

Abstract
Gap junction proteins (connexins) are required for myocardial function., since they allow intercellular transmission of current carrying ions and signaling molecules. Previous studies demonstrated that rat cardiac myocytes infected with Trypanosoma cruzi lost gap junctional communication and decreased automaticity. We infected mouse cardiac myocytes with trypomastigotes of the Y strain of T. cruzi and observed alterations in connexin43 (Cx43) distribution. One hour post infection Cx43 levels were significantly increased. However, at longer time points post infection there was a significant loss of Cx43 staining in membranes of infected cardiac myocytes. Interestingly, there was also a significant reduction in myocardial Cx43 protein levels during acute infection. These data indicate that T. cruzi infection alters Cx43 expression both in vitro and in vivo and is not strain specific. Disruptions in Cx43 may contribute to the pathogenesis of cardiac electrical alterations observed in T. cruzi infection.
Keywords: Trypanosoma cruzi, Chagas’ disease, cardiac myocytes, connexin43, gap junctions
Introduction
Trypanosoma cruzi is the causative agent of Chagas’ disease (American Trypanosomiasis). It is the major cause of heart disease in endemic areas of Mexico, Central and South America. Ten to 30% of infected individuals eventually develop clinically apparent heart disease including dilated cardiomyopathy, congestive heart failure, dysrhythmias and thrombo-embolic events.
This parasite has a complicated life cycle involving a mammalian host and an insect vector [1]. During in vitro infection of mammalian cells (host cells), T. cruzi alters intracellular calcium levels both in non-excitable cells [2] and in cardiac myocytes [3,4]. The parasites enter cells through a mechanism involving the formation of a parasitophorous vacuole, which arises from the fusion of lysosomes and endocytic vesicles [5]. After invasion, trypomastigotes escape from the vacuole and remain free within the cytoplasm where they differentiate to the multiplicative amastigote form [6]. Amastigotes multiply by binary fission and eventually escape the host cell as trypomastigotes capable of infecting adjacent uninfected cells and spreading by a hematogenous route to distant tissues resulting in the infection of new cells.
Studies of alterations in cardiac myocytes during in vitro infection with T. cruzi indicate that the parasite is capable of impairing host cell functioning through alterations in cell-cell communication [7]. Such an effect is of particular importance in the heart where maintenance of synchronous contractions requires functional gap junctions [see 8 and 9, for reviews]
Gap junction channels are composed of the connexin family of transmembrane proteins that assemble as end to end alignments of hexameric connexin subunits, thereby forming intercellular conduits for current-carrying ions and molecules Mr < 1000 Da such as Ca2+, IP3 and cyclic AMP. The connexin gene family in mammals includes more than twenty isoforms encoded by separate genes [10] and such isoforms are named according to the molecular weight (in kDa) of the protein predicted from its cDNA [11]. Gap junction channels are critical in maintaining cardiac homeostasis by allowing the free flow of ions and metabolites between cardiac myocytes. This contributes to the synchronized contraction of and signal exchange throughout the tissue. Connexin43 (Cx43) is the most abundant gap junction protein in ventricular myocytes, being localized at intercalated disks in normal myocardium [see 8]. Cx43 function and intercellular distribution is affected by its phosphorylation and other factors including intracellular pH and calcium concentration and protein-protein interactions [12]. In diverse cardiac disease states, such as infarct and ischemia, significant remodeling of the distribution of Cx43 occurs, resulting in disorganization of normal microconduction pathways and arrhythmias [13].
Since gap junctional communication is important in normal cardiac conduction and the report that intercellular communication and Cx43 distribution are altered in T. cruzi-infected rat cardiac myocytes [7], we examined the expression and distribution of Cx43 in widely used in vivo and in vitro mouse models of infection. The present study characterizes such changes in cultured cardiac myocytes during initial infection and during the intracellular cycle of this parasite and demonstrates that Cx43 expression is down-regulated in the hearts of infected mice. These alterations in Cx43 may contribute to the cardiac dysrhythmias observed in the acute phase of T. cruzi infection.
Material and Methods
1. Antibodies and reagents
Rabbit polyclonal antibody against Cx43 (C6219, amino acids 363-382), 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI); Phenylmethanesulfonyl fluoride (PMSF); 1,4-Diazabicyclo[2.2.2]octane (DABCO); Ethylene Diamine Tetraacetic Acid (EDTA); Sodium Bicarbonate (NaHCO3); Sodium Orthovanadate (Na3VO4); Ponceau Solution; Polyethylene glycol sorbitan monolaurate (Tween 20), Triton x-100, Bovine Serum Albumin (BSA) and polyclonal rabbit anti-connexin43 (amino acids 363-382) were purchased from Sigma-Aldrich (St. Louis, IL, USA). Rabbit polyclonal antibody against Cx43 181A (amino acids 346-360) was kindly provided by Professor Elliot Hertzberg, Department of Neuroscience, Albert Einstein College of Medicine. Secondary goat anti-rabbit IgG AlexaFluor 594 and Phalloidin AlexaFluor 488 were purchased from Molecular Probes (Invitrogen, Carlsbad, CA). Monoclonal anti-rabbit glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was obtained from RDI Division of Fitzgerald Industries Intl.
