

Abstract
Cerebrovascular reactivity to changes in the partial pressure of arterial carbon dioxide (Pa,CO2) via limiting changes in brain [H+] modulates ventilatory control. It remains unclear, however, how exercise-induced alterations in respiratory chemoreflex might influence cerebral blood flow (CBF), in particular the cerebrovascular reactivity to CO2. The respiratory chemoreflex system controlling ventilation consists of two subsystems: the central controller (controlling element), and peripheral plant (controlled element). In order to examine the effect of exercise-induced alterations in ventilatory chemoreflex on cerebrovascular CO2 reactivity, these two subsystems of the respiratory chemoreflex system and cerebral CO2 reactivity were evaluated (n= 7) by the administration of CO2 as well as by voluntary hypo- and hyperventilation at rest and during steady-state exercise. During exercise, in the central controller, the regression line for the Pa,CO2–minute ventilation 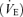 relation shifted to higher
relation shifted to higher 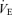 and Pa,CO2 with no change in gain (P = 0.84). The functional curve of the peripheral plant also reset rightward and upward during exercise. However, from rest to exercise, gain of the peripheral plant decreased, especially during the hypercapnic condition (−4.1 ± 0.8 to −2.0 ± 0.2 mmHg l−1 min−1, P = 0.01). Therefore, under hypercapnia, total respiratory loop gain was markedly reduced during exercise (−8.0 ± 2.3 to −3.5 ± 1.0 U, P = 0.02). In contrast, cerebrovascular CO2 reactivity at each condition, especially to hypercapnia, was increased during exercise (2.4 ± 0.2 to 2.8 ± 0.2% mmHg−1, P = 0.03). These findings indicate that, despite an attenuated chemoreflex system controlling ventilation, elevations in cerebrovascular reactivity might help maintain CO2 homeostasis in the brain during exercise.
and Pa,CO2 with no change in gain (P = 0.84). The functional curve of the peripheral plant also reset rightward and upward during exercise. However, from rest to exercise, gain of the peripheral plant decreased, especially during the hypercapnic condition (−4.1 ± 0.8 to −2.0 ± 0.2 mmHg l−1 min−1, P = 0.01). Therefore, under hypercapnia, total respiratory loop gain was markedly reduced during exercise (−8.0 ± 2.3 to −3.5 ± 1.0 U, P = 0.02). In contrast, cerebrovascular CO2 reactivity at each condition, especially to hypercapnia, was increased during exercise (2.4 ± 0.2 to 2.8 ± 0.2% mmHg−1, P = 0.03). These findings indicate that, despite an attenuated chemoreflex system controlling ventilation, elevations in cerebrovascular reactivity might help maintain CO2 homeostasis in the brain during exercise.
Numerous enzymes and ion channels which influence neural activity are modified by changes in pH (Chesler, 2003); therefore, the regulation of pH is a vital homeostatic function. The respiratory chemoreflex is an important feedback control system which keeps the partial pressure of arterial carbon dioxide (Pa,CO2) remarkably constant via ventilatory regulation. For example, the periodic nature of inspiration and expiration is carefully controlled by changes in Pa,CO2 via central and peripheral chemoreflexes so as to maintain pH nearly constant. The resulting hyper- or hypo-ventilation reduces or increases the CO2 in the blood, respectively, and therefore in the cerebrospinal fluid.
Pa,CO2 serves as an important controlled variable or mediator, especially in the brain. The blood–brain barrier is relatively impermeable to H+ and HCO3− ions; however, molecular CO2 diffuses across it readily, with the result that the CO2 in the cerebrospinal fluid parallels the arterial CO2. Therefore, CO2 diffuses freely to the cerebrospinal fluid and influences pH which drives ventilation via the central chemoreceptors (Severinghaus et al. 1963; Severinghaus & Carcelen, 1964). Moreover, the middle cerebral artery mean blood velocity (MCA Vmean), as an index of cerebral blood flow (CBF), is highly sensitive to direct changes in Pa,CO2 (Markwalder et al. 1984; Rasmussen et al. 2006). For example, hypocapnia causes cerebral vasoconstriction which reduces MCA Vmean and therefore, because of a reduced ‘washout’, attenuates the fall of brain tissue PCO2. In contrast, hypercapnia increases MCA Vmean by cerebral vasodilatation, which limits elevations in brain tissue PCO2.
