, 127.9, 128.0, 128.0, 128.0, 128.5, 128.5, 128.5, 128.5, 129.1, 129.1, 129.1, 129.1, 129.7, 129.7, 132.3, 132.4, 135.3, 135.3, 135.3, 135.3, 137.8, 137.8, 141.5, 142.9, 150.1, 151.1, 153.3, 155.9, 174.4. Mass (FAB): m/z 907 (MH+). Anal. Calcd for C51H54N6O8Si·H2O: C, 66.21; H, 6.10; N, 9.08. Found: C, 66.15; H, 5.93; N, 9.04.
3′-O-Benzyl-5′-O-tert-butyldiphenylsilyl-6-O-diphenylcarbamoyl-2-N-isobutyryl-2′-O,4′-C-(methylenoxymethylene)guanosine (20)
To a solution of 19 (123 mg, 0.136 mmol) in anhydrous DMSO (1.7 ml) was added NBS (145 mg, 0.816 mmol) at room temperature and the mixture was stirred at 60°C for 40 min. After addition of saturated aqueous NaHCO3 solution at 0°C, the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (3:1, v/v)] to give compound 20 (56 mg, 45%). White powder; m.p. 97–99°C. [α]D23 + 39.1 (c 0.94, CHCl3). IR νmax (KBr): 3313, 3062, 2962, 2865, 2246, 1958, 1890, 1745, 1588, 1500, 1451, 1405, 1328, 1176, 1116, 1068 cm−1. 1H NMR (CDCl3): δ 0.97 (9H, s), 1.11 (3H, d, J = 7 Hz), 1.17 (3H, d, J = 7 Hz), 2.69 (1H, m), 3.80 (1H, d, J = 12 Hz), 4.12 (1H, d, J = 12 Hz), 3.93 (2H, s), 4.79 (1H, d, J = 6 Hz), 4.82 (2H, s), 5.23 (1H, d, J = 6 Hz), 5.47 (1H, d, J = 6 Hz), 5.41 (1H, d, J = 6 Hz), 6.32 (1H, s), 7.05–7.10 (2H, m), 7.20–7.59 (23H, m), 7.84 (1H, brs), 8.07 (1H, s). 13C NMR (CDCl3): δ 19.2, 19.2, 19.3, 26.8, 26.8, 26.8, 36.2, 63.9, 71.6, 72.8, 76.4, 77.1, 89.1, 92.3, 96.0, 121.8, 127.2, 127.2, 127.5, 127.5, 127.5, 127.5, 127.7, 127.7, 127.7, 127.7, 127.7, 128.3, 128.3, 128.3, 128.3, 129.1, 129.1, 129.1, 129.1, 129.5, 129.6, 132.4, 132.8, 135.3, 135.3, 135.5, 135.5, 137.8, 137.8, 141.6, 143.3, 150.2, 151.3, 153.5, 155.9, 174.0. Mass (FAB): m/z 919 (MH+). Anal. Calcd for C52H54N6O8Si·3/4H2O: C, 66.97; H, 6.00; N, 9.01. Found: C, 66.92; H, 5.91; N, 8.94.
3′-O-Benzyl-2-N-isobutyryl-2′-O,4′-C-(methylenoxymethylene)guanosine (21)
To a solution of 20 (801 mg, 0.871 mmol) in anhydrous DMSO (7.2 ml) was added NaNO2 (1.20 g, 17.42 mmol) at room temperature and the mixture was stirred at 70°C for 19 h. After addition of saturated aqueous NaHCO3 solution at 0°C, the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was dissolved in THF (8.7 ml) and TBAF (1.0 M in THF, 1.05 ml, 1.05 mmol) was added. After stirring for 15 h at room temperature, the solvent was removed under reduce pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (1:2, v/v) and AcOEt/EtOH (50:1, v/v)] to give compound 21 (264 mg, 62% over two steps). Pale brown powder; m.p. 144–146°C. [α]D26 − 30.0 (c 1.04, CHCl3). IR νmax (KBr): 3157, 2975, 2923, 2248, 1682, 1607, 1559, 1473, 1406, 1315, 1252, 1169, 1117, 1074 cm−1. 1H NMR (CDCl3): δ 1.25 (6H, d, J = 5 Hz), 2.72 (1H, m), 3.75 (1H, d, J = 12 Hz), 3.97 (1H, d, J = 12 Hz), 3.83 (1H, d, J = 12 Hz), 3.92 (1H, d, J = 12 Hz), 4.44 (1H, d, J = 5 Hz), 4.62 (1H, d, J = 11 Hz), 4.72 (1H, d, J = 11 Hz), 4.94 (1H, d, J = 5 Hz), 5.16 (1H, d, J = 6 Hz), 5.36 (1H, d, J = 6 Hz), 6.18 (1H, s), 7.24–7.33 (5H, m), 8.04 (1H, s), 9.92 (1H, brs), 12.09 (1H, brs). 13C NMR (CDCl3): δ 19.1, 19.1, 36.1, 61.1, 71.7, 72.4, 75.9, 89.4, 91.6, 96.0, 120.6, 127.2, 127.2, 127.7, 128.3, 128.3, 128.3, 137.2, 138.4, 146.9, 147.7, 154.9, 179.3. Mass (FAB): m/z 486 (MH+). High-resolution MS (FAB): Calcd for C23H28N5O7 (MH+): 486.1989. Found: 486.1980.
