

Abstract
For microbial pathogens, phylogeographic differentiation seems to be relatively common. However, the neutral population structure of Salmonella enterica serovar Typhi reflects the continued existence of ubiquitous haplotypes over millennia. In contrast, clinical use of fluoroquinolones has yielded at least 15 independent gyrA mutations within a decade and stimulated clonal expansion of haplotype H58 in Asia and Africa. Yet, antibiotic-sensitive strains and haplotypes other than H58 still persist despite selection for antibiotic resistance. Neutral evolution in Typhi appears to reflect the asymptomatic carrier state, and adaptive evolution depends on the rapid transmission of phenotypic changes through acute infections.
Many bacterial taxa can be subdivided into multiple, discrete clonal groupings (clonal complexes, or ecotypes) that have diverged and differentiated as a result of clonal replacement, selective sweeps, periodic selection, and/or population bottlenecks (1). Geographic isolation and clonal replacement can also result in phylogeographic differences between bacterial pathogens from different parts of the world (2), even within young, genetically monomorphic pathogens (3) (supporting online material text) such as Mycobacterium tuberculosis (4) and Yersinia pestis (5). Typhi is a genetically monomorphic (6), human-restricted bacterial pathogen that causes 21 million cases of typhoid fever and 200,000 deaths per year, predominantly in southern Asia, Africa, and South America (7). Typhi also enters a carrier state in rare individuals [such as Mortimer’s example of “Mr. N the milker” (8)], who can shed high levels of these bacteria for decades in the absence of clinical symptoms. Genome sequences are available from strains CT18 (9) and Ty2 (10), but the global diversity, population genetic structure, and evolutionary history of Typhi were poorly understood. It has been speculated that Typhi evolved in Indonesia, which is the exclusive source of isolates with the z66 flagellar antigen (11).
We investigated the evolutionary history and population genetic structure of Typhi by mutation discovery (12) within 200 gene fragments (~500 base pairs each) from a globally representative strain collection of 105 strains. The 200 genes included 121 housekeeping genes; 50 genes encoding cell surface structures, regulation, and pathogenicity; and 29 pseudogenes. Size variation of a poly-T6-7 homopolymeric stretch within one gene fragment was inconsistent with other phylogenetic patterns (homoplasies) and this fragment was excluded from further analysis. The other 199 gene fragments cover 88,739 base pairs, or 1.85% of the genome. Sixty-six were polymorphic as a result of 88 alternative allelic states [biallelic polymorphisms (BiPs)], for a frequency of approximately one BiP per kilobase. Five of the 88 BiPs probably represent three independent recombination events: Four seem to reflect two similar imports spanning 24 to 25 kb from S. enterica serovar Typhimurium (fig. S1), and a gene fragment with six single-nucleotide polymorphisms (SNPs) is identical to the corresponding gene fragment in S. enterica serovar Paratyphi A. The other 83 BiPs consisted of 37 nonsynonymous SNPs, 3 of which resulted in premature stop codons; 33 synonymous SNPs; 12 SNPs in pseudogenes; and one deletion of 4 base pairs.
We anticipated that housekeeping genes would exhibit diminished levels of nucleotide diversity, π, as a result of purifying selection, and that pathogenicity genes would exhibit elevated levels as a result of diversifying selection. However, π did not differ significantly with gene category (P > 0.05, analysis of variance) (table S1). Purifying selection should result in Ka/Ks (the ratio of nonsynonymous substitutions per nonsynonymous site to synonymous substitutions per synonymous site) values that are less than 1.0 and diversifying selection should result in ratios higher than 1.0. A trend in this direction was observed (table S1), but it was not particularly strong. We therefore concluded that these 88 BiPs largely reflect the lack of strong selection and are markers of neutral population structure in Typhi. It was somewhat surprising that a supposedly obligate pathogen such as Typhi should possess a neutral population structure, but the population structure of several other bacterial species that occasionally cause disease can also be explained by neutral genetic drift (13).
