

Abstract
The effect of N-arachidonoyl l-serine (ARA-S), a recently discovered lipoamino acid found in the CNS, on N-type Ca2+ channels of rat sympathetic ganglion neurons was determined using whole cell patch clamp. Application of ARA-S produced a rapid and reversible augmentation of Ca2+ current that was voltage dependent and resulted from a hyperpolarizing shift in the activation curve. ARA-S did not influence G protein modulation of Ca2+ channels and appeared to act independently of G-protein-coupled receptors. These findings provide a foundation for investigating possible roles for ARA-S in nervous system function.
INTRODUCTION
Endocannabinoids comprise a group of endogenously produced lipid-derived molecules that bind to and affect CB1 and CB2 G-protein-coupled cannabinoid receptors. The best-characterized endocannabinoids, 2-arachidonoyl glycerol (2-AG) and N-arachidonoyl ethanolamine (anandamide), act as agonists at both CB1 and CB2 receptors and are implicated in a variety of physiological responses (Pacher et al. 2006). In addition to receptor-mediated effects, endocannabinoids interact directly with voltage- and ligand-gated ion channels producing alterations in gating usually manifested as channel inhibition (Oz 2006). Recently, a novel endocannabinoid-like compound, N-arachidonoyl l-serine (ARA-S), was identified in extracts of bovine brain and shown to produce endothelium-dependent vasodilation of rat mesenteric arteries (Millman et al. 2006). Synthetic ARA-S has minimal activity at traditional endocannabinoid targets (CB1R, CB2R, and TRPV1 channels) but appears to act via a novel, but as yet unidentified, G-protein-coupled non-CB1/CB2 cannabinoid receptor. ARA-S is also reported to be an antagonist at a non-CB1/CB2 cannabinoid receptor found in neutrophils (McHugh et al. 2007). While screening GPR35 (Guo et al. 2008), an orphan G-protein-coupled receptor, as a potential target of ARA-S, we found that ARA-S produced receptor-independent effects on the activation of N-type Ca2+ channels (CaV2.2) in rat sympathetic neurons leading to an enhancement of current especially at hyperpolarized potentials.
METHODS
Cell isolation and electrophysiology
Superior cervical ganglion (SCG) neurons from male Wistar rats (150–300 g) were enzymatically dissociated and placed in short-term (<24 h) culture as described previously (Ikeda 2004). For some experiments, the neurons were incubated overnight in tissue culture media containing 500 ng/ml Bordetella pertussis toxin (PTX; List Biological Laboratories, Campbell, CA) as previously described (Guo and Ikeda 2004). Rats were killed by decapitation after anesthesia with CO2 as approved by the Institutional Animal Care and Use Committee. Neurons were voltage-clamped using the whole cell patch-clamp technique as described previously (Guo and Ikeda 2004). ICa tail currents were filtered at 10 kHz prior to digitization at 50 kHz. Series resistance was electronically compensated ≥80%. Experiments were carried out at room temperature (22–26°C).
Solutions and chemicals
The external recording solution contained (in mM) 140 methanesulphonic acid, 145 tetraethylammonium hydroxide, 10 HEPES, 10 glucose, and 10 CaCl2 and 0.0003 tetrodotoxin (Alomone Labs, Jerusalem, Israel), pH 7.4 with TEA-OH. For tail current experiments, the CaCl2 was reduced to 5 mM. The pipette solution contained (in mM) 120 N-methyl-d-glucamine, 20 tetraethylammonium hydroxide, 11 EGTA, 10 HEPES, 10 sucrose, 10 HCl, 1 CaCl2, 4 MgATP, 0.3 Na2GTP, and 14 Tris creatine phosphate, pH 7.2 with methanesulphonic acid. The osmolalities of the bath and pipette solutions were adjusted with sucrose to 325 and 300 mOsmol/kg, respectively. N-arachidonoyl-l-alanine (ARA-A), N-arachidonoyl-l-glycine (ARA-G), N-arachidonoyl-l-Serine, and N-arachidonoyl-dopamine (ARA-DA) were purchased from Cayman Chemical (Ann Arbor, MI) as ethanol stock solutions (25–140 mM) and dissolved directly into the recording solution on the day of the experiment. Ethanol was <=0.1% in all solutions and at this concentration produced no discernable effect on ICa. Drugs were applied by positioning the outlet tube (200 μm ID) of a custom-designed gravity-fed microperfusion system ∼100 μm from the cell body.