2-N-Isobutyryl-2′-O,4′-C-(methylenoxymethylene)guanosine (22)
Twenty percent of Pd(OH)2-C (170 mg) was added to a solution of compound 21 (171 mg, 0.351 mmol) in MeOH (7.0 ml) at room temperature and the mixture was stirred under an H2 atmosphere for 40 h. After filtration of the mixture, the filtrate was concentrated under reduced pressure. The residue was purified by silica gel column chromatography [CHCl3/MeOH (15:1 to 10:1, v/v)] to give compound 22 (118 mg, 85%). White powder; m.p. 182–184°C. [α]D24 − 12.2 (c 1.09, MeOH). IR νmax (KBr): 3239, 2978, 2932, 1681, 1606, 1566, 1473, 1405, 1316, 1253, 1181, 1111 cm−1. 1H NMR (CD3OD): δ 1.22 (6H, d, J = 7 Hz), 2.71 (1H, sept, J = 7 Hz), 3.67 (1H, d, J = 12 Hz), 3.74 (1H, d, J = 12 Hz), 3.75 (2H, s), 4.39 (1H, d, J = 6 Hz), 4.73 (1H, d, J = 6 Hz), 5.07 (1H, d, J = 7 Hz), 5.35 (1H, d, J = 7 Hz), 6.23 (1H, s), 8.44 (1H, brs). 13C NMR (DMSO-d6): δ 19.1, 19.1, 34.9, 60.2, 68.3, 70.6, 79.0, 89.0, 89.3, 94.9, 120.2, 136.7, 147.8, 148.2, 154.8, 180.2. Mass (FAB): m/z 396 (MH+). High-resolution MS (FAB): Calcd for C16H22N5O7 (MH+): 396.1519. Found: 396.1530.
5′-O-(4,4′-Dimethoxytrityl)-2-N-isobutyryl-2′-O,4′-C-(methylenoxymethylene)guanosine (23)
To a solution of 22 (265 mg, 0.670 mmol) in anhydrous pyridine (5.2 ml) was added DMTrCl (454 mg, 1.340 mmol) at room temperature and the mixture was stirred for 9 h. After addition of saturated aqueous NaHCO3 solution at 0°C, the mixture was extracted with AcOEt. The organic extracts were washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [CHCl3/MeOH (50:1 to 20:1, v/v)] to give compound 23 (442 mg, 95%). White powder; m.p. 173–175°C. [α]D24 + 24.4 (c 0.97, CHCl3). IR νmax (KBr): 3146, 2973, 2250, 2051, 1899, 1680, 1605, 1560, 1507, 1407, 1305, 1250, 1179, 1115, 1072, 1033 cm−1. 1H NMR (CDCl3): δ 1.23 (3H, d, J = 7 Hz), 1.27 (3H, d, J = 7 Hz), 2.75 (1H, sept, J = 7 Hz), 3.37 (1H, d, J = 10 Hz), 3.56 (1H, d, J = 10 Hz), 3.70 (6H, s), 3.90 (1H, d, J = 13 Hz), 4.19 (1H, d, J = 13 Hz), 4.57 (1H, d, J = 5 Hz), 5.10 (1H, brd, J = 5 Hz), 5.14 (1H, brs), 5.17 (1H, d, J = 7 Hz), 5.37 (1H, d, J = 7 Hz), 6.19 (1H, s), 6.70 (4H, d, J = 9 Hz), 7.11–7.18 (3H, m), 7.24 (4H, d, J = 9 Hz), 7.32–7.36 (2H, m), 7.67 (1H, s), 9.58 (1H, brs), 12.34 (1H, brs). 13C NMR (CDCl3): δ 19.0, 19.1, 36.3, 55.1, 55.1, 64.6, 71.6, 72.5, 79.0, 86.2, 89.1, 91.3, 95.2, 112.8, 112.8, 112.8, 112.8, 121.1, 126.7, 127.5, 127.5, 128.1, 128.1, 129.9, 129.9, 129.9, 129.9, 135.4, 135.5, 138.8, 144.1, 147.7, 147.7, 155.5, 158.2, 158.2, 180.1. Mass (FAB): m/z 698 (MH+). High-resolution MS (FAB): Calcd for C37H40N5O9 (MH+): 698.2826. Found: 698.2813.
3′-O-[2-Cyanoethoxy(diisopropylamino)phosphino]-5′-O-(4,4′-dimethoxytrityl)-2-N-isobutyryl-2′-O,4′-C-(methylenoxymethylene)guanosine (24)
To a solution of 23 (440 mg, 0.630 mmol) in MeCN/THF (1:1, v/v, 5.0 ml) were added 4,5-dicyanoimidazole (0.25 M in MeCN, 3.0 ml, 0.75 mmol) and (i-Pr2N)2POCH2CH2CN (0.30 ml, 0.945 mmol) at room temperature and the mixture was stirred for 19 h. Then, 4,5-dicyanoimidazole (0.25 M in MeCN, 2.6 ml, 0.65 mmol) and (i-Pr2N)2POCH2CH2CN (0.30 ml, 0.945 mmol) were added, and the mixture was further stirred at room temperature for 6 h. After removal of the solvent under reduced pressure, the residue was diluted with AcOEt, washed with saturated aqueous NaHCO3 solution, water and brine, dried over Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography [hexane/AcOEt (2:3, v/v)] followed by precipitation from hexane/AcOEt to give compound 24 (360 mg, 64%). White powder; m.p. 124–127°C. 31P NMR (CDCl3): δ 150.5, 150.6. MS (FAB): m/z 898 (MH+). High-resolution MS (FAB): Calcd for C46H57N7O10P (MH+): 898.3905. Found: 898.3887.