The distribution pattern of the 88 BiPs within Typhi is highly unusual because it is fully parsimonious according to maximum parsimony analysis (homoplasy index = 0). The 88 BiPs defined 59 haplotypes that form a unique path within a single minimal spanning tree of length 88, except for three hypothetical nodes (Fig. 1). These observations suggest that each BiP was caused by a unique genetic event, either a single mutation (83 BiPs) or the three imports described above (5 BiPs). The tree contains 19 informative BiPs that mark the evolutionary history of Typhi plus 69 noninformative BiPs that are specific to single haplotypes. A second, highly unusual feature of this data set is that the ancestral node, haplotype H45, is represented by extant bacteria. H45 must be the ancestral “root” node, because it possesses the identical nucleotides for all 82 SNPs, as did eight genomes of S. enterica of other serovars, whereas all other haplotypes result from one or more mutations. The general appearance of the tree (Fig. 1) suggests descent from H45 in multiple lineages, followed by diversification during multiple, independent population expansions that resulted in radial clusters of haplotypes containing the noninformative BiPs. For example, one cluster contains all seven Indonesian isolates with the z66 flagellar variant. The z66 cluster radiates from a single haplotype, indicating that it has arisen only once. Hence, z66 isolates cannot represent the evolutionary source of Typhi (11), because the z66 cluster is distant from H45.
Fig. 1.
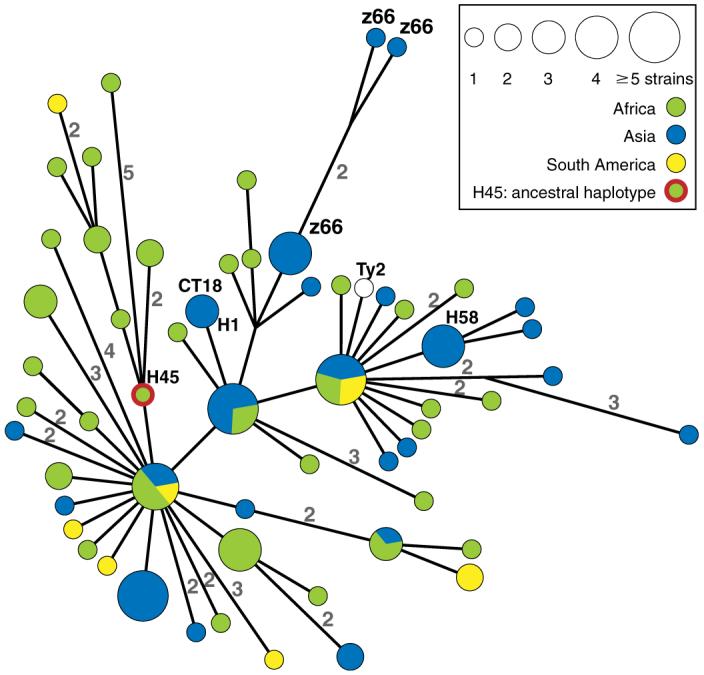
Minimal spanning tree of 105 global isolates based on sequence polymorphisms in 199 gene fragments (88,739 base pairs). The tree shows 59 haplotypes (nodes) based on 88 BiPs, the continental sources of which are indicated by colors within pie charts. The numbers along some edges indicate the number of BiPs that separate the nodes that they connect; unlabeled edges reflect single BiPs. The genomes of the CT18 and Ty2 strains have been sequenced (GenBank accession codes AL513382 and AE014613, respectively). z66 refers to a flagellar variant that is common in Indonesia (11).
The haplotype tree has a third, highly unusual feature: Most links between sequential haplotypes consist of single SNPs, and many longer edges, including one hypothetical node, were resolved into steps of single SNPs when additional strains were surveyed (fig. S2). Even within this initial sample of 105 isolates, almost half of the mutational steps during the evolutionary history of Typhi are represented by extant haplotypes, indicating long persistence of individual haplotypes. If ecotypes associated with periodic selection were to exist within Typhi, the genetic continuum between haplotypes implies that ecotypes are subdivisions of haplotypes. Furthermore, haplotypes and haplotype clusters were found in multiple continents. For example, it is unclear where H45 evolved, because it has been isolated from five locations in Asia, Africa, and North America (3). Because each BiP is associated with a single, genetic event, each haplotype or haplotype cluster that is present in multiple continents marks at least one independent wave of global transmission. Global transmission has not been previously described for Typhi but is a well-known phenomenon with other human pathogens.