Data analysis and statistics
Nonlinear least-squares curve fitting was performed using a Marquardt-Levenberg algorithm from Igor Pro version 6.02A (WaveMetrics, Lake Oswego, OR). Statistical comparisons, as indicated in the text, were determined with Prism 4 version 4.0c (GraphPad Software, San Diego, CA). P < 0.05 was considered significant. Summary data are presented as means ± SE.
RESULTS
We tested the effect of ARA-A, ARA-G (Huang et al. 2001), ARA-S, and ARA-DA (Huang et al. 2002) application on Ca2+ currents (ICa) recorded from dissociated adult rat sympathetic neurons using whole cell patch clamp. Under these recording conditions, ICa arises primarily from ω-conotoxin GVIA-sensitive N-type Ca2+ channels (Ikeda 1991). Currents were evoked with a 25-ms test pulse to −10 mV from a holding potential of −80 mV. Application of ARA-A (10 μM) or ARA-S (10 μM) produced a rapid and reversible enhancement of ICa amplitude (Fig. 1 A) without overt modification of ICa kinetics (Fig. 1B). Conversely, application of ARA-DA (10 μM) to the same neuron produced minimal effects. A second series of sequential drug applications produced similar effects indicating that the minuscule ARA-DA response did not result from tachyphylaxis. ARA-G (10 μM) was tested in a separate set of experiments and produced results similar to those observed following ARA-S application (Fig. 1C). Comparison of the average change in ICa amplitude (Fig. 1C) revealed that ARA-S produced the largest increase (132 ± 14%, n = 14) and thus further experiments focused on this compound.
FIG. 1.

Voltage-dependent enhancement of Ca2+ currents (ICa) by N-arachidonoyl l-serine and related compounds. Data are from whole cell patch-clamp recordings of rat sympathetic neurons obtained at room temperature (22–26°C). A: time course of ICa amplitude during exposure to 10 μM of N-arachidonoyl l-alanine (ARA-A, □), N-arachidonoyl l-serine (ARA-S, ▪), or N-arachidonoyl dopamine (ARA-DA,  ). B: ICa traces for the time course shown in A were evoked with 25-ms test pulses to +10 mV from a holding potential of −80 mV. Superimposed traces in the absence or presence of drug as indicated. - - -, the 0 current level. C: mean ± SE increase in ICa amplitude produced by application of ARA-A, ARA-S, N-arachidonoyl l-glycine (ARA-G), and ARA-DA. With the exception of ARA-S vs. ARA-G, all means differ significantly (P < 0.001) from each other as determined from 1-way ANOVA followed by Newman-Keuls post hoc test. D: effect of ARA-S (10 μM) on ICa at different test potentials. ICa was evoked from a holding potential of −80 mV with 70-ms pulses to the indicated potential in the absence or presence (•) of ARA-S. E: current-voltage (I-V) curves were obtained in the absence (○) or presence of ARA-S (10 μM, •) and then normalized to the ICa amplitude at +10 mV in the absence of ARA-S. The data represent the means ± SE for 10 neurons. F: concentration-response curve for ARA-S. Three concentrations of ARA-S were applied to each neuron (0.1, 1.0, and 10 or 0.3, 3.0, and 30 μM ARA-S). ICa was allowed to recover between applications.
). B: ICa traces for the time course shown in A were evoked with 25-ms test pulses to +10 mV from a holding potential of −80 mV. Superimposed traces in the absence or presence of drug as indicated. - - -, the 0 current level. C: mean ± SE increase in ICa amplitude produced by application of ARA-A, ARA-S, N-arachidonoyl l-glycine (ARA-G), and ARA-DA. With the exception of ARA-S vs. ARA-G, all means differ significantly (P < 0.001) from each other as determined from 1-way ANOVA followed by Newman-Keuls post hoc test. D: effect of ARA-S (10 μM) on ICa at different test potentials. ICa was evoked from a holding potential of −80 mV with 70-ms pulses to the indicated potential in the absence or presence (•) of ARA-S. E: current-voltage (I-V) curves were obtained in the absence (○) or presence of ARA-S (10 μM, •) and then normalized to the ICa amplitude at +10 mV in the absence of ARA-S. The data represent the means ± SE for 10 neurons. F: concentration-response curve for ARA-S. Three concentrations of ARA-S were applied to each neuron (0.1, 1.0, and 10 or 0.3, 3.0, and 30 μM ARA-S). ICa was allowed to recover between applications.