Synthesis of BNACOC 27 and 30
Syntheses of 2′,4′-BNACOC-modified oligonucleotides (BNACOC) 27 and 30 were performed at a 0.2 μmol scale on an automated DNA synthesizer (Applied Biosystems Expedite™ 8909) using the standard phosphoramidite protocol except that the coupling time was increased from the standard 1.5–20 min. As an activator, 5-ethylthio-1H-tetrazole was used for every coupling step. Oligonucleotide synthesis was performed on DMTr-OFF mode. Cleavage from the universal CPG support was accomplished by using a 2 M ammonia methanol solution at room temperature for 1.5 h and removal of the protecting groups was accomplished by using a 28% ammonia solution at 55°C for 15 h. The obtained crude oligonucleotides were purified with SephadexTM G-25 (Amersham Biosciences, NAPTM 10 Column) followed by reversed-phase HPLC (Waters XTerra® MS C18 2.5 μm, 10 × 50 mm) [buffer A, 0.1 M TEAA; buffer B, 0.1 M TEAA/MeCN = 1:1, B 12% to 24%/30 min, linear gradient; flow rate, 3.0 ml/min]. The composition of the oligonucleotides was confirmed by MALDI-TOF-MS analysis. MALDI-TOF-MS data ([M–H]–) for oligonucleotides 27 and 30: For 27, found 4354.19 (calcd 4354.71); for 30, found 4391.12 (calcd 4391.80).
UV melting experiments (Tm measurements)
UV melting experiments were carried out on a SHIMADZU UV-1650PC spectrometer equipped with Tm analysis accessory quartz cuvettes of 1 cm optical path length. The UV melting profiles were recorded in 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl at scan rate of 0.5°C/min with detection at 260 nm. The final concentration of each oligonucleotide was 4 μM. The melting temperatures were obtained as the maxima of the first derivative of the melting curves.
CD spectroscopy
CD spectra were measured with a JASCO J-720W spectrophotometer. Spectra were recorded at 12°C in a quartz cuvette of 0.1 cm optical path length. The samples were prepared in the same manner as described for the UV melting experiments. The molar ellipticity was calculated from the equation [θ] = θ/cl, where θ is the relative intensity, c is the sample concentration, and l is the cell path length in centimeters.
X-ray crystallographic data
Crystallographic data (excluding structure factors) of 13 has been deposited at the Cambridge Crystallographic Data Center as supplementary publication no. CCDC 634527. Copy of the data can be obtained, free of charge via www.ccdc.cam.ac.uk/cgi-bin/catreq.cgi (or from the Cambridge Crystallographic Data Center, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033; or email: deposit@ccdc.cam.ac.uk).
RESULTS
Synthesis of 2′,4′-BNACOC-A
As shown in Scheme 2, the synthesis of the protected 2′,4′-BNACOC-ABz (7), bearing a seven-membered bridged structure, from 4 (30) was performed. According to Vorbrüggen's procedure (32,33), triacetate 4 was coupled with N6-benzoyladenine to give 5. Two acetyl groups of 5 were removed to give diol 6 sequentially. At first, diol 6 was treated with paraformaldehyde under acidic conditions; the same reaction conditions for the synthesis of the pyrimidine congener (Scheme 1) (30). However, the methyleneacetal formation did not proceed, while cleavage of the glycosidic linkage was predominantly observed. Then, several reaction conditions were attempted for formation of the methylenoxymethylene (COC) linkage (Table 1). Basic reaction conditions were also employed; however, the desired product 7 was not obtained at all (Table 1, Run 2 and 3). At last, we found that the reaction of diol 6 with NBS in DMSO (34) slightly proceeded to give a trace amount of the target compound 7 bearing a seven-membered bridged structure (Table 1, Run 4). The methyleneacetal formation using NBS in DMSO was dramatically improved by protection with the benzyloxymethyl (BOM) group of the N6-amide moiety, which might cause many side reactions, to give the desired compound 10 bearing a seven-membered bridged structure in 74% yield as shown in Scheme 3. Because all our attempts for O3′-debenzylation of 2′,4′-BNACOC-adenine (2′,4′-BNACOC-A) in the presence of the N6-benzoyl group failed, we alternatively attempted to remove the N6-benzoyl group with 28% ammonia aqueous solution to afford 11. The tert-butyldiphenylsilyl (TBDPS) group was removed by treatment with TBAF to give 12. Hydrogenation of 12 with palladium hydroxide on carbon and ammonium formate resulted in removal of both benzyl and BOM groups to finally afford the desired 2′,4′-BNACOC-A monomer 13. As shown in Scheme 4, after perbenzoylation of 13, the obtained compound was treated with sodium methoxide to give diol 14. Reaction of a primary hydroxyl group in 14 with 4,4′-dimethoxytrityl trifluolomethanesulfonate (35) gave 15 in 68% yield from 13 (path A). We attempted another synthetic route to improve the yield of 15. We found that reaction of 2′,4′-BNACOC-A monomer 13 with 4,4′-dimethoxytrityl trifluolomethanesulfonate followed by treatment with benzoyl chloride by a one-pot operation gave 16. Selective removal of both the O3′-benzoyl group and one of the N6-benzoyl groups gave 15 in 92% yield from 13 (path B). At last, phosphitylation of 15 led to the corresponding amidite 17.
Scheme 2.

Reagents and conditions: (a) silylated N6-benzoyladenine, TMSOTf, DCE, reflux, 11 h, 76%; (b) K2CO3, MeOH, 0°C, 1.5 h, 98%; (c) p-TsOH·H2O, (CH2O)n, DCE, reflux.
Table 1.
Attempt at cyclization of compound 6
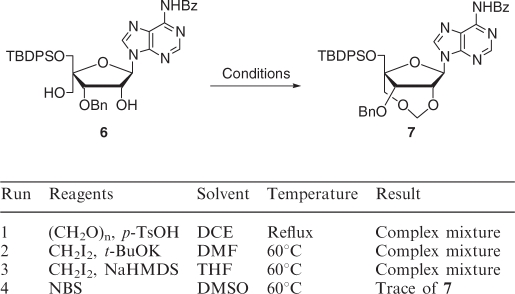 |
Scheme 3.