To place the time scale associated with neutral evolution in context, we calculated the time since the most recent common ancestor (tmrca) and the effective population size (Ne) from the selectively neutral data in Fig. 1. These calculations were performed with the use of two estimates of the molecular clock rate, a high rate corresponding to the long-term rate of accumulation of synonymous mutations between Escherichia coli and S. enterica (5) and a clock rate one-fifth as high, corresponding to the rate of accumulation of all mutations in conserved housekeeping genes between these species (14). For Typhi, tmrca is 10 to 43 thousand years (95% confidence limits of 5.7 to 15.8 thousand years for the high clock rate and 25.5 to 71 thousand years for the low rate) according to both Bayesian skyline plots (15) and maximum likelihood trees (fig. S3). Based on the same clock rates, Ne is currently 2.3 × 105 to 1.0 × 106 (confidence limits of 1.2 × 104 to 9.3 × 105 for the high clock rate and 5.3 × 104 to 4.1 × 106 for the low clock rate) (fig. S3A). Similar values were obtained from the nucleotide variation, θw, by an independent method (16) (table S1). The maximum likelihood tree also suggests that H45, the ancestral haplotype, and multiple descendent haplotypes arose after human migrations out of Africa but before the Neolithic period (fig. S3B).
The existence of an asymptomatic human carrier state for typhoid is formally similar to tuberculosis, for which the reactivation of granulomas after decades results in delays of centuries between initial new infections and subsequent epidemic peaks (17). Likewise, we propose that the human carrier state allowed persistence of infection with Typhi during periods of isolation and was essential for transmission between hunter-gatherer groups. Hence, the population structure and geographical distribution of Typhi may largely reflect the frequency of carriers.
The 55 polymorphic coding gene fragments (excluding pseudogenes) were screened by mutation discovery with 59 additional strains that were isolated between 1958 and 1967 from Africa and Vietnam. All but three strains were assigned to known haplotypes from the global sample (fig. S4). Twelve haplotypes were isolated on multiple occasions over a range of 22 to 44 years from eight countries (Table 1), demonstrating that Typhi haplotypes persist in single countries for decades, or longer. For example, CT18 (9) is a multidrug-resistant (MDR) strain of haplotype H1 that was isolated in Vietnam in 1993, soon after multidrug resistance emerged. However, a Vietnamese isolate from 1967 was also of haplotype H1, showing that H1 was present in Vietnam long before multidrug resistance emerged. The long-term persistence of Typhi may also reflect the carrier state and can help explain why Typhi remains endemic in regions of the world with poor drinking-water quality and limited sewage treatment (18).
Table 1.
Persistence of haplotypes over decades
| Haplotype | Persistence (years) | Years persisted | No. of isolates |
|---|---|---|---|
| Vietnam | |||
| H1 | 37 | 1967-2004 | 25 |
| H50 | 37 | 1959-1996 | 3 |
| Madagascar | |||
| H15 | 31 | 1965-1996 | 4 |
| H50 | 33 | 1967-2000 | 2 |
| Algeria | |||
| H36 | 34 | 1966-2000 | 3 |
| Ivory Coast | |||
| H39 | 34 | 1967-2001 | 4 |
| H81 | 35 | 1967-2002 | 2 |
| Senegal | |||
| H39 | 22 | 1966-1988 | 4 |
| H52 | 39 | 1962-2001 | 4 |
| Congo | |||
| H46 | 34 | 1966-2000 | 3 |
| Morocco | |||
| H52 | 34 | 1966-2000 | 2 |
| Cameroon | |||
| H77 | 44 | 1958-2002 | 2 |
Antibiotic-resistant typhoid fever has recently become an enormous public health problem in southern Asia because of the emergence of MDR Typhi followed by nalidixic acid resistance (NalR) with concomitant reduced susceptibility to fluoroquinolones (19). Fluoroquinolones were first used for antibiotic therapy in southeast Asia in 1989 (20) and NalR Typhi were reported in 1991 (21). Such strong selection should have led to a population expansion of NalR Typhi, and possibly to clonal replacement of existing haplotypes within southern Asia. We therefore performed mutation discovery with the 55 polymorphic coding fragments on 295 additional strains of Typhi that were isolated from southern Asia between 1986 and 2004. Again, most strains were assigned to known haplotypes and only a few defined new, peripheral haplotypes (Fig. 2B). However, the relative frequencies of isolates differed from those in the global set of 105 strains (Fig. 1), because most recent isolates from southern Asia, particularly NalR isolates, belonged to haplotype H58 (Fig. 2B). Thus, a recent population expansion of H58 seems to have resulted from the general use of fluoroquinolones.