For cardiac myocyte isolation and cultivation, trypsin was obtained from Difco Laboratories, type II collagenase from Worthington Biochemical Corporation (Lakewood, NJ), and Fetal Bovine Serum (FBS), L-glutamine, penicillin, streptomycin and Dulbecco’s Modified Eagle’s Medium (DMEM) from Sigma-Aldrich.
2. Primary cultures of mouse cardiac myocytes
Hearts from Swiss Webster mouse embryos (18-21 days of gestation) were harvested and digested with collagenase and trypsin using the method described by Meirelles [14]. Isolated cells were plated in gelatin coated dishes and maintained in DMEM medium supplemented (DMEMs) with 10% Fetal Bovine Serum, 1% Penicillin/Streptomycin, 2% L-Glutamine, 2% Chick Embryo Extract, 10% CaCl2 25mM [14, 4]. For immunoblotting, 2×106 cells were plated on 60mm cell culture plastic dishes (Corning Inc., Corning, NY) and for Giemsa microscopy experiments, 5 × 105 cells were plated on glass coverslips in 24-well plates.
3. Infection with Trypanosoma cruzi (Y strain)
Trypomastigotes of the Y strain (MHOM/BR/1950/Y) were obtained from infected Vero cell cultures. The parasites in the supernatant were collected and centrifuged at 3,000rpm for 30 minutes at 4°C. Cardiac myocytes were infected after 72 hours of plating. The multiplicity of infection was 20:1 (parasites: host cell) and parasites were diluted in DMEM without FBS in a final volume of 300μl for 24-well plates and 1ml for 60mm dishes. After two hours of interaction, extracellular parasites were washed out and fresh supplemented DMEM was added. For subsequent days, medium was replaced every 24 hours. Cardiac myocytes were fixed and stained with Giemsa stain for assessment of intracellular parasitism as previous described [15].
4. Immunofluorescence
Cells were washed with PBS and fixed with 4% Paraformaldehyde for 5min at 20°C at desired time points (1 and 72hpi). After washes in PBS, cells were permeabilized with 0.5% Triton and non-specific staining was blocked with 4% BSA. Primary anti-Cx43 antibody (Sigma) was incubated overnight 4°C, after which cells were washed and incubated with secondary polyclonal goat anti-rabbit Alexa Fluor 594 antibody (Invitrogen) for 1 hour at 37°C. F-actin filaments were stained with Alexa Fluor 488 Phalloidin (Invitrogen) for 30min at 37°C and DNA was stained with DAPI. Images were acquired with a Olympus Laser Scanning Confocal Microscope.
5. Immunoblotting
At desired intervals after infection (1, 2, 6, 24, 48 and 72 hours post infection), cells were washed 3 times with PBS and scraped with 300μl of lysis buffer [2mM PMSF, 5mM EDTA, 1mM Na3VO4, 1mM NaHCO3, 10% Protease Inhibitor Cocktail (Roche, Indianapolis, IN)]. Samples were frozen in -80°C until used, the lysates were sonicated and protein concentration was measured using the BCA protein quantification kit (Pierce, Rockford, IL). 5μg of protein was loaded and resolved in 10% SDS-Polyacrylamide gels. After resolving, proteins were transferred to nitrocellulose membranes (Whatman). Membranes were incubated with 5% skim milk in TBST (TBS and 0.5% Tween 20) for 30 minutes and with primary rabbit polyclonal anti-connexin43 181A antibody diluted in TBST with 5% skim milk overnight at 4°C. Loading control was done using mouse anti-GAPDH (36KDa) monoclonal antibody. Membranes were washed with TBST and incubated with secondary goat anti-rabbit IgG and secondary goat anti-mouse IgG HRP-labeled antibody (Santa Cruz) for 1h at 25°C followed by washes with TBST and incubation with chemoluminescent kit ECL (Pierce) and exposed to X-Ray film.
6. Infection of mice
The Y strain of T. cruzi was maintained by serial passage in mice. Swiss Webster mice (18-20g) were infected with 104 trypomastigotes. The parasitemia was monitored from day 4 to 10 post infection. Mice were sacrificed and hearts were obtained 11 days post infection (i.e. two days after the parasitemia peak). Atria and ventricles were obtained and processed for immunoblot analysis as described for the cell cultures.
7. Statistical Analysis
Films were scanned using a Microtek scanner and protein quantification was performed with Scion Image software. Western blot results were normalized by values of GAPDH for each sample in the same gel. For Cx43 phosphorylation, we considered the two upper bands in each lane as phosphorylated bands (P1 and P2) and the lower band as non-phosphorylated (P0). Statistical analyses were performed using the ANOVA test, with the level of significance set at p<0.05. Data represent the averages and variations obtained in three independent experiments.