Reagents and conditions: (a) BOMCl, DBU, DMF, 0°C, 1 h, 64%; (b) K2CO3, MeOH, 0°C, 50 min, 97%; (c) NBS, DMSO, 60°C, 1.5 h, 74%; (d) 28% NH3aq, THF, 50°C, 48 h, quant.; (e) TBAF, THF, rt, 3 h, 91%; (f) 20% Pd(OH)2-C, HCOONH4, EtOH-AcOH, reflux, 3.5 h, 55%.
Scheme 4.

Reagents and conditions: (a) (1) BzCl, pyridine, rt, 12 h, (2) NaOMe, MeOH-1,4-dioxane, rt, 15 min, 68% over two steps; (b) DMTrOTf, pyridine-CH2Cl2, rt, 1 h, quant.; (c) DMTrOTf then BzCl, pyridine-CH2Cl2, rt, 21.5 h; (d) LiOH·H2O, THF-H2O, rt, 5 h, 92% over two steps; (e) (i-Pr2N)2POCH2CH2CN, DCI, MeCN, rt, 16 h, 84%.
Synthesis of 2′,4′-BNACOC-G
As shown in Scheme 5, an amidite 24 was synthesized by using a common intermediate 4 as the starting material. To circumvent the formation of an N7/N9-diastereo-mixture during nucleosidation of 4, we chose O6-(diphenylcarbamoyl)-N2-isobutyrylguanine as the base moiety (36) to give 18 in 84% yield. After removal of the acetyl groups of 18, the bridge structure formation was accomplished by the same reaction conditions as for the synthesis of the adenine congener to afford 20 in a moderate yield. The O6-diphenylcarbamoyl group was removed by treatment with sodium nitrite (37) and subsequent desilylation by TBAF leading to 21. Hydrogenolysis of 21 with palladium hydroxide on carbon afforded the desired 2′,4′-BNACOC-Gi-Bu monomer 22. Finally, dimethoxytritylation of 22 with 4,4′-dimethoxytrityl chloride followed by phosphitylation gave the phosphoroamidite 24.
Scheme 5.

Reagents and conditions: (a) silylated O6-(diphenylcarbamoyl)-N2-isobutyrylguanine, TMSOTf, toluene, 80°C, 2 h, 84%; (b) K2CO3, MeOH, 0°C, 40 min, 96%; (c) NBS, DMSO, 60°C, 40 min, 45%; (d) (1) NaNO2, DMSO, 70°C, 19 h, (2) TBAF, THF, rt, 15 h, 62% over two steps; (e) 20% Pd(OH)2-C, H2, MeOH, rt, 40 h, 85%; (f) DMTrCl, pyridine, rt, 9 h, 95%; (g) (iPr2N)2POCH2CH2CN, DCI, MeCN-THF, rt, 25 h, 64%.
Structure of 2′,4′-BNACOC-A and 2′,4′-BNACOC -G
The structural features of 2′,4′-BNACOC-A monomer 13 were well confirmed by 1H NMR measurement and X-ray crystallography (Figure 3, Table 4 and also see experimental sections). No scalar coupling between H1′ and H2′ in the 1H NMR spectra of 13 and 22 strongly suggested the North-type (N-type) sugar puckers, as observed in a series of 2′,4′-BNA (LNA). The crystal structure of 2′,4′-BNACOC-A (13) revealed the pseudorotation phase angle P (32°) supporting its N-type sugar pucker and COC linkage configuration surrounded the O3′ atom. Moreover, the νmax and δ values of 13 were 38° and 78°, respectively. These values are in good agreement with the parameters of the C3′-endo structure which is the typical N-type seen in the A-form RNA duplex (4–6).
Figure 3.

X-ray structure of 2′,4′-BNACOC-A (13).
Table 4.
Selected parameters by X-ray analysis
| δ | νmax | P | ||
|---|---|---|---|---|
| 2′,4′-BNA7) | 66° | 57° | 17° | |
| ENA24) | 76° | 48° | 15° | |
| 2′,4′-BNANC[NMe]19) | 75° | 49° | 23° | |
| 2′,4′-BNAcoc-T (3)30) | 78° | 38° | 17° | |
| 2′,4′-BNAcoc-A (13) | this work | 78° | 38° | 32° |
| C3′-endo4-6) | (A-form RNA) | 83° | 38.6° ± 3° | 0°–36° |
BNACOC oligonucleotide synthesis
2′,4′-BNACOC phosphoramidites were incorporated into oligonucleotides by conventional phosphoramidite chemistry on an automated DNA synthesizer. The coupling efficiency of 2′,4′-BNACOC phosphoramidites was decreased by using 4,5-dicyanoimidazole as an activator due to steric hindrance of the seven-membered ring structure with a methylenoxymethylene linkage. After several trials, we found that the coupling efficiency was remarkably improved (over 95% yield) when the coupling time was prolonged to 20 min from 90 s for 2′,4′-BNACOC phosphoramidites and 5-ethylthio-1H-tetrazole was used as the activator [In the case of PrNA which also has a seven-membered ring structure, the coupling efficiency was not good (∼50–60%) even with the use of 5-ethylthio-1H-tetrazole. See ref. (25)].