Fig. 2.

Selection for mutations in gyrA versus a neutral population framework in 483 strains. The strains consisted of 105 global isolates (Fig. 1), 59 older isolates from Africa and Vietnam (1958 to 1967) (fig. S4 and Table 1), and 317 isolates from southeast Asia (1984 to 2004) and other sources. (A) Sequence of codons 81 to 89 of gyrA, showing all mutated nucleotides (bold) that were detected within a 489-base pair stretch. Each mutation is designated by the name of the resulting amino acid and codon position (left). NAS, nalidixic acid sensitive. (B) Minimal spanning tree of 85 haplotypes based on 97 BiPs within 55 polymorphic genes. Sizes of circles and arcs reflect numbers of isolates. Strains without mutations in gyrA are shown in white, whereas strains with mutations are indicated by colored arcs that correspond to the colors in (A). The 15 letters indicate independent mutations associated with resistance to nalidixic acid. (C) Time course of isolation of 118 isolates of haplotype H58 or its derivative haplotypes H34, H57, and H60 to H65. These isolates were selected for haplotyping and gyrA genotyping without prior knowledge of their susceptibility to nalidixic acid. Fifty-two other H58 isolates from Vietnam are not included because they were a nonrandom sample of NalR bacteria. The apparent increase of Phe83 in 2004 is based on a sample from the Mekong Delta province of Vietnam and may represent an outlier.
We also investigated the genetic diversity of a 489-base pair fragment of the gyrA gene encoding a DNA gyrase subunit, within which nonsynonymous mutations at codons 83 and 87 result in resistance to nalidixic acid (22). All 125 strains that were sensitive to nalidixic acid (Nal5) and all other strains with unknown resistance to nalidixic acid possessed the ancestral gyrA+ sequence. In contrast, all 119 NalR strains, most of which were isolated in south central or southeast Asia (table S2), possessed one of six nonsynonymous mutations at codons 83 and 87 of gyrA (Fig. 2A), and no other mutations were detected in gyrA (or parC). We identified 15 independent mutational events (A through O in Fig. 2B) in distinct haplotypes that also possessed gyrA+ alleles. Assuming that they all arose between 1991 and 2004 (13 years), the identification of ≥15 mutations in two codons (6 base pairs) yields a minimum frequency of 0.19 per base pair per year, ≥2.5 × 108 greater than the longterm mutation clock rate within E. coli (14).
For most haplotypes with gyrA mutations, NalR strains were detected only once or twice; however, NalR variants of haplotype H58 and its derivative haplotypes (H34, H57, and H60 to H65) were isolated in Vietnam, India, and Pakistan, and other countries in southern Asia (Table 2). These NalR variants represent at least five distinct gyrA mutations (K, L, M, N, and O), which arose during or before the mid-1990s (Fig. 2C). The frequency of gyrA+ and mutation L has remained fairly constant since the mid-1990s, but H58 isolates with mutation K seem to have become more common in recent years, particularly in Vietnam (Table 2).
Table 2.