The effect of ARA-S at different potentials was examined by evoking ICa with 70-ms test pulses over the range −60 to +80 mV from a holding potential of −80 mV in the presence or absence of 10 μM ARA-S (Fig. 1D). Under control conditions, ICa became apparent around −30 mV, reached a maximum amplitude near +10 mV, and thereafter declined asymptotically toward baseline. In the presence of ARA-S, ICa was augmented several-fold at negative test potentials with lesser increases at more depolarized potentials. At very depolarized test potentials (> +40 mV), ICa amplitude was minimally affected, suggesting that a shift in Ca2+ channel activation (Fig. 1E) rather than an increase in the number of available channels was responsible for the action of ARA-S. A concentration-response curve for ARA-S is illustrated in Fig. 1F. Three concentrations of ARA-S (0.1, 1.0, and 10 or 0.3, 3.0, and 30 μM) were applied to neurons allowing time to recover between each concentration. Potentiation of ICa amplitude was evident starting at 3 μM and increased monotonically without evidence of saturation ≤30 μM, the highest concentration examined.
To investigate shifts in channel activation, Ca2+ channel tail current amplitudes were determined in the absence or presence of ARA-S (10 μM). Tail currents result from the deactivation of channels on return to a hyperpolarized potential following a sojourn at a step potential that produces channel activation (Fig. 2 A). Because tail currents are measured at a constant potential (here −40 mV), driving force, which is nonlinear for large ionic gradients such as Ca2+, remains constant and thus ICa amplitude can be equated with conductance. To facilitate accurate tail current measurement, analog filter bandwidth (−3 dB) and digital sampling rate were increased to 10 and 50 kHz, respectively. External [Ca2+] was decreased to 5 mM, which served to decrease tail current amplitude and hence the effects of residual uncompensated series resistance. Under these conditions, tail current decays were well fit (following a 100-μs delay to allow uncompensated capacitive transients to settle) by a single-exponential function (Fig. 2B, —) with τ of ∼0.5 ms. Activation curves were plotted as tail current amplitude, normalized to maximum amplitude (e.g., +80-mV step potential) in the absence of drug, versus step potential (Fig. 2C) and fit (—) with a two-component modified Boltzmann equation
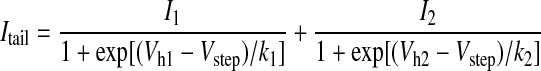 |
where Itail is the normalized tail current amplitude and Vstep is the step potential. In, Vhn, and kn are the fractional amplitude, half activation potential, and slope factor, receptively for each component. Under control conditions, Ca2+ channel activation curves from adult rat SCG neurons are composed of two components—a result of tonic G protein modulation (Ikeda 1991). In the presence of ARA-S, the activation was shifted toward more hyperpolarized potentials, retained a two-component profile, and achieved a similar maximum amplitude. Analysis of the activation parameters (Fig. 2E) revealed significant decreases in Vh1, Vh2, and k2 but no change in the fractional contribution of each component. The hyperpolarizing shift in the activation curve account for the voltage-dependent ICa increases as shown in Fig. 2D. Both the tail and step ICa were affected to a much greater extent by ARA-S at hyperpolarized voltages. The coincidence of the step and tail ICa data in Fig. 2D provide evidence for the fidelity of the tail current recordings.
FIG. 2.

ARA-S shifts ICa activation curves toward hyperpolarized potentials. A: tail current voltage protocol (bottom) and representative traces, recorded at a step potential of −10 mV, obtained in the absence or presence of ARA-S (10 μM). External [Ca2+] was decreased to 5 mM to reduce current amplitude thereby reducing series resistance error. B: tail current decay illustrated on an expanded time scale (same traces as in A). The initial 100 μs following the step pulse have been blanked. —, nonlinear least-square fits to single-exponential function. C: activation curves composed of mean ± SE (n = 5) normalized tail current amplitudes vs. step potential. Tail current amplitudes were determined from the exponential fit of the tail current decay phase. Amplitudes obtained in the absence (○) or presence (•) of ARA-S were normalized to the maximum amplitude in the absence of drug. —, the best fit of a 2-component Boltzmann equation (see text) to the data using nonlinear least-squares regression. D: voltage dependence of ARA-S effects illustrated by plotting the ratio of ICa amplitude in the presence of ARA-S to the control condition for step (□) and tail currents (▪) vs. step potential. E: table of mean ± SE Boltzmann equation parameters. A paired t-test was used for statistical comparison between the control and drug condition. ***, P < 0.001; *, P < 0.05.