Duplex formation abilities of fully modified BNACOC oligonucleotides with DNA and RNA
The duplex forming abilities of fully 2′,4′-BNACOC-modified oligonucleotides (BNACOC) containing all four bases (thymine, 5-methylcytosine, adenine and guanine) were examined by means of UV melting experiments. Base pairing experiments of BNACOC 27 and 30 were carried out with complementary DNA, RNA and BNACOC. The melting temperatures (Tm) for the duplexes are depicted in Table 2. In its own series, the BNACOC duplex showed an extremely high Tm value compared with the corresponding DNA duplex (▵Tm > +43°C) and the RNA duplex (▵Tm > +44°C). With the RNA complement, BNACOC constituted more stable pairing systems for 30/26 and 27/29 than the RNA/RNA duplex (▵Tm of +9°C and +27°C, respectively) and the DNA/RNA heteroduplex (▵Tm of +30°C each). On the other hand, BNACOC/DNA heteroduplexes 30/25 and 27/28 showed lower Tm values compared with the DNA/DNA duplex (▵Tm of −7°C and −13°C, respectively). BNACOC 27 formed stable duplex with complementary DNA 28 compared with the corresponding RNA/DNA duplex (26/28), probably due to an effect of 5-methyl group of 2′,4′-BNACOC-T in 27 [Probably, thymine in place of uracil nucleobase should cause higher thermal stability of BNACOC 27 with the DNA complement than that of natural RNA/DNA duplex. See ref. (7) describing the effect of displacement of thymine with uracil in 2′,4′-BNA duplex.] Remarkably, BNACOC clearly distinguished complementary RNA from complementary DNA (▵Tm of +39°C for 27 and +15°C for 30, respectively), whereas natural RNAs, 26 and 29, showed relatively poor discrimination between complementary RNA and complementary DNA (▵Tm of +21°C for 26 and +3°C for 29, respectively).
Table 2.
Tm values (°C) of BNACOC with complementary DNA, RNA and 2′,4′- BNACOC oligonucleotides
| Target strands |
|||
|---|---|---|---|
| 28 5′-d(AGCAAAAAACGC)-3′ | 29 5′-r(AGCAAAAAACGC)-3′ | 30 5′-agmcaaaaaamcgmc-3′ | |
| 25 5′-d(GCGTTTTTTGCT)-3′ | 52 | 48 | 45 |
| 26 5′-r(GCGUUUUU1UGCU)-3′ | 30 | 51 | 60 |
| 27 5′-g mc g t t t t t t g mc t-3′ | 39 | 78 | >95 |
The UV melting profiles were measured in 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl at a scan rate of 0.5°C/min at 260 nm. The concentration of the oligonucleotide used was 4 μM for each strand. BNACOC oligonucleotides are shown in small letters.
Mismatch discrimination of 2′,4′-BNACOC-modified oligonucleotides
To analyze the base selectivity of BNACOC, we investigated the duplex stabilities between BNACOC 27 and mismatched DNA or RNA complements. The melting temperatures (Tm) of duplexes are shown in Table 3. With the mismatched DNA complement, the Tm values of natural DNA 25 decreased by −11°C to −15°C compared to that of the fully-matched duplex, while both natural RNA 26 and BNACOC 27 showed Tm values lower than room temperature. In the case of duplexes with a mismatched RNA complement, BNACOC 27 recognized one base mismatch (▵Tm = −6°C to −21°C) by a similar manner to that observed in natural DNA 25 (▵Tm = −5°C to −16°C). The differences in Tm values between matched and mismatched duplexes were larger than those observed for duplexes containing RNA 26 (▵Tm = −3°C to −12°C). As expected, the T−G wobble base pair was more stable than the other mismatched base pairs in all cases investigated.
Table 3.
Tm values (°C) of oligonucleotides 25–27 with their complementary DNA or RNA with or without a one base mismatch
| Target sequence 5′-d(AGCAAANAACGC)-3′ |
Target sequence 5′-r(AGCAAANAACGC)-3′ |
|||||||
|---|---|---|---|---|---|---|---|---|
| N = A | T | G | C | N = A | U | G | C | |
| 25 5′-d(GCGTTTTTTGCT)-3′ | 52 | 38 (−14) | 41 (−11) | 37 (−15) | 48 | 33 (−15) | 43 (−5) | 32 (−16) |
| 26 5′-r(GCGUUUUUUGCU)-3′ | 30 | <25 | <25 | <25 | 51 | 42 (−9) | 48 (−3) | 39 (−12) |
| 27 5′-g mc g t t t t t t g mc t-3′ | 39 | <25 | <25 | <25 | 78 | 62 (−16) | 72 (−6) | 57 (−21) |
Conditions: 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl at a scan rate of 0.5°C/min at 260 nm. The concentration of the oligonucleotide used was 4 μM for each strand. The values in parentheses are differences in Tm values between mismatched and matched duplexes.
Analysis of the helical structure of 2′,4′-BNACOC-modified oligonucleotide
Circular dichroism spectroscopic analyses of BNACOC-hybridizing duplexes were performed to investigate the structural preferences. CD spectra were recorded at 10°C for the BNACOC duplex 27/30, the BNACOC/DNA heteroduplexes 27/28 and 30/25, the BNACOC/RNA heteroduplexes 27/29 and 30/26, as well as for the control DNA duplex 25/28, the RNA duplex 26/29 and the DNA/RNA duplexes 25/29 and 28/26 (Figure 4). The CD spectra of BNACOC duplex 27/30 and the BNACOC/RNA heteroduplexes 27/29 and 30/26 were similar to that of the natural RNA duplex. These spectra showed a large positive Cotton band around 265 nm and a large negative Cotton band at 210 nm, indicating a typical A-form helix formation (Figure 4A). In the case of the BNACOC/DNA and the RNA/DNA heteroduplexes, all spectra were very similar to each other and the shoulder band around 280 nm suggested an intermediate between A- and B-form geometry. This revealed that BNACOC formed duplexes with complementary DNA in the same manner the natural RNA did. Additionally, the CD profile of the natural DNA duplex belonged to no other types of duplexes in this study.
