

Abstract
Genotoxic stress activates the phosphatidylinositol 3-kinase like kinases (PIKKs) that phosphorylate proteins involved in cell cycle arrest, DNA repair, and apoptosis. Previous work showed that the PIKK ataxia telangiectasia mutated (ATM) but not ATR (ATM and Rad3-related) phosphorylates p53 (Ser15) during hyperoxia, a model of prolonged oxidative stress and DNA damage. Here, we show hSMG-1 is responsible for the rapid and early phosphorylation of p53 (Ser15) and that ATM helps maintain phosphorylation after 24 hours. Despite reduced p53 phosphorylation and abundance in cells depleted of hSMG-1 or ATM, levels of the p53 target p21 were still elevated and the G1 checkpoint remained intact. Conditional over-expression of p21 in p53-deficient cells revealed that hyperoxia also stimulates wortmannin-sensitive degradation of p21. SiRNA depletion of hSMG-1 or ATM restored p21 stability and the G1 checkpoint during hyperoxia. These findings establish hSMG-1 as a proximal regulator of DNA damage signaling and reveal that the G1 checkpoint is tightly regulated during prolonged oxidative stress by both PIKK-dependent synthesis and proteolysis of p21.
INTRODUCTION
Cell cycle checkpoints are vital in preserving genomic integrity and protecting cells from adverse environmental conditions, including that caused by reactive oxygen species (ROS). ROS may be produced acutely, such as when ionizing radiation causes radiolysis of water. They may also form and accumulate over time as normal byproducts of aerobic respiration. Indeed, cancer and aging are thought to be caused by the persistent production of toxic ROS that damage DNA and other macromolecules. While much is known about how checkpoints stall cell growth following acute damage, it is less clear how checkpoints are activated and maintained over hours or even days of persistent oxidative damage.
Cell cycle checkpoints are activated by members of the phosphatidylinositol 3-kinase like kinase (PIKK) family of proteins that respond to genotoxic stress. The PIKKs ATM (ataxia telangiectasia mutated) and ATR (ATM and Rad3 related) function in parallel pathways to coordinate the cellular response to DNA damage. ATM plays a crucial role in regulating checkpoint activation in response to DNA double strand breaks as seen with ionizing radiation (Suzuki et al., 1999). While ATM can directly bind damaged DNA in vitro, it can also be recruited to sites of damage by a complex of Mre11, Rad50 and Nbs1 (MRN complex) (Abraham & Tibbetts, 2005; Lee & Paull, 2005; Smith et al., 1999). ATM becomes activated in response to DNA lesions or perhaps by alterations in chromatin structure through intermolecular autophosphorylation on Ser1981 (Bakkenist & Kastan, 2003). On the other hand, ATR regulates the cellular response to ultraviolet (UV) radiation induced damage and stalled DNA replication forks and also participates in the late response to IR (Guo et al., 2000; Tibbetts et al., 1999). Activation of ATR proceeds through its association with ATR-interacting protein (ATR-IP) (Cortez et al., 2001). More recently, an additional PIKK family member termed hSMG-1 or ATX was also shown to regulate cell cycle checkpoints in response to IR and UV irradiation (Brumbaugh et al., 2004). hSMG-1 was first recognized for its role in regulating non-sense mediated mRNA decay (NMD), an mRNA quality control pathway allowing for degradation of transcripts containing premature termination codons (Denning et al., 2001; Yamashita et al., 2001). While some overlap in function exists, PIKKs are generally not thought of as being redundant and further studies seeking to elucidate how PIKKs respond to different signals are warranted.
Activated PIKKs phosphorylate various downstream targets involved in controlling cell cycle checkpoints, DNA repair and apoptotic pathways. These include p53, Chk1, Chk2, Brca-1, Rad9, Rad17, H2AX, and others (Bao et al., 2001; Bartek & Lukas, 2003; Canman et al., 1998; Chen et al., 2001; Cortez et al., 1999; Stiff et al., 2004). Of particular importance to checkpoint activation is p53 phosphorylation on serine 15. ATM can directly phosphorylate p53 (Ser15) in response to IR while ATR is critical in the same response to UV (Canman et al., 1998; Tibbetts et al., 1999). Additionally, p53 is phosphorylated on Ser20 by Chk1 or Chk2, which are targets for ATR and ATM signaling respectively, although some cross-talk exists between the pathways (Bartek & Lukas, 2003). Phosphorylation of p53 on Ser15 or Ser20 stabilizes p53 against proteasomal degradation (Brooks & Gu, 2003). Stabilized p53 serves as a transcription factor and stimulates expression of the cyclin-dependent kinase inhibitor p21 which activates cell cycle checkpoints by inhibiting cyclin/cdk complexes and PCNA, and also promotes cell survival in response to oxidative stress (el-Deiry et al., 1993; O'Reilly et al., 2001; Podust et al., 1995). Tight regulation of the p53 pathway in response to genotoxic stress is critical in ensuring the optimal outcome since it controls whether cells undergo programmed cell death or survive in response to DNA damage (O'Connor, 1997).
While checkpoint signaling has been characterized relatively well utilizing acute models of DNA damage, the ability to activate and maintain cell cycle checkpoints in response to prolonged periods of stress is less well understood. We previously reported that hyperoxia (95% oxygen), a model of chronic oxidative stress and DNA damage that occurs over several days, stimulate ATM-dependent phosphorylation of p53 (Ser15) and Chk2 (Thr68) (Helt et al., 2005). Intriguingly, some p53 (Ser15) phosphorylation was still detected in ATM (−/−) lymphoblasts exposed to hyperoxia. Since p53 (Ser15) phosphorylation was not blocked by dominant-negative ATR, the current study elucidates the involvement of hSMG-1 in phosphorylation of p53 during hyperoxia and determines how activated PIKKs maintain the G1 checkpoint over several days of persistent oxidative stress.
RESULTS
ATM is not required for early p53 phosphorylation in hyperoxia
We previously reported ATM, but not ATR, regulates p53 (Ser15) phosphorylation and abundance after 24 hours of hyperoxia (Helt et al., 2005). To further define p53 activation during hyperoxia, ATM (+/+) and ATM (−/−) lymphoblasts were cultured in room air (0 hours) or in hyperoxia. In ATM (+/+) cells, p53 (Ser15) phosphorylation and expression increased within 3 hours of hyperoxia and continued to increase over the first 24 hours of exposure (Figure 1a). While p53 (Ser15) phosphorylation and abundance also increased in ATM (−/−) cells, it was not maintained after 24 hours. Consistent with ATM activation between 12–24 hours, Chk2 (Thr68) phosphorylation was detected after 12 hours of exposure in ATM (+/+), but not in ATM (−/−) cells. To determine whether other wortmannin-sensitive kinases signal to p53 at early time-points in hyperoxia, ATM (−/−) lymphoblasts were cultured in hyperoxia and wortmannin for 12 hours. Hyperoxia stimulated phosphorylation of p53 in ATM (−/−) cells, but not in cells treated with wortmannin (Figure 1b). Therefore, additional wortmannin-sensitive kinases initiate p53 (Ser15) phosphorylation and abundance during hyperoxia while ATM maintains p53 activation over time.
Figure 1. ATM is not required for p53 activation at early times in hyperoxia.