Geographic sources of haplotypes with gyr mutations by haplotype. Where more than one isolate was found in a country, the number of isolates is indicated in parentheses
| Haplotype | gyrA mutation (isolates) | gyrA+ (isolates) |
|---|---|---|
| H45 | A: India | Global (4) |
| H50 | B: India | Global (55) |
| C: China | ||
| D: Mexico | ||
| H42 | E: India; F: Pakistan | Global (24) |
| H1 | G: Vietnam | Vietnam (23); Laos (7); Bangladesh; |
| H: Vietnam | Indonesia | |
| H85 | I: India | Morocco, Pakistan, Indonesia |
| H52 | J: India (2) | Global (53) |
| H58 | K: Vietnam (68), Pakistan (5), India (4), Cambodia, Nepal, central Africa; | Vietnam (31), India (12), Laos (5), Pakistan (3), Hong Kong (2), Bangladesh, Sri Lanka, Morocco |
| L: India (6), Bangladesh (6), Vietnam (5), Indonesia | ||
| M: Pakistan (2) | ||
| N: Vietnam | ||
| O: Pakistan | ||
| H34, H57, and H60-65 | K: Vietnam (8), Bangladesh (2) | Nepal, Laos |
| L: India (2) |
These results show that selection for antibiotic resistance has probably led to clonal expansion of H58 and its NalR derivatives in southern Asia. These strains have now also reached Africa, given that one MDR H58 strain (isolated in Morocco in 2003) was included among nine rare, recent MDR isolates from Africa and that the sole MDR NalR isolate from Africa that was tested (mutation K, isolated in central Africa in 2004) also belonged to H58 (Table 2). Thus, H58 is probably not ethnically restricted to southern Asians, and nalidixic acid-resistant typhoid fever may soon present an additional public health problem in Africa.
Despite the selection for resistance to nalidixic acid in southern Asia, the data do not show complete clonal replacement, which would be expected from periodic selection; about 20% of Typhi isolated in recent years in northern Vietnam and 5% of Typhi from southern Vietnam remain susceptible to nalidixic acid, as are many other recent H58 isolates (Fig. 2C). Furthermore, recent isolates from southern Asia also belong to other haplotypes, where mutations in gyrA are rare (Fig. 2B). Thus, the population structure indicative of neutral evolution has not been disrupted by strong selection for resistance to nalidixic acid during the past 15 years, except for the clonal expansion of H58. Possibly gyrA mutations in many haplotypes reduce fitness (23) or some cases of typhoid fever have not been treated with fluoroquinolones. But still another alternative is that the population structure of Typhi reflects two distinct epidemiological dynamics associated with different time scales: first, the human carrier state permitting slow neutral evolution (millennia), and second, infectious transmission facilitating a rapid response to selection in real time. Outbreaks of infections, similar to the recent expansion of H58 in southeast Asia, may be responsible for independent chains of intercontinental transmission. These, in turn, create a global distribution of carriers for multiple haplotypes. According to this interpretation, it is exactly because the environment selects that everything is everywhere in Typhi, thus inverting a hotly disputed (24) tenet of microbial ecology that was proposed by L. G. M. Baas Becking in 1934 (25).
The results presented here open multiple avenues for future research. Long-term epidemiology with larger strain collections is now possible on the basis of neutral SNPs (fig. S2), whereas classical microbiological methods do not seem to provide reliable markers for such purposes (table S3). Surveillance of haplotypes is particularly appropriate to provide early warning of the continued spread of NalR H58. Our overview of the current global population diversity in Typhi will allow comparisons of genomic sequences from representative strains without the risk of phylogenetic discovery bias (26). Finally, we suggest that the human carrier state may be of much greater importance for neutral evolution and genetic buffering than had been previously appreciated, an interpretation that would demand major changes in public health campaigns to reduce the incidence of typhoid.
Supplementary Material
Acknowledgments
We thank M. Feldkamp and L. Fabre-Berland for technical assistance; K. E. Sanderson, M. Torpdahl, J. M. Collard, and S. Porwollik for DNA; and G. Morelli, B. Linz, Y. Moodley, F. Balloux, and D. Falush for helpful discussions. Support was provided by The Wellcome Trust, UK. Work at IVI was supported by the Diseases of the Most Impoverished Program, funded by the Bill and Melinda Gates Foundation. IVI thanks M. Ali, A.-L. Page, C. M. Galindo, J. D. Clemens, R. L. Ochiai (IVI); B. Dong (Guangxi Center for Prevention and Disease Control, CDC, China); Z. A. Bhutta (Aga Khan University, Pakistan); S. A. Bhattacharya (National Institute of Cholera and Enteric Disease, India); M. Agniti (National Institute of Health Research and Development, Indonesia); V. D. Thiem (National Institute of Hygiene and Epidemiology, Vietnam); A. Naheed (International Centre for Diarrhoeal Disease Research, Bangladesh). GenBank accession numbers: EF070864 to EF070980.