Figure 4.

(A) CD spectra of BNACOC duplex 27/30 (red line), BNACOC/RNA heteroduplexes 27/29 (orange line) and 26/30 (green line), DNA duplex 25/28 (black line), RNA duplex 26/29 (blue line); (B) CD spectra of duplexes the BNACOC/DNA heteroduplexes 27/28 (red line) and 25/30 (green line) and DNA/RNA duplexes 25/29 (black line) and 26/28 (blue line). The spectra were recorded in 10 mM sodium phosphate buffer (pH 7.2) containing 100 mM NaCl at 12°C. The concentration of each strand was 4 μM.
DISCUSSION
X-ray crystallography established the relationship between the sugar conformation and the ring size of the bridged structure between the 2′- and 4′-positions. The values of selected torsion angles are summarized in Table 4. In making a comparison between 2′,4′-BNACOC-A monomer 13 and 2′,4′-BNACOC-T monomer 3, even though the pseudorotation phase angle P are slightly different from each other, they are in the range preferred by the C3′-endo conformation (0° ≤ P ≤ 36°) which is the typical N-type geometry fit in a natural A-form RNA duplex (4–6). From another point of view, non-observed H1′–H2′ coupling in both 1H NMR spectra suggested very similar N-type sugar puckering in the solution phase. These data indicate that the sugar conformation of 2′,4′-BNACOC is fixed to an N-type, but still slightly flexible. Molecular dynamics simulations supported its conformational features showing larger P-amplitude of 2′,4′-BNACOC than that of 2′,4′-BNA (LNA) and 2′,4′-BNANC (Figure S1). Concerning the νmax values, which represent the maximum degree of the sugar puckering mode (N/S-type), the sugar conformation of 2′,4′-BNACOC (νmax 38°) is very similar to that of a natural A-form RNA duplex (νmax 38°) compared with that of 2′,4′-BNA (LNA), ENA and 2′,4′-BNANC[NMe] [νmax 57° (7), 48° (23) and 49° (19), respectively]. These results showed that the sugar conformation of 2′,4′-BNACOC is closest to that of A-form RNA among nucleic-acid analogues with a different type of bridged structure between the 2′- and 4′-positions.
NMR and molecular dynamics studies of the 2′,4′-BNA(LNA)-modified oligonucleotide (BNA) demonstrated that the conformation of the complementary DNA in BNA/DNA heteroduplexes is changed adaptively into a suitable one to form A-form duplexes and results in larger populations of N-type sugar puckers (38,39). On the other hand, the 2′,4′-BNACOC-modified oligonucleotide (BNACOC) duplex and the BNACOC/RNA heteroduplexes showed the typical A-form features on the CD spectra, while the BNACOC/DNA heteroduplexes showed the same aspect with the natural RNA/DNA duplex (Figure 4). These differences between BNA and BNACOC probably correlate with the above-mentioned conformational features of each monomer, that is to say, the oligonucleotide bearing a fixed N-type sugar conformation and larger νmax value than that of the typical A-form duplex could force the complementary DNA to take an N-type sugar conformation in the duplex.
Concerning the thermal stability, BNACOC forms a very stable duplex with complementary RNA, while it moderately stabilizes the duplex with complementary DNA. On the other hand, BNA formed extremely stable duplexes with both complementary DNA and RNA (9,20–22), and PrNA, which possesses an alternative –CH2CH2CH2– group to –CH2OCH2– of 2′,4′-BNACOC within the bridge structure, leads to a decrease in duplex stabilities with both complementary DNA and RNA (24). These differences in hybridization properties would probably result from a balance between the entropic advantages derived from the conformational restriction of the sugar moiety (10) and the disadvantages caused by the additional bridge structure. BNA possesses a rigid N-type sugar restricted with a small bridge to make a great entropic advantage over any other disadvantages. In the case of BNACOC, the entropic advantage by the sugar restriction would be smaller than that of BNA because of the large perturbation of 2′,4′-BNACOC come from the larger bridge structure compared with 2′,4′-BNA (LNA) (40; Supplementary Figure S1). However, the hydrophilic bridge structure of BNACOC could somewhat positively influence the network of hydration around the duplex. Consequently, BNACOC makes the duplex more thermally stable than PrNA bearing a hydrophobic bridge structure.