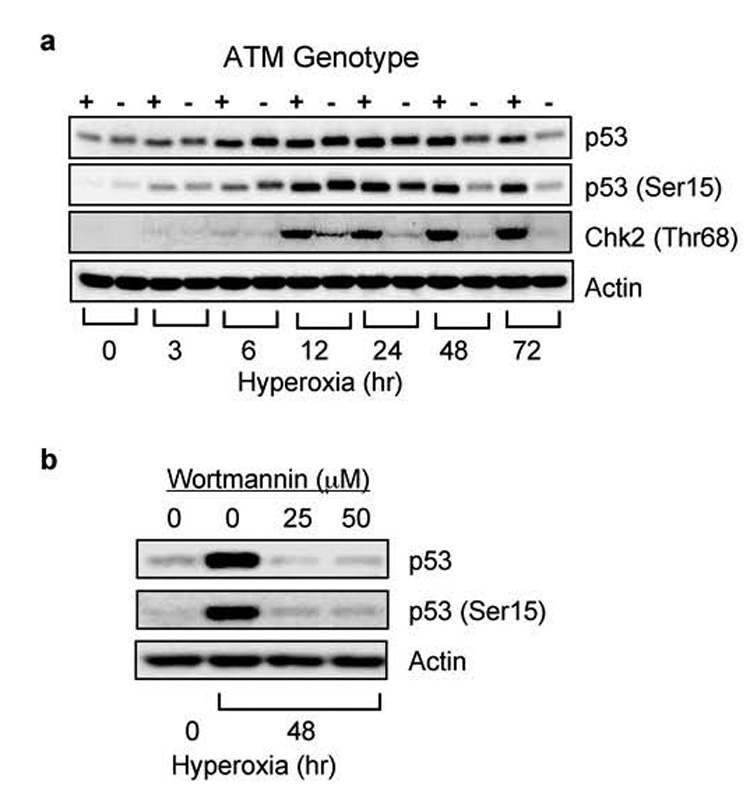
(a) GM00536 (ATM +/+) and GM01526 (ATM −/−) cells were exposed to room air (0) or hyperoxia for 3, 6, 12, 24, 48, 72 hours. Cell lysates were immunoblotted for p53, p53 (Ser15), Chk2 (Thr68), and actin. (b) GM01526 ATM (−/−) cells were treated with wortmannin (25 and 50 µM) or vehicle (0 µM) and exposed to hyperoxia for 12 hours. Cell lysates were immunoblotted for p53, p53 (ser15), and actin.
hSMG-1 and ATM phosphorylate p53 (Ser15) during hyperoxia
Further studies were carried out using an RNAi approach in A549 cells because we were not able to successfully transfect the lymphoblast cell lines with siRNA oligonucleotides. A549 cells were mock transfected and with siRNAs oligonucleotides targeting ATM, hSMG-1, ATR, or luciferase as a non-PIKK targeting control. Each siRNA specifically inhibited expression of its target protein by at least 70% while oligonucleotides against luciferase did not affect PIKK expression (Figure 2a). A549 cells were transfected with siRNA oligonucleotides, exposed to hyperoxia for 48 hours, and p53 phosphorylation assessed by western blot analysis (Figure 2b). Quantitation of band intensities revealed knockdown of ATM and hSMG-1 significantly reduced p53 (Ser15) phosphorylation (Figure 2c). In contrast, knockdown of ATR did not affect p53 (Ser15) phosphorylation. To distinguish PIKK-dependent changes in p53 phosphorylation from abundance, A549 cells were transfected with increasing doses of hSMG-1 siRNA and exposed to hyperoxia for 48 hours. While 50 nM of oligonucleotide was sufficient to reduce p53 (Ser15) phosphorylation and abundance, 1 nM was sufficient to reduce p53 (Ser15) phosphorylation by 63% without lowering p53 abundance (Figure 2d). Similarly, siRNA knockdown of hSMG-1 inhibited p53 (Ser15) phosphorylation in HCT116 colon carcinoma cells exposed to hyperoxia (Figure 2d). For unknown reasons, higher doses of targeting oligonucleotides in HCT116 cells had minimal effects on p53 abundance. These findings reveal hSMG-1 as another PIKK controlling p53 (Ser15) phosphorylation during hyperoxia.
Figure 2. hSMG-1 and ATM phosphorylate p53 (Ser15) during hyperoxia.
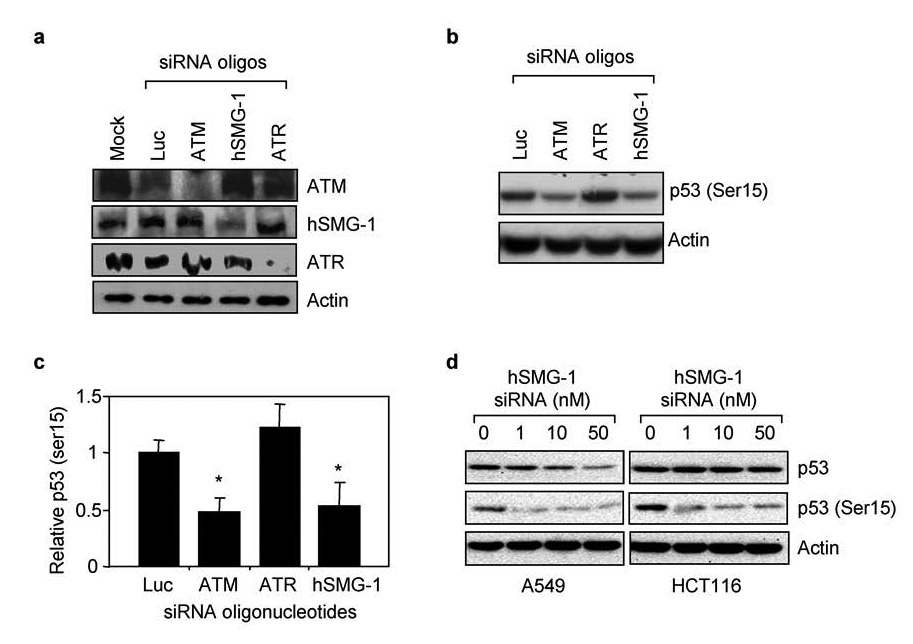
(a) A549 cells were mock transfected or with siRNA oligonucleotides against luciferase, ATM, hSMG-1, or ATR. Cell lysates obtained 24 hours later were immunoblotted for ATM, hSMG-1, ATR or actin. (b) A549 cells were transfected with siRNA oligonucleotides against luciferase, ATM, hSMG-1 or ATR and exposed to hyperoxia for 48 hours. Cell lysates were immunoblotted for p53 (Ser15) or actin. (c) p53 (Ser15) abundance in cells was quantified and graphed relative to values obtained with cells transfected with luciferase siRNA. Values represent mean +/− SEM (n =3–4 experiments). (d) A549 and HCT116 cells were transfected with increasing concentrations of siRNA oligonucleotides against hSMG-1 and exposed to hyperoxia for 48 hours. Cell lysates were immunoblotted for p53, p53 (Ser15) and actin.
The PIKK hSMG-1 mediates early p53 phosphorylation in hyperoxia
To determine if hSMG-1 is responsible for activating p53 at early ATM-independent time-points in hyperoxia, A549 cells were transfected with siRNA oligonucleotides targeting ATM, hSMG-1 or control luciferase and exposed to hyperoxia for 1, 3, 6, and 12 hours. Knockdown of hSMG-1 but not ATM or control luciferase reduced p53 phosphorylation at 3 hours (Figure 3a). After 6 hours, siRNA knockdown of hSMG-1 reduced both p53 phosphorylation and abundance. To determine the role of hSMG-1 and ATM at later times in hyperoxia, A549 cells were transfected with targeting oligonucleotides and exposed to hyperoxia for 24, 48, and 72 hours of hyperoxia. siRNA knockdown of ATM and hSMG-1 reduced both p53 (Ser15) and p53 abundance at these later times (Figure 3b).
Figure 3. The PIKK hSMG-1 mediates early p53 phosphorylation in hyperoxia.