Results
Cx43 expression is increased during the initial phase of infection
Immunoblots demonstrated that cultured cardiac myocytes displayed Cx43 phosphorylation similar to cardiac tissue in vivo, with two upper phosphorylated bands (P1+P2) and one lower non-phosphorylated band (P0) (Figure 1A). During interaction of cultured cardiac myocytes with the parasite the levels of Cx43 were increased 68% during the first hour and then decreased, reaching levels close to what was observed in uninfected cultures at two and six hours post infection (Figure 1B and C). One hour post infection the phosphorylated P1+P2 forms of Cx43 were 60% higher compared with controls, whereas the level of the unphosphorylated P0 form was not significantly altered (Figure 1B,D)
Figure 1. Initiation of infection induced a transient increase in Cx43 in cardiac myocytes.
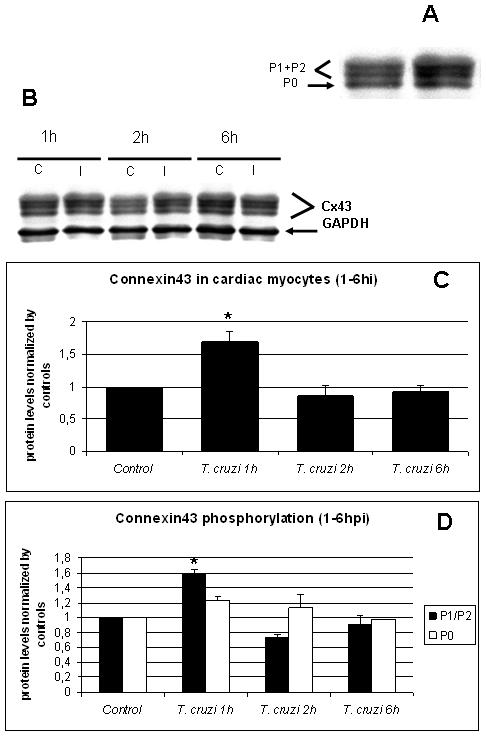
A. Immunoblots for Cx43 of mouse cardiac myocytes had two upper phosphorylated bands (P1+P2) and one lower non-phosphorylated band (P0). B. Immunoblot analysis of non-infected (C) or T. cruzi-infected (Tc) cultured myocytes. GAPDH was used as loading control for all the experiments. C. Whole protein quantification of Cx43 showed that at one hour PI there was a 68% increase in Cx43 expression and at two and six hours, there was a 13.4% and 7.9% decrease respectively. D. Differential quantification for protein phosphorylation analyses of P1+P2 (phosphorylated) and P0 (non-phosphorylated) showed that both forms of Cx43 were transiently increased during the first hour of infection (60% and 20%, respectively). After this period, all three bands returned to levels near baseline (*: p<0.05, ANOVA).
Immunostaining revealed that after three days in culture, cardiac myocytes displayed myofibrils as determined by Phalloidin staining (Figure 2C and 2F) and large gap junction plaques and annular junctions when stained with anti-Cx43 antibody (Figure 2B). Cultured myocytes infected for 1 hour showed remodeling of Cx43 distribution and increased perinuclear staining (Figure 2D and 2E). There was no labeling of trypomastigotes when cells were stained with anti-Cx43 antibody recognizing amino acids 363-382 (Sigma) (Figure 2E).
Figure 2. T. cruzi alters cardiac myocyte Cx43 distribution at initial times of infection.
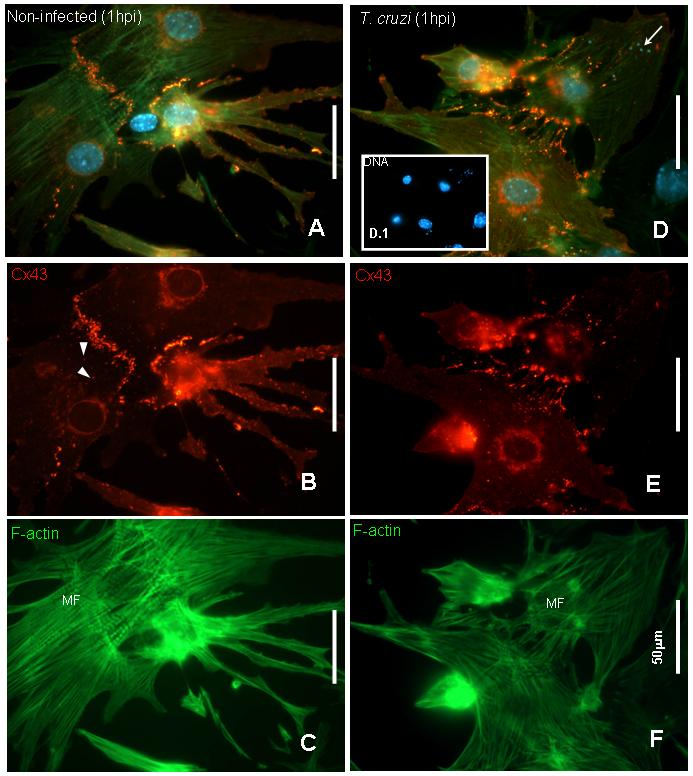
Mouse cardiac myocytes infected after 72 hours of cultivation were fixed at one hour post infection and stained with anti-connexin43 (Sigma antibody), Phalloidin-FITC for F-actin filaments and DAPI for DNA. A. Merged image of a non-infected culture showing that cardiac myocytes were connected by Cx43 and well differentiated. B. Cx43 was localized at cell-cell contacts and also displayed large vesicular structures (arrowheads). C. Phalloidin staining showed that cells were differentiated into myocytes with well defined polarity and presence of myofibrils (MF). D. and E: After one hour of infection Cx43 lost its plaque distribution compared with uninfected cells and was evident in stress fibers. In addition, there was more perinuclear staining indicating intense synthesis of Cx43. F. Phalloidin staining remained similar to observed in controls (C). D and D.1 show in detail DAPI staining corresponding to host cell and trypomastigotes’ DNA (arrows). Bars= 50.0μm.