Next, we asked whether ARA-S affects ICa modulation by G-protein-coupled receptors. In SCG neurons, norepinephrine (NE) acts by binding to α2-adrenergic receptors (Schofield 1990) thereby liberating Gβγ from the heterotrimeric G protein complex resulting in voltage-dependent modulation (Bean 1989; Ikeda 1996). This form of ICa modulation is detected using a double-pulse voltage protocol (Elmslie et al. 1990) (Fig. 3 B) in which two identical test pulses to +10 mV are separated by a depolarizing conditioning pulse to +80 mV. Facilitation, defined as the ratio of postpulse to prepulse ICa amplitude, provides a convenient index of Gβγ-mediated modulation (Fig. 3A). Increases in facilitation result from relief of Gβγ inhibition produced by the conditioning pulse. Application of NE produced ICa inhibition with a coincidental increase in facilitation that was similar in the presence of ARA-S. The mean inhibition of ICa, basal (i.e., in the absence of agonist) facilitation, and NE-induced facilitation were significantly different in the presence of ARA-S (Fig. 3C). However, the magnitude of the changes were small and possibly arose from effects of ARA-S on minor components of ICa that do not arise from N-type Ca2+ channels. Thus ARA-S does not appear to greatly influence either receptor-mediated or tonic modulation of N-type Ca2+ channels by Gβγ. The effects of ARA-S (10 μM) on neurons treated overnight with 500 ng/ml PTX were also examined. ICa potentiation following ARA-S application was not significantly altered by pretreatment with PTX (210 ± 4, n = 3 vs. 290 ± 31%, n = 9 for control and PTX-treated, respectively). Conversely, NE-mediated ICa inhibition was decreased (13 ± 6, n = 3 vs. 52 ± 3%, n = 9, for PTX and control, respectively) following PTX treatment providing evidence for toxin effectiveness. Thus activation of PTX-sensitive G proteins, namely Gi/o-containing heterotrimers, were not essential for ICa increases resulting from ARA-S application.
FIG. 3.

G-protein-mediated ICa modulation in the presence of ARA-S. A: time course of ICa amplitude during superfusion with norepinephrine (NE, 10 μM; □) and ARA-S (10 μM; ▪). ICa was evoked at 0.1 Hz with the voltage protocol shown in B. ○ and • (top), prepulse and postpulse ICa amplitudes, respectively. Facilitation (bottom, ▪) is the ratio of postpulse to prepulse current amplitudes. B: representative ICa traces from the time course shown in A. Numbers next to the traces refer to corresponding time points labeled in A. C, left: mean ± SE facilitation determined in the absence (□) or presence (▪) of ARA-S. Basal facilitation was determined prior to NE administration. Right: mean ± SE ICa inhibition produced by application of NE. Inhibition was determined from the prepulse current. Number of neurons used for all analyses indicated in parentheses. Means were compared with a paired Student's t-test. *, P < 0.05; **, P < 0.01.
DISCUSSION
Our data show that ARA-S produces a rapid and reversible augmentation of ICa in sympathetic neurons that is voltage dependent and results from a hyperpolarizing shift in the activation curve. At depolarized potentials, the same maximal conductance is attained in the presence of ARA-S arguing against a recruitment of covert channels or a change in the maximum probability of opening. ARA-S did not interfere with or contribute to G protein modulation of N-type Ca2+ channels. We thus conclude that ICa enhancement by ARA-S occurs independently of G-protein-coupled receptors and possibly results from direct interaction with the CaV2.2 channel or alteration of plasma membrane surface potential.