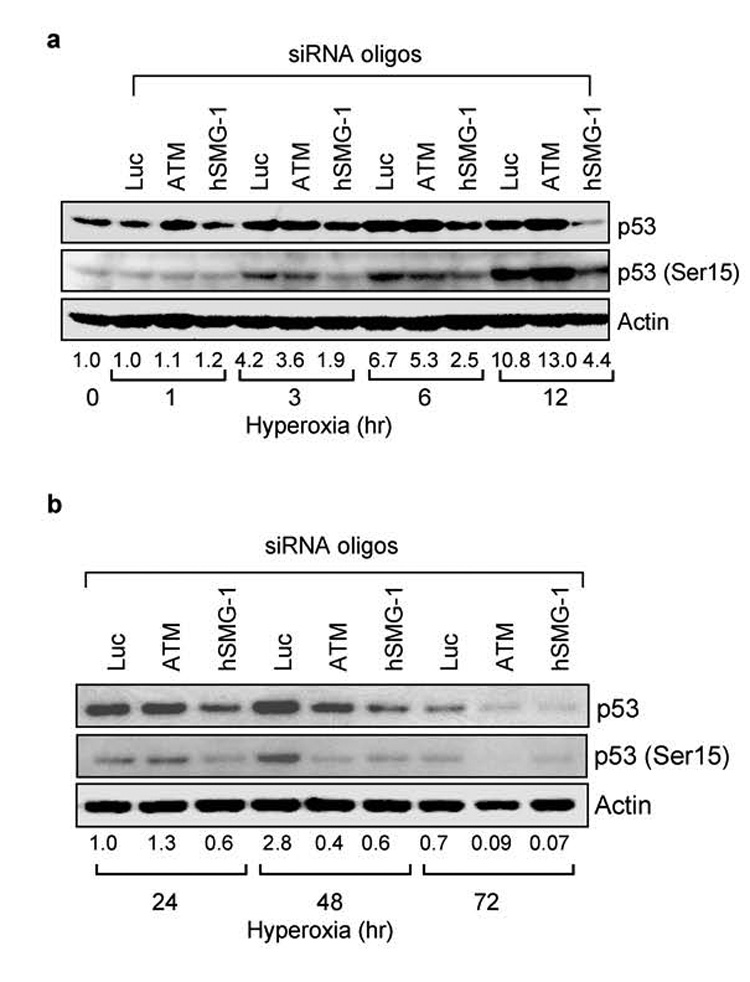
(a) A549 cells were transfected with siRNA against luciferase, ATM or hSMG-1 and immediately harvested (0 hours) or after 1, 3, 6, and 12 hours of hyperoxia. Cell lysates were immunoblotted for p53, p53 (Ser15) and actin. (b) A549 cells were transfected with siRNA against luciferase, ATM, or hSMG-1 and after 24, 48, and 72 hours of hyperoxia. Cell lysates were immunoblotted for p53, p53 (Ser15) and actin. After normalizing band intensities to levels of actin, the fold induction of p53 (Ser 15) during hyperoxia was determined and listed below each lane. Blots are representative of 3–4 experiments with similar results.
hSMG-1 activity is not required for hyperoxia to activate ATM-dependent phosphorylation of Chk2
Since hyperoxia sequentially stimulated hSMG-1 and ATM-dependent phosphorylation of p53, it was of interest to determine whether ATM activity required the earlier activation of hSMG-1. Since Chk2 (Thr68) phosphorylation was dependent upon ATM kinase activity (Figure 1), Chk2 phosphorylation was investigated in A549 cells transfected with siRNA against hSMG-1, ATM, or luciferase as a non-targeting control. As expected, Chk2 (Thr68) phosphorylation increased during hyperoxia and was dependent upon ATM (Figure 4). SiRNA knockdown of hSMG-1 or luciferase did not inhibit Chk2 phosphorylation. ATM undergoes autophosphorylation on serine 1981 as it becomes active (Bakkenist & Kastan, 2003). siRNA oliognucleotides against ATM, but not hSMG-1 reduced ATM (Ser1981) expression (data not shown). Together these findings reveal that hSMG-1 is not required to activate ATM.
Figure 4. hSMG-1 is not required for ATM activation in hyperoxia.
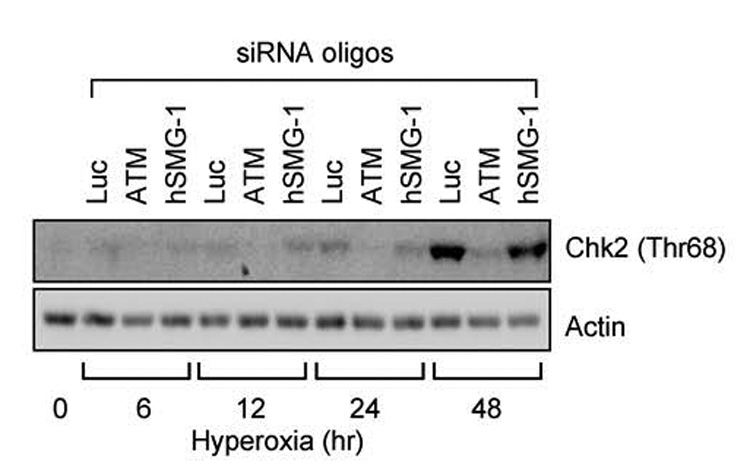
A549 cells were transfected with siRNA oligonucleotides against luciferase, ATM, or hSMG-1 and immediately harvested (0 hours) or after 6, 12, 24 and 48 hours of hyperoxia. Cell lysates were immunoblotted for Chk2 (Thr68) and actin.
PIKKs control p21 and the G1 checkpoint during hyperoxia
Previous studies established that induction of p21 and the G1 checkpoint during hyperoxia are entirely dependent upon p53 activation (Helt et al., 2004). Cells lacking p53 exit G1 during hyperoxia and arrest in S phase through a presently undefined mechanism that involves progressive reduction in BrdU incorporation and elongation of BrdU labeled DNA. To confirm that PIKK dependent inhibition of p53 activation also inhibited the induction of p21 and the G1 checkpoint, A549 cells were transfected with siRNA oligonucleotides against ATM, hSMG-1, ATR, and luciferase. Cells were then exposed to room air or hyperoxia for 48 hours and the expression of p21 determined. Intriguingly, p21 levels increased two-fold higher in hyperoxic cells transfected with siRNA against ATM or ATR while siRNA against hSMG-1 or luciferase did not affect p21 expression (Figure 5a). Increased levels of p21 were also associated with fewer cells arresting in S phase (Figure 5b,c). Thus, PIKK inhibition and subsequent reduction in p53 phosphorylation was not associated with a failure to upregulate p21 and execute the G1 checkpoint. To test whether PIKKs also control p21 stability, A549 cells were exposed to room air or hyperoxia for 48 hours and then treated with cyclohexamide to inhibit new protein synthesis. As predicted, p21 levels declined faster in cells exposed to hyperoxia compared to room air (Figure 5d). These findings reveal that oxidative stress stimulates proteolysis of p21.
Figure 5. Knockdown of PIKKs does not reduce p21 and the G1 checkpoint.