Footnotes
Supporting Online Material, www.sciencemag.org/cgi/content/full/314/5803/1301/DC1, Materials and Methods, SOM Text, Figs. S1 to S4, Tables S1 to S9, References
References and Notes
- 1.Gevers D, et al. Nat. Rev. Microbiol. 2005;3:733. doi: 10.1038/nrmicro1236. [DOI] [PubMed] [Google Scholar]
- 2.Zhu P, et al. Proc. Natl. Acad. Sci. U.S.A. 2001;98:5234. [Google Scholar]
- 3.Materials and methods are available as supporting material on Science Online.
- 4.Gutacker MM, et al. J. Infect. Dis. 2006;193:121. doi: 10.1086/498574. [DOI] [PubMed] [Google Scholar]
- 5.Achtman M, et al. Proc. Natl. Acad. Sci. U.S.A. 2004;101:17837. doi: 10.1073/pnas.0408026101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kidgell C, et al. Infect. Genet. Evol. 2002;2:39. doi: 10.1016/s1567-1348(02)00089-8. [DOI] [PubMed] [Google Scholar]
- 7.Crump JA, Luby SP, Mintz ED. Bull. World Health Organ. 2004;82:346. [PMC free article] [PubMed] [Google Scholar]
- 8.Mortimer PP. Lancet. 1999;353:1354. doi: 10.1016/S0140-6736(98)11315-6. [DOI] [PubMed] [Google Scholar]
- 9.Parkhill J, et al. Nature. 2001;413:848. doi: 10.1038/35101607. [DOI] [PubMed] [Google Scholar]
- 10.Deng W, et al. J. Bacteriol. 2003;185:2330. doi: 10.1128/JB.185.7.2330-2337.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frankel G, Newton SM, Schoolnik GK, Stocker BA. EMBO J. 1989;8:3149. doi: 10.1002/j.1460-2075.1989.tb08468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Underhill PA, et al. Genome Res. 1997;7:996. doi: 10.1101/gr.7.10.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fraser C, Hanage WP, Spratt BG. Proc. Natl. Acad. Sci. U.S.A. 2005;102:1968. doi: 10.1073/pnas.0406993102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wirth T, et al. Mol. Microbiol. 2006;60:1136. doi: 10.1111/j.1365-2958.2006.05172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drummond AJ, Rambaut A, Shapiro B, Pybus OG. Mol. Biol. Evol. 2005;22:1185. doi: 10.1093/molbev/msi103. [DOI] [PubMed] [Google Scholar]
- 16.Funk DJ, Wernegreen JJ, Moran NA. Genetics. 2001;157:477. doi: 10.1093/genetics/157.2.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blower SM, et al. Nat. Med. 1995;1:815. doi: 10.1038/nm0895-815. [DOI] [PubMed] [Google Scholar]
- 18.Young D, Hussell T, Dougan G. Nat. Immunol. 2002;3:1026. doi: 10.1038/ni1102-1026. [DOI] [PubMed] [Google Scholar]
- 19.Bhan MK, Bahl R, Bhatnagar S. Lancet. 2005;366:749. doi: 10.1016/S0140-6736(05)67181-4. [DOI] [PubMed] [Google Scholar]
- 20.Rodrigues C, Mehta A, Joshi VR. Clin. Infect. Dis. 2002;34:126. doi: 10.1086/323019. [DOI] [PubMed] [Google Scholar]
- 21.Threlfall EJ, Ward LR. Emerg. Infect. Dis. 2001;7:448. doi: 10.3201/eid0703.010315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hopkins KL, Davies RH, Threlfall EJ. Int. J. Antimicrob. Agents. 2005;25:358. doi: 10.1016/j.ijantimicag.2005.02.006. [DOI] [PubMed] [Google Scholar]
- 23.Gagneux S, et al. PLoS Pathog. 2006;2:e61. doi: 10.1371/journal.ppat.0020061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martiny JB, et al. Nat. Rev. Microbiol. 2006;4:102. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- 25.Baas Becking LGM. Geobiologie of Inleiding Tot de Milieukunde. W. P. Van Stockum & Zoon; The Hague, Netherlands: 1934. [Google Scholar]
- 26.Pearson T, et al. Proc. Natl. Acad. Sci. U.S.A. 2004;101:13536. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.