Proliferation of intracellular parasites decreases Cx43 levels and immunostaining
We analyzed Cx43 expression in cultured cardiac myocytes 24, 48 and 72 hours post infection (PI) (Figure 3A). Immunoblotting demonstrated that in infected cultures 24 and 48 hours PI the Cx43 levels comparable to controls (Figure 3B) which was then followed by a significant reduction (61%) at 72 hours PI (Figure 3B). The internal control with GAPDH showed no variation in this protein levels during the infection even after 72 hour PI (Figure 3A). The proportion of Cx43 in phosphorylated P1 and P2 states remained higher from 24 to 72 hours (Figure 3C). Figure 3D illustrates the effects of infection on Cx43 expression in cultured myocytes from the time of parasite adhesion/invasion (one hour post infection) to differentiation from amastigote to trypomastigote (72 hours PI).
Figure 3. Cx43 levels were reduced during parasite proliferation.
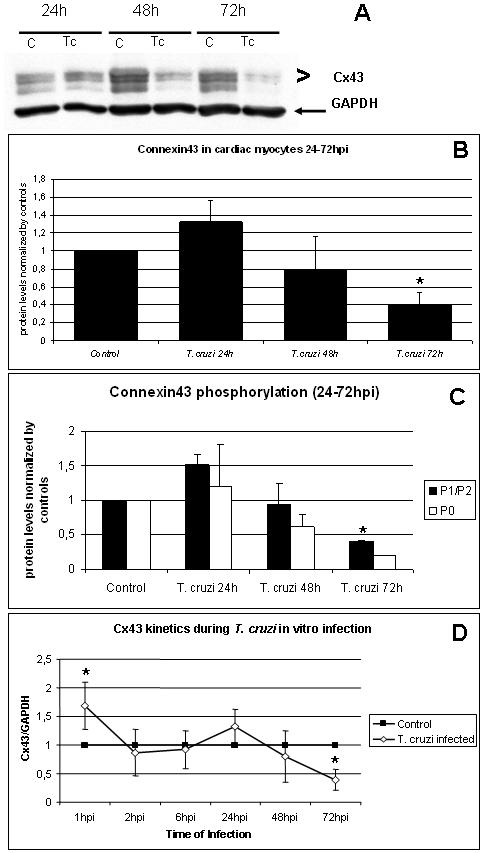
In vitro infection of mouse cardac myocytes was analyzed from 24 to 72hr. A and B. Immunoblots showed that Cx43 was increased after 24 hours PI (+32%±0.24) and was reduced at 48 (-21% ± 0.36) and 72 hours PI (-61% ± 0.14). C. Differential quantification of Cx43 phospho-bands revealed that 72 hours PI, P1+P2 bands had intensity higher then non-phosphorylated P0 band. (*: p<0.05, ANOVA). D. Biphasic effect of infection on mouse cardiac myocytes Cx43. There was a transient increase in Cx43 levels (68%) after one hour PI but it returned to baseline levels between 2hpi until 48hpi. Thereafter, Cx43 expression was reduced 61% at 72 hours PI (*: p<0,05, ANOVA).
Immunostaining of cardiac myocytes (Figures 4A and 4B) for Cx43 demonstrated gap junction plaques at their margins (Figures 4C and 4D). Highly infected cells at 72hpi (Figure 4C), which had many intracellular amastigotes, had no Cx43 immunoreactivity at cell membranes, whereas non-infected cells in an infected dish maintained strong antibody signal, showing plaques similar to control cells (Figure 4D).
Figure 4. Immunostaining of mouse embryo cardiac myocyte gap junction protein Cx43.