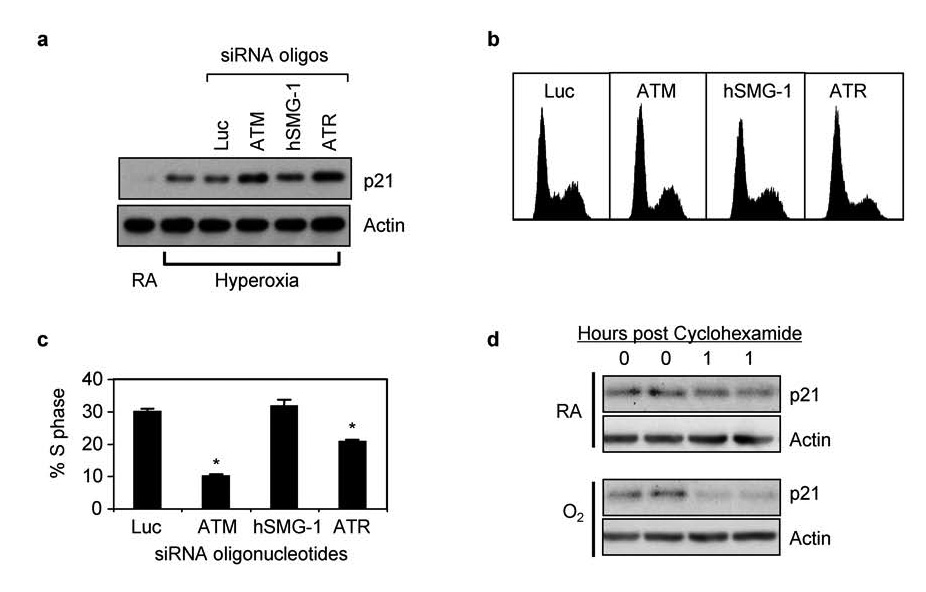
(a) A549 cells were exposed mock transfected or with siRNA oligonucleotides against luciferase, ATM, hSMG-1, or ATR and immediately harvested (0 hours) or after 48 hours of hyperoxia. Cell lysates were immunoblotted for p21 and actin. (b) Representative DNA histograms of cells transfected with siRNA oligonucleotides against luciferase, ATM, hSMG-1, or ATR and exposed to hyperoxia for 48 hours. (c) The percentage of transfected cells in S phase was determined and graphed. Values represent mean % +/− SEM (n = 4 experiments). (d) A549 cells were cultured in room air or hyperoxia for 48 hours and then treated with cyclohexamide. Cells were immediately harvested in duplicate and after one hour. Lysates were immunoblotted for p21 and actin.
EGFp21 stability is regulated by the proteasome
To further investigate how oxidative stress controls p21 stability, we took advantage of p53-deficient H1299 cells with conditional expression of enhanced green fluorescent protein (EGFP) fused to p21 (EGFp21). Because these cells lack p53, they fail to express p21 and therefore growth arrest in S phase during hyperoxia (Helt et al., 2004). Conditional over-expression of p21 restores the G1 checkpoint during hyperoxia. Thus, these cells allowed us to investigate how oxidative stress controlled p21 stability independent of its ability to induce p21 via p53 activation. Since endogenous p21 is degraded by the proteasome, we first investigated whether EGFp21 stability could be enhanced when the proteasome is inhibited. H1299-EGFp21 cells were treated with doxycycline to induce expression of EGFp21 and then cultured with cyclohexamide in the absence or presence of the proteasome inhibitor MG132. As predicted, inhibition of the proteasome with MG132 prevented the loss of EGFp21 over time (Figure 6). These findings confirm that the proteasome controls EGFp21 stability in H1299 cells.
Figure 6. Proteasomal processing of EGFp21 fusion protein.
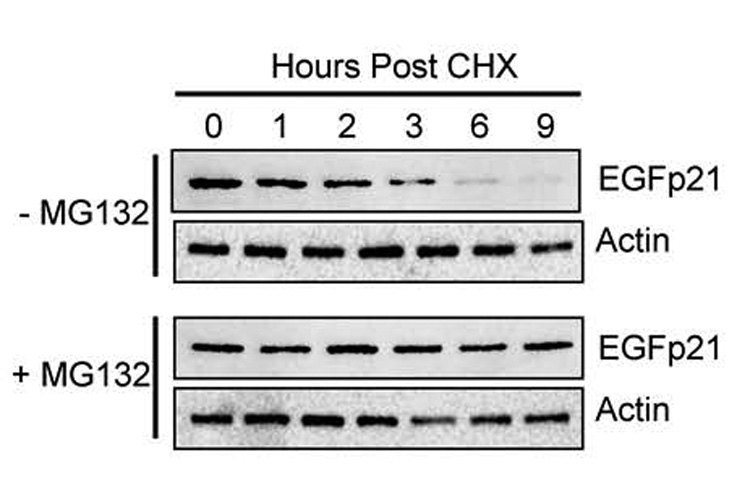
H1299-EGFp21 cells were treated for 24 hours with doxycycline (2µg/mL) to induce EGFp21 expression and then with cyclohexamide (50µg/mL) in the absence or presence of the proteasome inhibitor MG132 (50μM). Protein lysates were harvested at 1, 2, 3, 6 and 9 hours post treatment and immunoblotted for EGFp21 and actin. Blots are representative of 3–4 experiments.
Hyperoxia stimulates proteasome-dependent p21 degradation through a wortmannin-sensitive pathway
To determine whether hyperoxia affects stability of EGFp21, H1299/EGFp21 cells were treated with or without doxycycline and simultaneously exposed to hyperoxia or room air for 24 hours. Cyclohexamide (50 µg/ml) was added to inhibit the synthesis of new proteins and the cells returned to room air or hyperoxia. Cell lysates were collected over the next 24 hours and immunoblotted for EGFp21. Expression of EGFp21 declined over time and the protein was more rapidly lost in cells exposed to hyperoxia (Figure 7a). Quantitation of blots revealed that exposure to hyperoxia destabilized EGFp21 by approximately 35 % from 4.1 +/− 0.46 hours in room air to 2.7 +/− 0.11 hours in hyperoxia treated cells (n=3, p = 0.002) (Figure 7b). To determine whether increased p21 proteolysis during hyperoxia was mediated by PIKKs, cells were treated with doxcycline to induce EGFp21, wortmannin to block PIKKs, and exposed to hyperoxia for 24 hours. Cyclohexamide was added to inhibit new protein synthesis and expression of EGFp21 assessed in lysates prepared over the next 9 hours. Consistent with PIKKs regulating p21 stability, wortmannin delayed loss of EGFp21 (Figure 7c). Quantitation of blots revealed that EGFp21 stability increased from 2.1 +/− 0.28 hours without wortmannin to 3.6 +/−1.1 hours with wortmannin (n=3, p = 0.035) (Figure 7d). These data support a role for wortmannin-sensitive kinases in the hyperoxia-induced degradation of EGFp21 since treatment with wortmannin essentially reversed the destabilizing effects of hyperoxia on p21.
Figure 7. Oxidative stress stimulates wortmannin sensitive proteolysis of p21.