There are two main implications of these findings. First, the ARA-S concentration (10 μM) used here falls well within the concentration range used to probe potential physiological roles of ARA-S and related endocannibinoids (Milman et al. 2006). Given the well-established role played by N-type Ca2+ channels in providing Ca2+ for synaptic transmission combined with the nonlinear relationship between [Ca2+] and transmitter release (Xu et al. 2007), one can easily envision changes in synaptic transmission produced by applying ARA-S. Thus receptor-independent effects, such as the one demonstrated here, will need to be considered before meaningful interpretations of ARA-S action are entertained. Although agents such as Bordetella pertussis toxin are useful for separating receptor-dependent (at least in terms of GPCRs using Gi/oα-containing heterotrimers) from receptor-independent effects, this ability degrades as the complexity of the system increases (e.g., in vivo experiments). Second, it is possible that direct actions of ARA-S on ion channels underlie physiological processes. For example, a related compound, anandamide, is proposed to influence physiological function by binding to CB1R/CB2R and subsequently activating downstream signaling cascades or directly activating TRPV channels without the aid of signaling intermediates (Smart et al. 2000; van der Stelt and Di Marzo 2005; van der Stelt et al. 2005). Although endocannabinoids and related lipid compounds (Bradshaw and Walker 2005) often have direct effects on ion channel function (Oz 2006), the effect of ARA-S on N-type Ca2+ channels is somewhat unique. Closely related compounds such as anandamide, 2-AG (Guo and Ikeda 2004) and ARA-DA (Fig. 1C) either have no effect or produced inhibition at similar concentrations, whereas both ARA-A and ARA-G were capable of augmenting ICa to varying degrees (Fig. 1C). From this series of compounds, the presence of a carboxylic acid group was common to the substances that enhanced ICa amplitude. We could not find literature values for the pKa of ARA-S or related lipoamino acids thus the charge status of the carboxylic acid group at physiological pH is unclear. Given the pKa of the carboxylic acid group in the free amino acids (2.1–2.4), it seems likely that ARA-S, ARA-G, and ARA-A are negatively charged at pH 7.4. Thus a possible explanation for the effects of ARA-S and related lipoamino acids is alteration of the membrane surface potential (Hille 2001) following incorporation of negative charges into the outer leaflet of the plasma membrane. The net result would be a negative shift in channel activation as seen following ARA-S application. It should be noted, however, that enhancement of L-type ICa in ventricular myocytes by long chain fatty acids occurred without shifting activation or inactivation along the voltage axis (Huang et al. 1992) thus arguing against this explanation as a universal mechanism for Ca2+ channel modulation by negatively charged lipophilic agents.
The actions of ARA-S are somewhat reminiscent of the effects of arachidonic acid on N-type Ca2+ channels in SCG neurons (Liu and Rittenhouse 2003; Liu et al. 2001). Although ARA-S, a conjugate of arachidonic acid and serine, likely breaks down to arachidonic acid, ARA-S lacks a Ca2+ channel inhibitory component characteristic of arachidonic acid effects. Therefore, it is unlikely that arachidonic acid mediates the effects of ARA-S although we cannot conclusively rule out this possibility. The stimulatory effects of ARA-S on N-type Ca2+ channels are strikingly similar to those observed for the fatty acid analogs palmitoyl coenzyme A and arachidonoyl coenzyme A (Barrett et al. 2001). Thus the lipid moiety in these compounds may share a common mechanism for Ca2+ channel stimulation that is conferred by the charge on the head group. At present, many details relevant to the biology of ARA-S, including synthetic and degradative pathways, partition coefficient, and sites of action have not been investigated. This void in our knowledge limits speculation as even fundamental facts, such as the relevant physiological concentration of ARA-S, remain unknown.
GRANTS
This work was supported by the intramural program of the National Institutes of Health, National Institute on Alcohol Abuse and Alcoholism, Bethesda, MD.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
REFERENCES
- Barrett et al. 2001.Barrett CF, Liu L, Rittenhouse AR. Arachidonic acid reversibly enhances N-type calcium current at an extracellular site. Am J Physiol Cell Physiol 280: C1306–1318, 2001. [DOI] [PubMed] [Google Scholar]
- Bean 1989.Bean BP Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage dependence. Nature 340: 153–156, 1989. [DOI] [PubMed] [Google Scholar]
- Bradshaw and Walker 2005.Bradshaw HB, Walker JM. The expanding field of cannabimimetic and related lipid mediators. Br J Pharmacol 144: 459–465, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmslie et al. 1990.Elmslie KS, Zhou W, Jones SW. LHRH and GTP-γ-S modify calcium current activation in bullfrog sympathetic neurons. Neuron 5: 75–80, 1990. [DOI] [PubMed] [Google Scholar]
- Guo and Ikeda 2004.Guo J, Ikeda SR. Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potassium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol Pharmacol 65: 665–674, 2004. [DOI] [PubMed] [Google Scholar]
- Guo et al. 2008.Guo J, Williams DJ, Puhl HL, Ikeda SR. Inhibition of N-type calcium channels by activation of GPR35, an orphan receptor, heterologously expressed in rat sympathetic neurons. J Pharmacol Exp Ther 324: 342–351, 2008. [DOI] [PubMed] [Google Scholar]
- Hille 2001.Hille B Modification of gating in voltage-sensitive channels. In: Ion Channels of Excitable Membranes (3rd ed.). Sunderland, MA: Sinauer Associates, 2001, p. 646–656.