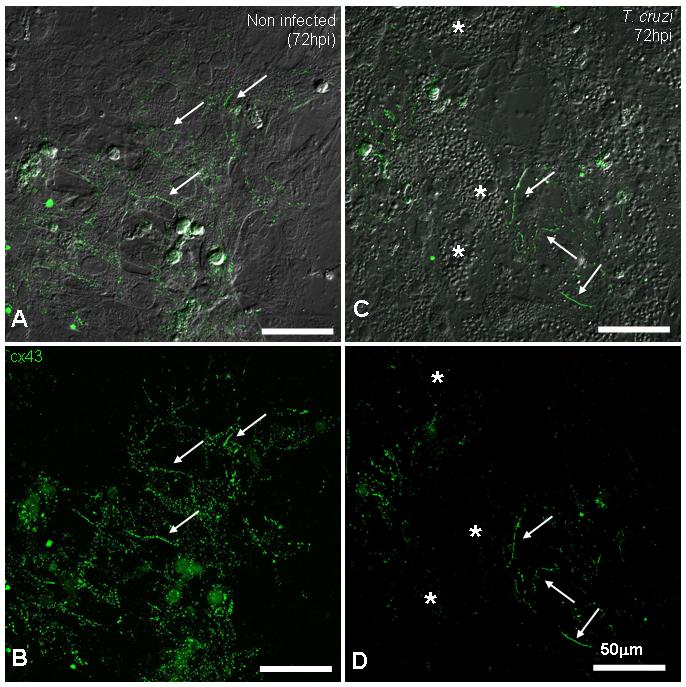
A and C show the merged image of DIC and anti-Cx43 antibody. Non-infected (A and B) cultures presented abundant gap junction plaques (arrows) between coupled cardiac myocytes. After 72 hours of infection (C and D), heavily infected cells (*) lost Cx43 staining and non-infected cells in the same field displayed strong junction plaques, similar to controls (arrows). Note in C, amastigote forms of the parasite revealed by DIC microscopy. (Scale bars = 50.0μm).
Cx43 expression is reduced in hearts of acutely infected mice
In the myocardium of mice at 11 days PI there were many pseudocysts (Figure 5A) and tissue inflammation was prominent (not shown). Immunoblotting analysis of atria and ventricles demonstrated a significant reduction of Cx43 levels (28 and 24% respectively) as compared with controls (Figure 5B and 5C). The ratio between phosphorylated and nonphosphorylated Cx43 in the infected hearts were not significantly different.
Figure 5. Infection reduced Cx43 expression in the heart.
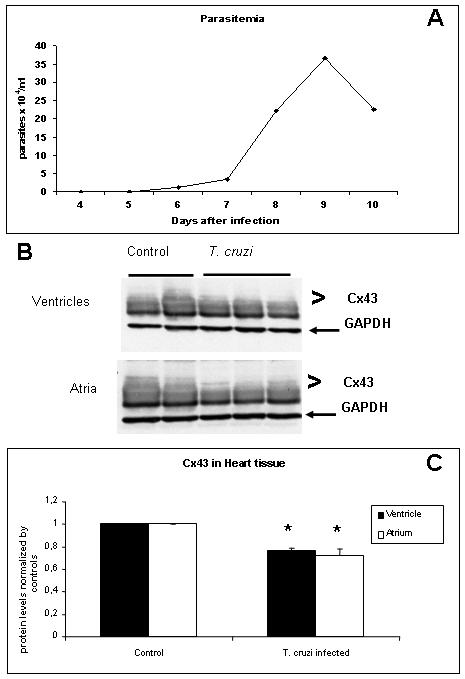
Mice infected with 104 trypomastigotes and reached the peak of parasitemia at day 9 PI (A). At day 11 PI hearts were collected and atria and ventricles were analyzed separately. B and C: Western blot analysis demonstrated a significant reduction in the expression of Cx43 in both atria (28%) and ventricles (24%), (* p<0.05, ANOVA).
Discussion
Investigations into the alterations in T. cruzi-associated reduction in gap junction expression and function has been a continuing interest to our laboratories because Chagas’ disease is an important cause of heart disease. Chagas disease is associated with both an acute myocarditis and chronic dilated cardiomyopathy. Cardiac dysrhythmias are common and alterations in gap junctions have been linked to disruption in the electrical activity of the heart. In the present report we sought to the further understanding of the role of T. cruzi infection and alteration on cardiac gap junction protein. For this purpose we used an in vitro model of cultured mouse embryonic cardiac myocytes, which exhibit typical cardiac myocyte features such as prominent myofibrils and spontaneous contractility [14]. When infected with Y strain trypomastigotes these myocytes responded with accelerated beating when compared to non-infected cells [16].