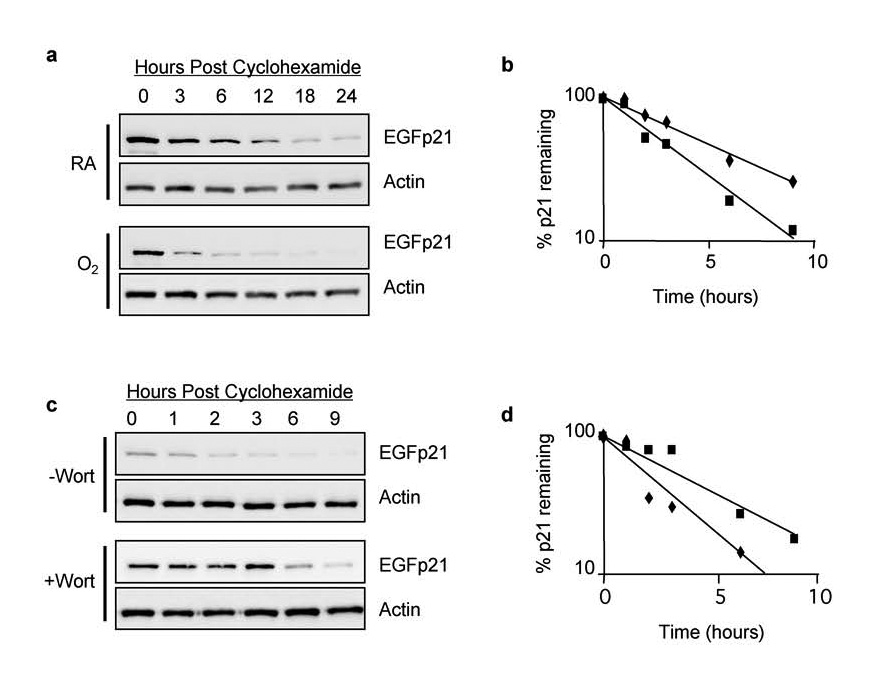
H1299-EGFp21 cells were exposed to room air or hyperoxia for 24 hours in the presence of doxycycline (2 µg/ml) to induce EGFp21 fusion protein. Cycloheximide (50 µg/ml) was added to inhibit new protein synthesis and cells were returned to room air or hyperoxia. (a) Cell lysates harvested following cycloheximide were immunoblotted with antibodies against EGFP or actin. (b) Graph of EGFp21 band intensity from a representative experiment in which cells were exposed to room air (filled triangles) or hyperoxia (open triangles). (c) H1299-EGFp21 cells were exposed to hyperoxia for 24 hours, doxycycline (2 µg/ml) to induce EGFp21 fusion protein, and the absence or presence of wortmannin to inhibit PIKK activity. Cycloheximide (50 µg/ml) was added to inhibit new protein synthesis and EGFp21 protein levels determined by western blot analysis. (d) Graph of EGFp21 band intensity from a representative experiment in which cells were exposed to hyperoxia (filed squares) or hyperoxia and wortmannin (open squares).
hSMG-1 and ATM regulate p21 stability during hyperoxia
To identify the PIKKs involved in regulating p21 stability during hyperoxia, H1299/EGFp21 cells were transfected with siRNA oligonucleotides targeting hSMG-1, ATM, ATR, and luciferase. Transfected cells were then treated with doxycycline to induce EGFp21 and exposed to hyperoxia for 48 hours. Expression of EGFp21 was assessed by western blot at that time (0 hours) or 9 hours after treatment with cyclohexamide (Figure 8a). Consistent with a role for ATM and hSMG-1 in mediating the loss of p21 in hyperoxia, siRNAs targeting ATM or hSMG-1 restored EGFp21 stability in hyperoxia (Figure 8b). While siRNAs targeting ATR enhanced expression of p21, it was not a significant change. To assess whether PIKK-dependent loss of p21 stability correlates with alterations in cell cycle progression, doxycycline treated H1299/EGFp21 cells were transfected with siRNA oliognucleotides against luciferase, ATM, hSMG-1, or ATR and exposed to hyperoxia for 48 hours. Interestingly, knockdown of ATM, hSMG-1, but not ATR, enhanced the G1 checkpoint (Figure 8c) and reduced the number of cells arrested in S phase (Figure 8d). Thus, ATM and hSMG-1 stimulate proteolysis of p21 during hyperoxia and therefore diminish G1 checkpoint activation. To confirm that cell cycle changes were dependent upon p21, the effects of PIKK inhibition was investigated in cells cultured in the absence of doxycycline. Since these cells lack p53, they fail to express p21 and therefore arrest in S phase during hyperoxia (Figure 8c). SiRNA knockdown of ATM, hSMG-1 or ATR had no effect on S phase growth arrest (Figure 8c,d). Taken together, these findings reveal that ATM and hSMG-1, and to a lesser extent ATR, control p53-independent proteolysis of p21 during oxidative stress.
Figure 8. hSMG-1 and ATM regulate p21 stability during oxidative stress.
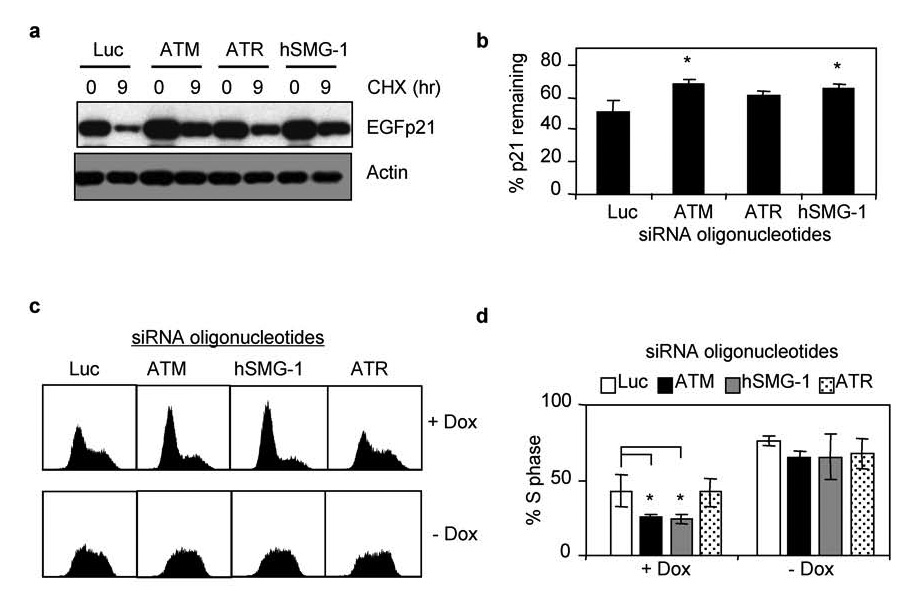
(a) H1299-EGFp21 cells were transfected with siRNA oligonucleotides against luciferase, ATM, ATR, or hSMG-1 and then exposed to hyperoxia and doxycycline for 24 hours. Cycloheximide (50 µ g/ml) was added to inhibit new protein synthesis and cells immediately harvested (0 hours) or 9 hours later. Lysates were immunblotted for EGFp21 and actin. (b) The percent of EGFp21 remaining 9 hours after cycloheximide was determined for each treatment and graphed. Values represent mean +/− SEM (n = 3 experiments). (c). Representative DNA histograms of H1299-EGFp21 cells transfected with siRNA oligonucleotides against luciferase, ATM, hSMG-1, or ATR and exposed to hyperoxia for 48 hours in the presence (+dox) or absence (−dox) of doxycycline. (d) The percentage of cells in S phase was determined and graphed. Values represent mean % +/− SEM (n = 4 experiments).
DISCUSSION
Cell cycle checkpoints activated in response to genotoxic stress prevent replication of damaged DNA presumably to allow time for repair or apoptosis. As loss of these checkpoints results in cell transformation or death, checkpoints are vital in preserving genomic integrity and protecting cells from a variety of adverse environmental conditions. Here, we present evidence that hSMG-1 and ATM, but not ATR, control p53 phosphorylation and abundance (Figure 9). Through the use of ATM-deficient lymphoblasts and siRNA knockdown in epithelial cells, we found that hSMG-1 initiates p53 phosphorylation during hyperoxia while ATM helps to maintain phosphorylation over time. While p53 in turn stimulates p21 and executes the G1 checkpoint, we surprisingly discovered ATM, hSMG-1 and to a lesser extent ATR also control p21 stability. Thus, p21 abundance over time is regulated by PIKKs controlling both p53-dependent synthesis and p53-independent proteolysis of p21. P21 is a critical mediator of cellular responses to DNA damage as low levels are required to inhibit cell growth while higher levels are required to protect against hyperoxia-induced cell death (Vitiello et al., 2006). As such, the dual functions of PIKKs to control both synthesis and proteolysis of p21 may allow levels of p21 and cell fate to be tightly coordinated with the extent of DNA damage.
Figure 9. Model of p21-dependent cell cycle arrest during oxidative stress.