BNACOC has interesting features, such as high binding affinity towards the RNA complement and discrimination between complementary RNA and DNA. Moreover, it has been revealed that the modification of 2′,4′-BNACOC leads to excellent nuclease resistance (30). These results indicate a high potential for 2′,4′-BNACOC in genome technology. Further biological studies of 2′,4′-BNACOC are now in progress.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This work was supported in part by a Grant-in-Aid for Science Research from the Japan Society for the Promotion of Science and the Molecular Imaging Research Program from the Ministry of Education, Culture, Sports, Science and Technology, Japan. Funding for open access charge: Japan Society of Promotion of Science.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Stein CA, Krieg AM. Applied Antisense Oligonucleotide Technology. Wiley-Liss, New York: 1998. [Google Scholar]
- 2.Schlingensiepen R, Brysch W, Schlingensiepen KH. Antisense—from Technology to Therapy. Berlin: Black Sciences Ltd; 1997. [Google Scholar]
- 3.Jepsen JS, Wengel J. LNA-antisense rivals siRNA for gene silencing. Curr. Opin. Drug Discov. Dev. 2004;7:188–194. [PubMed] [Google Scholar]
- 4.Saenger W. Principles of Nucleic Acid Structure. New York: Springer-Verlag; 1984. [Google Scholar]
- 5.Altona C, Sundaralingam M. Conformational analysis of the sugar ring in nucleosides and nucleotides. A new description using the concept of pseudorotation. J. Am. Chem. Soc. 1972;94:8205–8212. doi: 10.1021/ja00778a043. [DOI] [PubMed] [Google Scholar]
- 6.Arnott S, Hukins DWL, Dover SD. Optimised parameters for RNA double-helices. Biochem. Biophys. Res. Commun. 1972;48:1392–1399. doi: 10.1016/0006-291x(72)90867-4. [DOI] [PubMed] [Google Scholar]
- 7.Imanishi T, Obika S. Synthesis and properties of novel conformationally restrained nucleoside analogs. J. Synth. Org. Chem. 1999;57:969–980. [Google Scholar]
- 8.Imanishi T, Obika S. BNAs: novel nucleic acid analogs with a bridged sugar moiety. Chem. Commun. 2002:1653–1659. doi: 10.1039/b201557a. [DOI] [PubMed] [Google Scholar]
- 9.Obika S, Nanbu D, Hari Y, Morio K, In Y, Ishida T, Imanishi T. Synthesis of 2′-O,4′-C-methyleneuridine and -cytidine. Novel bicyclic nucleosides having a fixed C3′-endo sugar puckering. Tetrahedron Lett. 1997;38:8735–8738. [Google Scholar]
- 10.Obika S, Nanbu D, Hari Y, Andoh J, Morio K, Doi T, Imanishi T. Stability and structural features of the duplexes containing nucleoside analogues with a fixed N-type conformation, 2′-O,4′-C-methylene ribonucleosides. Tetrahedron Lett. 1998;39:5401–5404. [Google Scholar]
- 11.Obika S, Sekiguchi M, Somjing R, Imanishi T. Adjustment of the γ dihedral angle of an oligonucleotide P3′→N5′ phosphoramidate enhances its binding affinity towards complementary strands. Angew. Chem. Int. Ed. 2005;44:1944–1947. doi: 10.1002/anie.200461942. [DOI] [PubMed] [Google Scholar]
- 12.Sekiguch M, Obika S, Harada Y, Osaki T, Somjing R, Mitsuoka Y, Shibata N, Masaki M, Imanishi T. Synthesis and properties of trans-3′,4′-bridged nucleic acids having typical S-type sugar conformation. J. Org. Chem. 2006;71:1306–1316. doi: 10.1021/jo051187l. [DOI] [PubMed] [Google Scholar]
- 13.Osaki T, Obika S, Harada Y, Mitsuoka Y, Sugaya K, Sekiguchi M, Roongjang S, Imanishi T. Development of a novel nucleoside analogue with S-type sugar conformation: 2′-deoxy-trans-3′,4′-bridged nucleic acids. Tetrahedron. 2007;63:8977–8986. [Google Scholar]
- 14.Kodama T, Sugaya K, Harada Y, Mitsuoka Y, Imanishi T, Obika S. Synthesis and properties of 2′-deoxy-trans-3′,4′-BNA with S-type sugar puckering. Heterocycles. 2009;77 doi: 10.1093/nass/nrm078. in press. [DOI] [PubMed] [Google Scholar]
- 15.Kodama T, Sugaya K, Baba T, Imanishi T, Obika S. Synthesis of a novel trans-3′,4′-BNA monomer bearing a 4,8-dioxa-5-azabicyclo[5.3.0]decane skeleton. Heterocycles. 2009;79 in press. [Google Scholar]
- 16.Miyashita K, Rahman SMA, Seki S, Obika S, Imanishi T. N-Methyl substituted 2′,4′-BNANC: a highly nuclease-resistance nucleic acid analogue with high-affinity RNA selective hybridization. Chem. Commun. 2007:3765–3767. doi: 10.1039/b707352f. [DOI] [PubMed] [Google Scholar]
- 17.Rahman SMA, Seki S, Utsuki K, Obika S, Miyashita K, Imanishi T. 2′,4′-BNANC: a novel bridged nucleic acid analogue with excellent hybridizing and nuclease resistance profiles. Nucleosides Nucleotides Nucleic Acids. 2007;26:1625–1628. doi: 10.1080/15257770701548980. [DOI] [PubMed] [Google Scholar]
- 18.Rahman SMA, Seki S, Obika S, Haitani S, Miyashita K, Imanishi T. Highly stable pyrimidine-motif triplex formation at physiological pH values by a bridged nucleic acid analogue. Angew. Chem. Int. Ed. 2007;46:4306–4309. doi: 10.1002/anie.200604857. [DOI] [PubMed] [Google Scholar]
- 19.Rahman SMA, Seki S, Obika S, Yoshikawa H, Miyashita K, Imanishi T. Design, synthesis, and properties of 2′,4′-BNANC: a bridged nucleic acid analogue. J. Am. Chem. Soc. 2008;130:4886–4896. doi: 10.1021/ja710342q. [DOI] [PubMed] [Google Scholar]
- 20.Singh SK, Nielsen P, Koshkin AA, Wengel J. LNA (locked nucleic acids): synthesis and high-affinity nucleic acid recognition. Chem. Commun. 1998:455–456. [Google Scholar]