Our previous observations [3,4] and those of others [2], demonstrated that intracellular calcium concentrations in the host cell were increased during initial times of infection with trypomastigote forms of the parasite and that these calcium waves could propagate in a cluster of neighboring cells [4]. To verify whether T. cruzi invasion could modulate gap junctional communication we examined Cx43 expression/distribution in mouse cardiac myocytes during initial infection with the Y strain. In cultured myocytes, immunoblots for Cx43 showed two phosphorylated bands (P1 and P2) and one lower non-phosphorylated band (P0), as previously described for heart tissue [see 17], and Cx43 immunostaining was present predominantly at appositional membranes of cell-cell contacts. At 1 hour PI we observed increased levels of both total Cx43 and Cx43 phosphorylated bands. This observation is consistent with the transmission of calcium waves between clustered myocytes as described by Garzoni et al [4] and helps the elucidation of mechanisms triggered in the host cell during initial steps of T. cruzi infection. Although Cx43 phosphorylation can lead to closure of gap junction channels [18], the compensatory increased production of Cx43 presumably accounts for the propagation of calcium waves during the invasion of the host cell by the parasite. Cx phosphorylation can occur due to rises in intracellular calcium levels [18] and this rise can be induced in mammalian cell cultures such as cardiac myocytes [3,4] and fibroblasts [2] by T. cruzi infection. Moreover, the increased expression of Cx43 may be explained by the fact that cyclic AMP, which is increased during T. cruzi invasion [19], can up-regulate Cx43 expression [20]. The immunostaining for Cx43 one hour PI also revealed increased perinuclear staining, indicating increased Cx43 in the sarcoplasmic reticulum, presumably reflecting increased synthesis.
Further analysis of Cx43 at 24-72 hours PI revealed a major alteration at the final time point, when infected cells had many intracellular amastigotes and no Cx43 staining. Our observation that non-infected cardiac myocytes in infected culture plates displayed Cx43 immunostaining comparable to controls is consistent to findings described by Campos de Carvalho [7,21] that that uninfected cardiac myocytes had normal junctional coupling. The decrease in Cx43 level that we detected at 72 hr PI in mouse cardiac myocytes is in contrast with previous observations in rat cardiac myocytes infected with Tulahuen strain, which showed no alteration in levels of either Cx43 or its phosphorylation state after 72 hours PI [21]. These results may be the due to different combinations of host cell types and parasite strains. That different combinations of host cell- T. cruzi strain give different results has been reported previously. For example, when mouse cardiac myocytes were infected with Y strain there was a reduced response to norepinephrine [16] and there were no alterations in myogenesis [14]. However, when rat cardiac myocytes were infected with Tulahuen there was an increased response to norepinephrine [3]. Interestingly, when rat myoblasts were infected with Brazil strain there was inhibition of myogenesis [22]. The increased phosphorylation of Cx43 at 72 hours PI could also explain why cardiac myocytes lost cell-cell communication as described by de Carvalho et al [7]. In addition, it was reported that T. cruzi infected murine peritoneal macrophages displayed up-regulation of tyrosine kinases [23], which could also promote Cx43 phosphorylation in cardiac myocytes [24]. Theories have emerged regarding Cx43-vesicles trafficking to the plasma membrane suggesting that this process relies on microtubule transport [25,26], which during T. cruzi infection could be impaired by cytoskeleton breakdown caused by the parasite at late times of infection [27].
During in vivo infection with T. cruzi, we found that atria and ventricles of infected mice displayed a 28 to 24% reduction in Cx43 levels, respectively. These results are consistent with our data with infected cultured cells. Such a reduction in overall Cx43 abundance in the infected heart presumably reflects in homogenous distribution; a condition that is a prominent feature of ventricular conduction disorders (13). The reduction in the expression of Cx43 levels in the infected heart may be induced by the parasite per se. However, it should be noted that infection is accompanied by a robust inflammatory response and a significant increase in cytokines such as TNF-α, IFN-γ and TGF-β [28] which may down regulate Cx expression in the cardiovascular system [29]. Gap junctional communication between cardiac myocytes has also been demonstrated to be modulated by soluble factors present in sera from chronic chagasic patients [30]. These observations indicate that the inflammatory process triggered by T. cruzi infection can also modulate Cx43 expression in cardiac tissue.
In summary, our data demonstrate redistribution of Cx43 expression in Y strain infected mouse cardiac myocytes. This is consistent with our previous published observation regarding infection of rat myocytes with the Tulahuen strain. However, unlike the observations that infection of rat myocytes with the Tulahuen strain did result in alterations in the expression of Cx43 by immunoblot in the current report there are alterations in Cx43 expression. In addition, we now show for the first time by immunoblot analysis that acute infection is associated with a reduction in Cx43 expression. Taken together these data may explain in part the dysrhythmias and conduction abnormalities that attend this infection.
Acknowledgments
This work was supported in part by CNPq; PAPES IV, Fiocruz; FAPERJ and NIH AI- 052739 (HBT). Daniel Adesse was supported a Fogarty International Training Grant (D43TW007129). The authors thank Marcia U. Maldonado and Dr. Mia M. Thi for assistance with the Western blots experiments and Carlos Bizarro and Rodrigo Mechas for assistance with the Confocal Imaging.