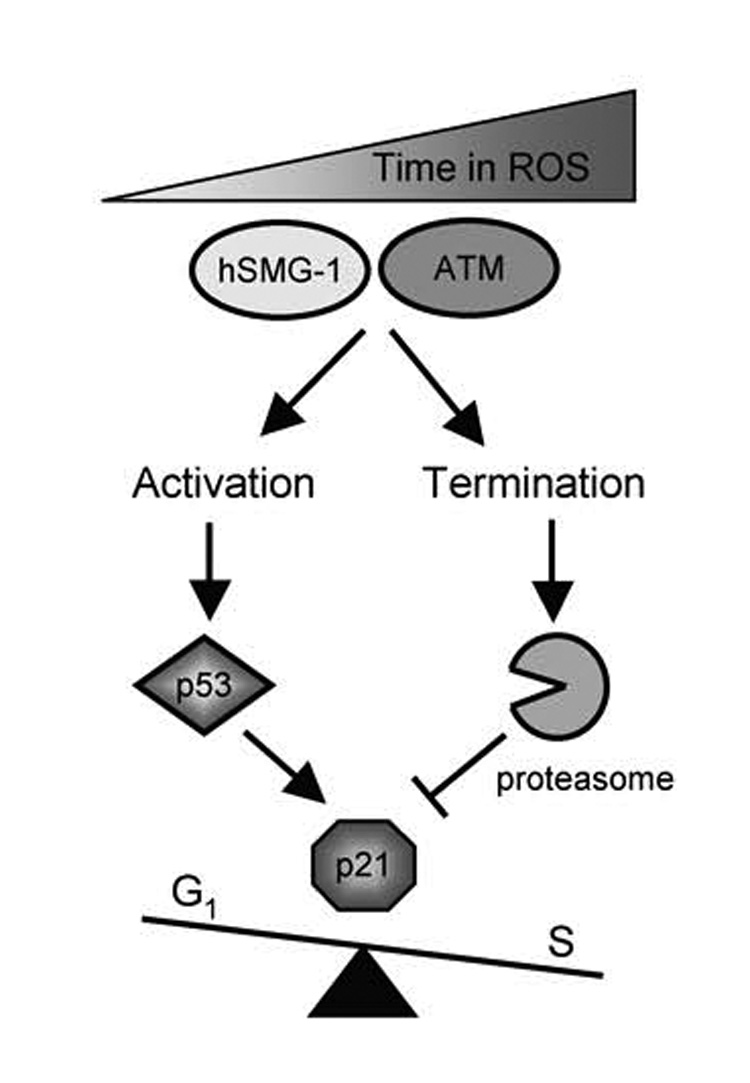
Oxidative stress and damage sequentially activates hSMG-1 and ATM to phosphorylate and activate p53-dependent transcription of p21. Active hSMG-1 and ATM also stimulate p53-independent proteolysis of p21 via the proteasome. Levels of p21 are ultimately dictated by PIKKs controlling p53-dependent synthesis versus p53-independent proteolysis of p21.
Using A549 cells and ATM (+/+) and ATM (−/−) lymphoblasts, we made the novel finding that hSMG-1 mediated phosphorylation of p53 on serine15 occurs prior to ATM-dependent phosphorylation. Moreover, early hSMG-1 signaling in hyperoxia was not necessary to activate ATM signaling at later times. hSMG-1 was previously shown to regulate p53 (Ser15) phosphorylation in response to ionizing radiation (Brumbaugh et al., 2004). The current study extends the role of hSMG-1 by showing it to be a proximal mediator of checkpoint signaling in response to chronic oxidative stress. The rapid activation of hSMG-1 over ATM appears to be unique to hyperoxia as both hSMG-1 and ATM were required to phosphorylate p53 in response to low dose (19cGy) ionizing radiation (data not shown).
Sequential activation of hSMG-1 and ATM may be due to the chronic and progressive nature of hyperoxia as a damaging agent. Differences in spatiotemporal localization of substrates could account for the differences in hSMG-1 and ATM activation. For instance, ATM physically interacts with damaged DNA in a complex containing Mre11, Rad50 and Nbs1 (MRN complex) (Abraham & Tibbetts, 2005; Lee & Paull, 2005; Smith et al., 1999). Thus, signals to recruit and phosphorylate p53 could take longer using a slow progressive model of damage like hyperoxia relative to a bolus of ionizing radiation (Abraham & Tibbetts, 2005). The dynamics of hSMG-1 localization in response to damage have also not been elucidated, so it is possible that hSMG-1 could diffuse more readily to DNA lesions. Alternatively, hSMG-1 and ATM may be responding to different lesions that are sequentially produced during hyperoxia but simultaneously produced with IR. Consistent with this, hSMG-1 somehow recognizes and responds to aberrantly spliced mRNAs while ATM binds DNA double strand breaks. Precedent for PIKKs to sequentially respond to different lesions comes from studies showing that ATM rapidly responds to ionizing radiation while ATR responds later presumably due to replication fork stalling caused by damaged DNA (Bakkenist & Kastan, 2003; Tibbetts et al., 1999). Additional studies are required to clarify why hSMG-1 activation precedes that of ATM in hyperoxia.
This study also found that endogenous and conditionally expressed p21 are destabilized in response to hyperoxia. The loss of p21 stability was reversed by wortmannin, suggesting the involvement of PIKKs. Targeting of PIKK family members ATM and hSMG-1, and perhaps ATR to a lesser extent, restored p21 stability in hyperoxia indicating that p21 stability is regulated through DNA damage signaling. P21 destabilization could affect replication and/or repair through its interactions with PCNA, a component of both processes. Consistent with p21 destabilization affecting DNA repair, the loss of p21 stability in response to low doses of UV was shown to alleviate p21 inhibition of repair through effects on PCNA (Bendjennat et al., 2003). Alternatively, decreased p21 stability could be important in regulating proliferation. We and others have shown that p21 expression alters the expression or stability of PCNA (Engel et al., 2003; Gehen et al., 2007). The enhanced degradation of the more abundant PCNA allows the less abundant p21 to bind and inhibit sliding-clamp functions of remaining PCNA more completely, thereby enhancing the G1 checkpoint. P21 has also been shown to block apoptosis at expression levels much higher than that needed to inhibit cell proliferation (Vitiello et al., 2006). Hypothetically, extensive or prolonged PIKK signaling would enhance p21 proteolysis, thereby reducing levels below the protective threshold and allowing apoptosis to ensue. The ability of PIKKs to control both synthesis and destruction of p21 may therefore allow cells to finely tune their response to DNA damage.
It is likely that PIKKs control proteasome activity because p21 proteolysis was inhibited by wortmannin and with siRNAs targeting ATM or hSMG-1. In addition, p21 stability is known to be regulated by the proteasome (Blagosklonny et al., 1996; Maki & Howley, 1997). Although ubiquitination is a signal for protein degradation, surprisingly, the modification of all six lysines in p21 to arginine did not alter the proteasome-dependent nature of p21 degradation (Sheaff et al., 2000). Instead, p21 was shown to physically interact with the C8 α subunit of the proteasome and over-expression of mutant forms of p21 unable to bind the proteasome showed greatly enhanced stability. Therefore, proteasomal degradation of p21 can proceed through unique mechanisms including direct binding to the proteasome (Coulombe et al., 2004; Touitou et al., 2001). Since PIKKs are activated when cells are damaged and the proteasome is responsible for degrading proteins, understanding how PIKKs control proteasome function may provide insight into how cells remove damaged proteins when injured.