- Huang et al. 1992.Huang JM, Xian H, Bacaner M. Long-chain fatty acids activate calcium channels in ventricular myocytes. Proc Natl Acad Sci USA 89: 6452–6456, 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang et al. 2001.Huang SM, Bisogno T, Petros TJ, Chang SY, Zavitsanos PA, Zipkin RE, Sivakumar R, Coop A, Maeda DY, De Petrocellis L, Burstein S, Di Marzo V, Walker JM. Identification of a new class of molecules, the arachidonoyl amino acids, and characterization of one member that inhibits pain. J Biol Chem 276: 42639–42644, 2001. [DOI] [PubMed] [Google Scholar]
- Huang et al. 2002.Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci USA 99: 8400–8405, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda 1991.Ikeda SR Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. J Physiol 439: 181–214, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda 1996.Ikeda SR Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature 380: 255–258, 1996. [DOI] [PubMed] [Google Scholar]
- Ikeda 2004.Ikeda SR Expression of G-protein signaling components in adult mammalian neurons by microinjection. Methods Mol Biol 259: 167–181, 2004. [DOI] [PubMed] [Google Scholar]
- Liu et al. 2001.Liu L, Barrett CF, Rittenhouse AR. Arachidonic acid both inhibits and enhances whole cell calcium currents in rat sympathetic neurons. Am J Physiol Cell Physiol 280: C1293–1305, 2001. [DOI] [PubMed] [Google Scholar]
- Liu and Rittenhouse 2003.Liu L, Rittenhouse AR. Arachidonic acid mediates muscarinic inhibition and enhancement of N-type Ca2+ current in sympathetic neurons. Proc Natl Acad Sci USA 100: 295–300, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh et al. 2007.McHugh D, Tanner C, Mechoulam R, Pertwee RG, Ross RA. Inhibition of human neutrophil chemotaxis by endogenous cannabinoids and phytocannabinods: evidence for a site distinct from CB1 and CB2. Mol Pharmacol Oct 26 [Epub ahead of print], 2007. [DOI] [PubMed]
- Milman et al. 2006.Milman G, Maor Y, Abu-Lafi S, Horowitz M, Gallily R, Batkai S, Mo FM, Offertaler L, Pacher P, Kunos G, Mechoulam R. N-arachidonoyl l-Serine, an endocannabinoid-like brain constituent with vasodilatory properties. Proc Natl Acad Sci USA 103: 2428–2433, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oz 2006.Oz M Receptor-independent actions of cannabinoids on cell membranes: focus on endocannabinoids. Pharmacol Ther 111: 114–144, 2006. [DOI] [PubMed] [Google Scholar]
- Pacher et al. 2006.Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev 58: 389–462, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield 1990.Schofield GG Norepinephrine blocks a calcium current of adult rat sympathetic neurons via an α2-adrenoceptor. Eur J Pharmacol 180: 37–47, 1990. [DOI] [PubMed] [Google Scholar]
- Smart et al. 2000.Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, Chambers JK, Randall AD, Davis JB. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1). Br J Pharmacol 129: 227–230, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stelt and Di Marzo 2005.van der Stelt M, Di Marzo V. Anandamide as an intracellular messenger regulating ion channel activity. Prostaglandins Other Lipid Mediat 77: 111–122, 2005. [DOI] [PubMed] [Google Scholar]
- van der Stelt et al. 2005.van der Stelt M, Trevisani M, Vellani V, De Petrocellis L, Schiano Moriello A, Campi B, McNaughton P, Geppetti P, Di Marzo V. Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J 24: 3026–3037, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu et al. 2007.Xu J, He L, Wu LG. Role of Ca2+ channels in short-term synaptic plasticity. Curr Opin Neurobiol 17: 352–359, 2007. [DOI] [PubMed] [Google Scholar]