All authors have no commercial or other association that might pose a conflict of interest. This work was supported by FIOCRUZ/FAPERJ, PAPES IV-FIOCRUZ, CNPq and NIH AI- 052739 (HBT), PO1 (HD 32573) (DCS), Daniel Adesse is a PhD student and was supported by IOC-FIOCRUZ and a Fogarty International Training Grant (D43TW007129).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tanowitz HB, Kirchhoff LV, Simon D, Morris SA, Weiss LM, Wittner M. Chagas’ disease. Clin Microbiol Rev. 1992;5(4):400–19. doi: 10.1128/cmr.5.4.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rodriguez A, Rioult MG, Ora A, Andrews NW. A trypanosome-soluble factor induces IP3 formation, intracellular Ca2+ mobilization and microfilament rearrangement in host cells. J Cell Biol. 1995;129(5):1263–73. doi: 10.1083/jcb.129.5.1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergdolt BA, Tanowitz HB, Wittner M, Morris SA, Bilezikian JP, Moreno AP, Spray DC. Trypanosoma cruzi: effects of infection on receptor-mediated chronotropy and Ca2+ mobilization in rat cardiac myocytes. Exp Parasitol. 1994;78(2):149–60. doi: 10.1006/expr.1994.1015. [DOI] [PubMed] [Google Scholar]
- 4.Garzoni LR, Masuda MO, Capella MM, Lopes AG, de Meirelles Mde N. Characterization of [Ca2+]i responses in primary cultures of mouse cardiomyocytes induced by Trypanosoma cruzi trypomastigotes. Mem Inst Oswaldo Cruz. 2003;98(4):487–93. doi: 10.1590/s0074-02762003000400010. [DOI] [PubMed] [Google Scholar]
- 5.Burleigh BA. Host cell signaling and Trypanosoma cruzi invasion: do all roads lead to lysosomes? Sci STKE. 2005;19(293):pe36. doi: 10.1126/stke.2932005pe36. 2005. [DOI] [PubMed] [Google Scholar]
- 6.Andrade LO, Andrews NW. Lysosomal fusion is essential for the retention of Trypanosoma cruzi inside host cells. J Exp Med. 2004;1;200(9):1135–43. doi: 10.1084/jem.20041408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Carvalho AC, Tanowitz HB, Wittner M, Dermietzel R, Roy C, Hertzberg EL, Spray DC. Gap junction distribution is altered between cardiac myocytes infected with Trypanosoma cruzi. Circ Res. 1992;70(4):733–42. doi: 10.1161/01.res.70.4.733. [DOI] [PubMed] [Google Scholar]
- 8.Duffy HS, Fort A, Spray DC. Cardiac connexins: genes to nexus. Adv Cardiol. 2006;42:1–17. doi: 10.1159/000092550. [DOI] [PubMed] [Google Scholar]
- 9.Severs NJ, Dupont E, Thomas N, Kaba R, Rothery S, Jain R, Sharpey K, et al. Alterations in cardiac connexin expression in cardiomyopathies. Adv Cardiol. 2006;42:228–42. doi: 10.1159/000092572. [DOI] [PubMed] [Google Scholar]
- 10.Söhl G, Willecke K. Gap junctions and the connexin protein family. Cardiovasc Res. 2004;1;62(2):228–32. doi: 10.1016/j.cardiores.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 11.Goodenough DA, Goliger JA, Paul DL. Connexins, connexons, and intercellular communication. Annu Rev Biochem. 1996;65:475–502. doi: 10.1146/annurev.bi.65.070196.002355. [DOI] [PubMed] [Google Scholar]
- 12.Hervé JC, Bourmeyster N, Sarrouilhe D. Diversity in protein-protein interactions of connexins: emerging roles. Biochim Biophys Acta. 2004;23;1662(12):22–41. doi: 10.1016/j.bbamem.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 13.Severs NJ. Gap junction remodeling and cardiac arrhythmogenesis: cause or coincidence? J Cell Mol Med. 2001;5(4):355–66. doi: 10.1111/j.1582-4934.2001.tb00170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meirelles MN, de Araujo-Jorge TC, Miranda CF, de Souza W, Barbosa HS. Interaction of Trypanosoma cruzi with heart muscle cells: ultrastructural and cytochemical analysis of endocytic vacuole formation and effect upon myogenesis in vitro. Eur J Cell Biol. 1986;41(2):198–206. [PubMed] [Google Scholar]
- 15.Garzoni LR, Caldera A, Meirelles Mde N, de Castro SL, Docampo R, Meints GA, Oldfield E, et al. Selective in vitro effects of the farnesyl pyrophosphate synthase inhibitor risedronate on Trypanosoma cruzi. Int J Antimicrob Agents. 2004;23(3):273–85. doi: 10.1016/j.ijantimicag.2003.07.020. [DOI] [PubMed] [Google Scholar]