Despite the clear capacity of hSMG-1 and ATM to phosphorylate p53 and regulate p21 stability during hyperoxia, the role of ATR remains less clear. Consistent with our previous study using U2OS cells with stable expression of dominant-negative ATR, siRNA knockdown of ATR failed to inhibit p53 (Ser15) phosphorylation during hyperoxia (Helt et al., 2005). These findings however do not agree with another study showing that over-expression of dominant-negative ATR in HEK293 cells blocks hyperoxia-induced p53 (Ser15) phosphorylation (Das & Dashnamoorthy, 2004). Since hyperoxia activates ATR-dependent phosphorylation of Chk1, unknown genetic differences between these cell lines might be affecting whether p53 is a substrate for ATR. Although ATR may not phosphorylate p53, this study shows that ATR modestly destabilizes p21 and affects the G1 checkpoint in A549 but not in H1299 cells (Figure 5 versus Figure 8). While these differences might be attributed to the presence or absence of p53, it is clear that ATM regulates p21 stability in both cell lines. Likewise, hSMG-1 regulates p21 stability in H1299 cells while having a minimal effect on p21 levels in A549 cells. Evidence suggests that hSMG-1 may be the primary regulator of p21 synthesis since the knockdown of hSMG-1 had dramatic effects on p53 expression levels (Figure 3). Therefore, in cells with wild-type p53, diminished p21 synthesis appears to balance with enhanced p21 stability upon knockdown of hSMG-1 resulting in little change in overall p21 expression. Thus, the role of hSMG-1 in regulating p21 stability was clearly revealed in p53-deficient H1299 cells conditionally over-expressing p21. Taken together, these findings reveal differential capacity of PIKKs to precisely control synthesis versus destruction of the cyclin-dependent kinase inhibitor p21 with hSMG-1 and ATM being the primary regulators of p21 production during hyperoxia.
In summary, this study establishes hSMG-1 as a critical regulator of cell cycle checkpoint signaling under oxidative stress. Furthermore, the results presented support that the G1 checkpoint is carefully and precisely regulated by PIKK-dependent signaling to allow for the optimal response to DNA damage. By regulating both the synthesis and proteolysis of p21, PIKKs can titer p21 levels required to inhibit cell proliferation, affect DNA repair, and block apoptosis. This dual function of PIKKs may therefore allow cells to respond appropriately to the extent of DNA damage present.
MATERIALS AND METHODS
Cell Culture
ATM (+/+) and ATM (−/−) lymphoblasts designated GM00536 and GM01526, respectively, were obtained from the Coriell Cell Repositories (Camden, NJ) and cultured in Roswell Park Memorial Institute medium 1640 with L-glutamine and 15% fetal bovine serum, 50 units/ml penicillin, and 50 µg/ml streptomycin (Invitrogen). A549 and H1299 cells were purchased from the American Type Culture Collection (Manassas, VA) and maintained in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum, 50 units/ml penicillin, and 50 µg/ml streptomycin (Invitrogen). Stable clones of H1299 cells with doxycycline-inducible expression of EGFP fused amino-terminal to human p21 (EGFp21) were created with the pBig2i expression plasmid (Helt et al., 2004). HCT116 colon carcinoma cells were obtained from Dr. Burt Vogelstein at Johns Hopkins School of Medicine. All cell lines were maintained at 37°C under normoxic conditions (room air with 5% CO2) or exposed to hyperoxia (95% O2 / 5% CO2) using a plexiglass exposure chamber (Bellco, Vineland, NJ). Cycloheximide (Sigma Chemical Company, St. Louis, MO), wortmannin (Sigma) and MG132 (Calbiochem, San Diego, CA) were resuspended in DMSO and used to treat cells.
Western blot analysis
Cells were lysed in 50 mM Tris (pH 7.4), 120 mM NaCl, and 0.5% Nonidet P-40 supplemented with 2 µg/ml of aprotinin and 100 µg/ml phenylmethylsulfonyl fluoride. The lysate was cleared by centrifugation, quantified, and boiled for five minutes in Laemmli Buffer (50 mM Tris pH 6.8, 1% β-mercaptoethanol, 2% SDS, 0.1% bromophenol blue, and 10% glycerol). Proteins were separated by polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membrane (PVDF, Millipore, Bedford, MA). The membranes were then incubated overnight at 4°C in antibodies for Chk2 (Thr 68) (Cell Signaling), p53 (Ser15) (Cell Signaling), p53 DO-1 (VisonBioscience), ATM (Ab-3, calbiochem), hSMG-1 (Ab-1) (a generous gift from Dr. Robert Abraham), ATR (Ab-2, Calbiochem), p21 (SX118 PharMingen, San Diego, CA), and anti-β- actin (Sigma, St. Louis, MO). Membranes were washed and then incubated in the appropriate secondary antibodies for 1 hour at room temperature. After washing the membranes with TBST, specific antibody interactions were visualized by chemiluminescence (Amersham Biosciences). Images were captured on a FluorChem SP (Alpha Innotech, San Leandro, CA) and band intensities were quantified.
RNAi Treatment
Cells were plated in 6 well plates overnight and then transfected with luciferase (5’-CGU ACG CGG AAU ACU UCG ATT-3’), ATM (GCG CCU GAU UCG AGA UCC UUU), hSMG-1 (CCA GGA CAC GAG GAA ACU GTT), or ATR (GCC AAG ACA AAU UCU GUG UTT) siRNA (Brumbaugh et al., 2004; Casper et al., 2002; Yoshida et al., 2003). All siRNA transfections were carried out using 100nM of a given siRNA oligonucleotide unless specified. Cells were transfected using lipofectamine2000 transfection reagent according to the manufacturer’s instructions (Invitrogen, Carlsbad, California). A high level of transfection efficiency was verified 12–24 hours post transfection using a fluorescently labeled siRNA (Invitrogen, Carlsbad, California).
Protein stability
Stability of p21 or EGFp21 was determined by treating A549 or H1299+EGFp21 cells with hyperoxia followed by 50µg/mL of the protein synthesis inhibitor cycloheximide. Protein lysates were harvested at the indicated times and immunoblotted for p21 or EGFP . Relative levels of p21 or EGFp21 expression was determined for each time-point following cycloheximide treatment. Stability of p21 or EGFp21 in hyperoxia was compared to that in normoxia-treated cells.
Statistical Analysis
All experiments were repeated 3–4 times. Where appropriate values are means +/− SD. Group means were compared by ANOVA using Fisher’s procedure post hoc analysis with Excel software (Microsoft Corporation, WA) with P < 0.05 considered significant.
ACKNOWLEDGEMENTS
We thank Peter Vitiello, Melissa Wu, and Jennifer Gewandter for critiquing this manuscript. This work was funded in part by National Institutes of Heath Grants HL-67392 (M. A. O’Reilly) and ES-01247. NIH training grant ES-07026 supported S. Gehen.