- 21.Koshkin AA, Singh SK, Nielsen P, Rajwanshi VK, Kumar R, Meldgaard M, Olsen CE, Wengel J. LNA (locked nucleic acids): synthesis of the adenine, guanine, 5-methylcytosine, thymine and uracil bicyclonucleoside monomers, oligomerisation, and unprecedented nucleic acid recognition. Tetrahedron. 1998;54:3607–3630. [Google Scholar]
- 22.Jepsen JS, Wengel J. LNA (locked nucleic acid): high-affinity targeting of complementary RNA and DNA. Biochemistry. 2004;43:13233–13244. doi: 10.1021/bi0485732. [DOI] [PubMed] [Google Scholar]
- 23.Morita K, Hasegawa C, Kaneko M, Tsutsumi S, Sone J, Ishikawa T, Imanishi T, Koizumi M. 2′-O,4′-C-ethylene-bridged nucleic acids (ENA): highly nuclease-resistant and thermodynamically stable oligonucleotides for antisense drug. Bioorg. Med. Chem. Lett. 2002;12:73–76. doi: 10.1016/s0960-894x(01)00683-7. [DOI] [PubMed] [Google Scholar]
- 24.Morita K, Takagi M, Hasegawa C, Kaneko M, Tsutsumi S, Sone J, Ishikawa T, Imanishi T, Koizumi M. Synthesis and properties of 2′-O,4′-C-ethylene-bridged nucleic acids (ENA) as effective antisense oligonucleotides. Bioorg. Med. Chem. 2003;11:2211–2226. doi: 10.1016/s0968-0896(03)00115-9. [DOI] [PubMed] [Google Scholar]
- 25.Wang G, Gunic E, Giratdet J.-L, Stoisavljevic V. Conformationally locked nucleosides. Synthesis and hybridization properties of oligodeoxynucleotides containing 2′,4′-C-bridged 2′-deoxynucleosides. Bioorg. Med. Chem. Lett. 1999;9:1147–1150. doi: 10.1016/s0960-894x(99)00146-8. [DOI] [PubMed] [Google Scholar]
- 26.Wang G, Giratdet J.-L, Gunic E. Conformationally locked nucleosides. Synthesis and stereochemical assignments of 2′,4′-C-bridged bicyclonucleosides. Tetrahedron. 1999;55:7707–7724. [Google Scholar]
- 27.Freier SM, Altmann K.-H. The ups and downs of nucleic acid duplex stability: structure–stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997;25:4429–4443. doi: 10.1093/nar/25.22.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Albæk N, Petersen M, Nielsen P. Analogues of a locked nucleic acid with three-carbon 2′,4′-linkages: Synthesis by ring-closing metathesis and influence on nucleic acid duplex stability and structure. J. Org. Chem. 2006;71:7731–7740. doi: 10.1021/jo061225g. [DOI] [PubMed] [Google Scholar]
- 29.Srivastava P, Barman J, Pathmasiri W, Plashkevych O, Wenska M, Chattopadhyaya J. Five- and six-membered conformationally locked 2′,4′-carbocyclic ribo-thymidines: synthesis, structure, and biochemical studies. J. Am. Chem. Soc. 2007;129:8362–8379. doi: 10.1021/ja071106y. [DOI] [PubMed] [Google Scholar]
- 30.Hari Y, Obika S, Ohnishi R, Eguchi K, Osaki T, Ohishi H, Imanishi T. Synthesis and properties of 2′-O,4′-C-methyleneoxymethylene bridged nucleic acid. Bioorg. Med. Chem. 2006;14:1029–1038. doi: 10.1016/j.bmc.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 31.Zoltewicz JA, Clark DF, Sharpless TW, Grahe G. Kinetics and mechanism of the acid catalyzed hydrolysis of some purine nucleosides. J. Am. Chem. Soc. 1970;92:1741–1750. doi: 10.1021/ja00709a055. [DOI] [PubMed] [Google Scholar]
- 32.Vorbrüggen H, Krolikiewicz K, Bennua B. Nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalysts. Chem. Ber. 1981;114:1234–1255. [Google Scholar]
- 33.Vorbrüggen H, Höfle G. On the mechanism of nucleoside synthesis. Chem. Ber. 1981;114:1256–1268. [Google Scholar]
- 34.Hanessian S, Yang-Chung G, Lavallee P, Pernet AG. Chemistry of α-alkoxy sulfoxides. Formation of methylene acetals from dimethyl sulfoxide and alcohols. J. Am. Chem. Soc. 1972;94:8929–8931. [Google Scholar]
- 35.Tärkoy M, Bolli M, Leumann C. Synthesis, pairing properties, and calorimetric determination of duplex and triplex stability of decanucleotides from [(3′S,5′R)-2′-deoxy-3′,5′-ethano-β-D-ribofuranosyl]adenine and -thymine. Helv. Chim. Acta. 1994;77:716–744. [Google Scholar]
- 36.Robins MJ, Zou R, Guo Z, Wnuk SF. A solution for the historic problem of regioselective sugar–base coupling to produce 9-glycosylguanines or 7-glycosylguanines. J. Org. Chem. 1996;61:9207–9212. [Google Scholar]
- 37.Ackermann D, Pitsch S. Synthesis and pairing properties of 3′-deoxyribopyranose (4′→2′)-oligonucleotides (‘p-DNA’) Helv. Chim. Acta. 2002;85:1443–1462. [Google Scholar]
- 38.Nielsen KE, Rasmussen J, Kumar R, Wengel J, Jacobsen JP, Petersen M. NMR studies of fully modified locked nucleic acid (LNA) hybrids: Solution structure of an LNA:RNA hybrid and characterization of an LNA:DNA hybrid. Bioconjugate Chem. 2004;15:449–457. doi: 10.1021/bc034145h. [DOI] [PubMed] [Google Scholar]
- 39.Pande V, Nilsson L. Insights into structure, dynamics and hydration of locked nucleic acid (LNA) strand-based duplexes from molecular dynamics simulations. Nucleic Acids Res. 2008;36:1508–1516. doi: 10.1093/nar/gkm1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Plashkevych O, Chatterjee S, Honcharenko D, Pathmasiri W, Chattopadhyaya J. Chemical and structural implications of 1′,2′- versus 2′,4′-conformational constraints in the sugar moiety of modified thymine nucleosides. J. Org. Chem. 2007;72:4716–4726. doi: 10.1021/jo070356u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.