- 16.Aprigliano O, Masuda MO, Meirelles MN, Pereira MC, Barbosa HS, Barbosa JC. Heart muscle cells acutely infected with Trypanosoma cruzi: characterization of electrophysiology and neurotransmitter responses. J Mol Cell Cardiol. 1993;25(10):1265–74. doi: 10.1006/jmcc.1993.1137. [DOI] [PubMed] [Google Scholar]
- 17.King TJ, Lampe PD. Temporal regulation of connexin phosphorylation in embryonic and adult tissues. Biochim Biophys Acta. 2005;20;1719(12):24–35. doi: 10.1016/j.bbamem.2005.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spray DC, Burt JM. Structure-activity relations of the cardiac gap junction channel. Am J Physiol. 1990;258(2 Pt 1):C195–205. doi: 10.1152/ajpcell.1990.258.2.C195. [DOI] [PubMed] [Google Scholar]
- 19.Caler EV, Morty RE, Burleigh BA, Andrews NW. Dual role of signaling pathways leading to Ca(2+) and cyclic AMP elevation in host cell invasion by Trypanosoma cruzi. Infect Immun. 2000;68(12):6602–10. doi: 10.1128/iai.68.12.6602-6610.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhein S, Polontchouk L, Salameh A, Haefliger JA. Pharmacological modulation and differential regulation of the cardiac gap junction proteins connexin 43 and connexin 40. Biol Cell. 2002;94(78):409–22. doi: 10.1016/s0248-4900(02)00018-7. [DOI] [PubMed] [Google Scholar]
- 21.Campos de Carvalho AC, Roy C, Hertzberg EL, Tanowitz HB, Kessler JA, Weiss LM, Wittner M, et al. Gap junction disappearance in astrocytes and leptomeningeal cells as a consequence of protozoan infection. Brain Res. 1998;20;790(12):304–14. doi: 10.1016/s0006-8993(97)01523-0. [DOI] [PubMed] [Google Scholar]
- 22.Rowin KS, Tanowitz HB, Wittner M, Nguyen HT, Nadal-Ginard B. Inhibition of muscle differentiation by trypanosoma cruzi. Proc Natl Acad Sci U S A. 1983;80(20):6390–4. doi: 10.1073/pnas.80.20.6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ruta S, Plasman N, Zaffran Y, Capo C, Mege JL, Vray B. Trypanosoma cruzi-induced tyrosine phosphorylation in murine peritoneal macrophages. Parasitol Res. 1996;82(6):481–4. doi: 10.1007/s004360050149. [DOI] [PubMed] [Google Scholar]
- 24.Lin R, Martyn KD, Guyette CV, Lau AF, Warn-Cramer BJ. v-Src tyrosine phosphorylation of connexin43: regulation of gap junction communication and effects on cell transformation. Cell Commun Adhes. 2006;13(4):199–216. doi: 10.1080/15419060600848516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shaw RM, Fay AJ, Puthenveedu MA, von Zastrow M, Jan YN, Jan LY. Microtubule plus-end-tracking proteins target gap junctions directly from the cell interior to adherens junctions. Cell. 2007;9;128(3):547–60. doi: 10.1016/j.cell.2006.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martin PE, Evans WH. Incorporation of connexins into plasma membranes and gap junctions. Cardiovasc Res. 2004;1;62(2):378–87. doi: 10.1016/j.cardiores.2004.01.016. [DOI] [PubMed] [Google Scholar]
- 27.Pereira MC, Costa M, Chagas Filho C, de Meirelles MN. Myofibrillar breakdown and cytoskeletal alterations in heart muscle cells during invasion by Trypanosoma cruzi: immunological and ultrastructural study. J Submicrosc Cytol Pathol. 1993;25(4):559–69. [PubMed] [Google Scholar]
- 28.Huang H, Chan J, Wittner M, Jelicks LA, Morris SA, Factor SM, Weiss LM, et al. Expression of cardiac cytokines and inducible form of nitric oxide synthase (NOS2) in Trypanosoma cruzi-infected mice. J Mol Cell Cardiol. 1999;31(1):75–88. doi: 10.1006/jmcc.1998.0848. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez-Cobo M, Gingalewski C, Drujan D, De Maio A. Downregulation of connexin 43 gene expression in rat heart during inflammation. The role of tumour necrosis factor. Cytokine. 1999;11(3):216–24. doi: 10.1006/cyto.1998.0422. [DOI] [PubMed] [Google Scholar]
- 30.Costa PC, Fortes FS, Machado AB, Almeida NA, Olivares EL, Cabral PR, Pedrosa RC, et al. Sera from chronic chagasic patients depress cardiac electrogenesis and conduction. Braz J Med Biol Res. 2000;33(4):439–46. doi: 10.1590/s0100-879x2000000400010. [DOI] [PubMed] [Google Scholar]