REFERENCES
- Abraham RT, Tibbetts RS. Cell biology. Guiding ATM to broken DNA. Science. 2005;308:510–511. doi: 10.1126/science.1112069. [DOI] [PubMed] [Google Scholar]
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. doi: 10.1038/nature01368. [DOI] [PubMed] [Google Scholar]
- Bao S, Tibbetts RS, Brumbaugh KM, Fang Y, Richardson DA, Ali A, et al. ATR/ATM-mediated phosphorylation of human Rad17 is required for genotoxic stress responses. Nature. 2001;411:969–974. doi: 10.1038/35082110. [DOI] [PubMed] [Google Scholar]
- Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell. 2003;3:421–429. doi: 10.1016/s1535-6108(03)00110-7. [DOI] [PubMed] [Google Scholar]
- Bendjennat M, Boulaire J, Jascur T, Brickner H, Barbier V, Sarasin A, et al. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell. 2003;114:599–610. doi: 10.1016/j.cell.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV, Wu GS, Omura S, el-Deiry WS. Proteasome-dependent regulation of p21WAF1/CIP1 expression. Biochem Biophys Res Commun. 1996;227:564–549. doi: 10.1006/bbrc.1996.1546. [DOI] [PubMed] [Google Scholar]
- Brooks CL, Gu W. Ubiquitination, phosphorylation and acetylation: the molecular basis for p53 regulation. Curr Opin Cell Biol. 2003;15:164–171. doi: 10.1016/s0955-0674(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Brumbaugh KM, Otterness DM, Geisen C, Oliveira V, Brognard J, Li X, et al. The mRNA surveillance protein hSMG-1 functions in genotoxic stress response pathways in mammalian cells. Mol Cell. 2004;14:585–598. doi: 10.1016/j.molcel.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677–1679. doi: 10.1126/science.281.5383.1677. [DOI] [PubMed] [Google Scholar]
- Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell. 2002;111:779–789. doi: 10.1016/s0092-8674(02)01113-3. [DOI] [PubMed] [Google Scholar]
- Chen MJ, Lin YT, Lieberman HB, Chen G, Lee EY. ATM-dependent phosphorylation of human Rad9 is required for ionizing radiation-induced checkpoint activation. J Biol Chem. 2001;276:16580–16586. doi: 10.1074/jbc.M008871200. [DOI] [PubMed] [Google Scholar]
- Cortez D, Guntuku S, Qin J, Elledge SJ. ATR and ATRIP: partners in checkpoint signaling. Science. 2001;294:1713–1716. doi: 10.1126/science.1065521. [DOI] [PubMed] [Google Scholar]
- Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science. 1999;286:1162–1166. doi: 10.1126/science.286.5442.1162. [DOI] [PubMed] [Google Scholar]
- Coulombe P, Rodier G, Bonneil E, Thibault P, Meloche S. N-Terminal ubiquitination of extracellular signal-regulated kinase 3 and p21 directs their degradation by the proteasome. Mol Cell Biol. 2004;24:6140–6150. doi: 10.1128/MCB.24.14.6140-6150.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das KC, Dashnamoorthy R. Hyperoxia activates the ATR-Chk1 pathway and phosphorylates p53 at multiple sites. Am J Physiol Lung Cell Mol Physiol. 2004;286:L87–L97. doi: 10.1152/ajplung.00203.2002. [DOI] [PubMed] [Google Scholar]
- Denning G, Jamieson L, Maquat LE, Thompson EA, Fields AP. Cloning of a novel phosphatidylinositol kinase-related kinase: characterization of the human SMG-1 RNA surveillance protein. J Biol Chem. 2001;276:22709–22714. doi: 10.1074/jbc.C100144200. [DOI] [PubMed] [Google Scholar]
- el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- Engel FB, Hauck L, Boehm M, Nabel EG, Dietz R, von Harsdorf R. p21(CIP1) Controls proliferating cell nuclear antigen level in adult cardiomyocytes. Mol Cell Biol. 2003;23:555–565. doi: 10.1128/MCB.23.2.555-565.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehen SC, Vitiello PF, Bambara RA, Keng PC, O'Reilly MA. Downregulation of PCNA potentiates p21-mediated growth inhibition in response to hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2007;292:L716–L724. doi: 10.1152/ajplung.00135.2006. [DOI] [PubMed] [Google Scholar]
- Guo Z, Kumagai A, Wang SX, Dunphy WG. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev. 2000;14:2745–2756. doi: 10.1101/gad.842500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helt CE, Cliby WA, Keng PC, Bambara RA, O'Reilly MA. Ataxia Telangiectasia Mutated (ATM) and ATM and Rad3-related Protein Exhibit Selective Target Specificities in Response to Different Forms of DNA Damage. J Biol Chem. 2005;280:1186–1192. doi: 10.1074/jbc.M410873200. [DOI] [PubMed] [Google Scholar]
- Helt CE, Staversky RJ, Lee YJ, Bambara RA, Keng PC, O'Reilly MA. The CDK and PCNA domains on p21 Cip1 both function to inhibit G1/S progression during hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2004;286:L506–L513. doi: 10.1152/ajplung.00243.2003. [DOI] [PubMed] [Google Scholar]
- Lee JH, Paull TT. ATM activation by DNA double-strand breaks through the Mre11-Rad50-Nbs1 complex. Science. 2005;308:551–554. doi: 10.1126/science.1108297. [DOI] [PubMed] [Google Scholar]
- Maki CG, Howley PM. Ubiquitination of p53 and p21 is differentially affected by ionizing and UV radiation. Mol Cell Biol. 1997;17:355–363. doi: 10.1128/mcb.17.1.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor PM. Mammalian G1 and G2 phase checkpoints. Cancer Surv. 1997;29:151–182. [PubMed] [Google Scholar]
- O'Reilly MA, Staversky RJ, Watkins RH, Reed CK, de Mesy Jensen KL, Finkelstein JN, et al. The cyclin-dependent kinase inhibitor p21 protects the lung from oxidative stress. Am J Respir Cell Mol Biol. 2001;24:703–710. doi: 10.1165/ajrcmb.24.6.4355. [DOI] [PubMed] [Google Scholar]
- Podust VN, Podust LM, Goubin F, Ducommun B, Hubscher U. Mechanism of inhibition of proliferating cell nuclear antigen-dependent DNA synthesis by the cyclin-dependent kinase inhibitor p21. Biochemistry. 1995;34:8869–8875. doi: 10.1021/bi00027a039. [DOI] [PubMed] [Google Scholar]
- Sheaff RJ, Singer JD, Swanger J, Smitherman M, Roberts JM, Clurman BE. Proteasomal turnover of p21Cip1 does not require p21Cip1 ubiquitination. Mol Cell. 2000;5:403–410. doi: 10.1016/s1097-2765(00)80435-9. [DOI] [PubMed] [Google Scholar]
- Smith GC, Cary RB, Lakin ND, Hann BC, Teo SH, Chen DJ, et al. Purification and DNA binding properties of the ataxia-telangiectasia gene product ATM. Proc Natl Acad Sci U S A. 1999;96:11134–11139. doi: 10.1073/pnas.96.20.11134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiff T, O'Driscoll M, Rief N, Iwabuchi K, Lobrich M, Jeggo PA. ATM and DNA-PK function redundantly to phosphorylate H2AX after exposure to ionizing radiation. Cancer Res. 2004;64:2390–2396. doi: 10.1158/0008-5472.can-03-3207. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Kodama S, Watanabe M. Recruitment of ATM protein to double strand DNA irradiated with ionizing radiation. J Biol Chem. 1999;274:25571–25575. doi: 10.1074/jbc.274.36.25571. [DOI] [PubMed] [Google Scholar]
- Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–157. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touitou R, Richardson J, Bose S, Nakanishi M, Rivett J, Allday MJ. A degradation signal located in the C-terminus of p21WAF1/CIP1 is a binding site for the C8 alpha-subunit of the 20S proteasome. Embo J. 2001;20:2367–2375. doi: 10.1093/emboj/20.10.2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello PF, Staversky RJ, Gehen SC, Johnston CJ, Finkelstein JN, Wright TW, et al. p21Cip1 protection against hyperoxia requires Bcl-XL and is uncoupled from its ability to suppress growth. Am J Pathol. 2006;168:1838–1847. doi: 10.2353/ajpath.2006.051162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A, Ohnishi T, Kashima I, Taya Y, Ohno S. Human SMG-1, a novel phosphatidylinositol 3-kinase-related protein kinase, associates with components of the mRNA surveillance complex and is involved in the regulation of nonsense-mediated mRNA decay. Genes Dev. 2001;15:2215–2228. doi: 10.1101/gad.913001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Wang HG, Miki Y, Kufe D. Protein kinase Cdelta is responsible for constitutive and DNA damage-induced phosphorylation of Rad9. Embo J. 2003;22:1431–1441. doi: 10.1093/emboj/cdg134. [DOI] [PMC free article] [PubMed] [Google Scholar]