Cerebrovascular reactivity and ventilatory response to CO2 seems to be tightly linked (Chapman et al. 1979; Dempsey, 2005; Xie et al. 2005, 2006; Ainslie et al. 2007; Peebles et al. 2007). Changes in CBF might have an important role in stabilizing the breathing pattern during fluctuating levels of chemical stimuli, especially to Pa,CO2 (Xie et al. 2006). In fact, an increase in CBF increases diffusion of CO2 from the cerebrospinal fluid and the brain extracellular fluid to the cerebral vessels. Therefore, [H+] decreases at the level of the central chemoreceptors when CBF increases. Early work by Severinghaus et al. (1963) investigated the regulation of cerebrospinal fluid pH during acclimatization from sea level to high altitude. They proposed three mechanisms for regulating cerebrospinal fluid pH. In addition to active transport across the blood–brain barrier and chemoreflexes, they suggested that cerebral arterioles, which dilate with high PCO2 and constrict with low PCO2, also reduce the pH variations of cerebrospinal fluid and may be regarded as a third homeostatic means to regulate cerebrospinal fluid pH and therefore central ventilatory control. In goats, Chapman et al. (1979) reported that severe brain ischaemia blunted ventilatory responses to CO2. In addition, reports indicate that cerebrovascular responsiveness to CO2 is an important determinant of eupnoeic and hypercapnic ventilatory responsiveness in otherwise healthy humans (Xie et al. 2006) and those with congestive heart failure and central sleep apnoea (Xie et al. 2005), primarily via its effects at the level of the central chemoreceptors. Such reductions in cerebrovascular CO2 reactivity affect the stability of the breathing pattern by causing ventilatory overshooting during hypercapnia and undershooting during hypocapnia (Xie et al. 2005). Therefore, changes in cerebrovascular CO2 reactivity play a critical role in the ventilatory control of Pa,CO2.
High altitude-induced hyperventilation via peripheral chemoreflex activation reduces Pa,CO2 and modifies cerebrospinal fluid pH and central chemoreceptor drive (Severinghaus et al. 1963). Therefore, it is possible that exercise-induced hyperpnoea also modifies the respiratory chemoreflex. In fact, exercise increases ventilation via the respiratory chemoreflex, and also modifies the ventilatory response to CO2 (Asmussen & Nielsen, 1957; Bhattacharyya et al. 1968; Poon & Greene, 1985); however, it remains unclear how exercise-induced alterations in the respiratory chemoreflex might influence CBF regulation, in particular cerebrovascular CO2 reactivity. The potential interactions between cerebrovascular reactivity and ventilatory responsiveness to CO2 during exercise have not been examined. In order to examine the effect of exercise-induced alterations in ventilatory chemoreflex on cerebrovascular CO2 reactivity, we evaluated two subsystems of the respiratory chemoreflex system using a new equilibrium diagram model (Miyamoto et al. 2004) and cerebral CO2 reactivity by the administration of CO2 as well as by voluntary hypo- and hyperventilation at rest and during steady-state exercise. Under a closed-loop condition of the respiratory chemoreflex system, ventilatory output is determined by chemical and metabolic drives (I: central controller), but this ventilatory loading alters these drives in the lung system which feeds back to ventilatory output (II: peripheral plant). We hypothesized that, during exercise, an increase in cerebrovascular CO2 reactivity will compensate for reductions in the peripheral plant of the respiratory chemoreflex system.
Methods
Seven healthy non-athletic men aged 20 ± 2 years, height 173 ± 8 cm, weight 64 ± 10 kg (mean ±s.d.) were recruited to participate in the study as approved by the Human Subjects Committee of Morinomiya University of Medical Sciences (No. 001). In addition, they were free of any known cardiovascular and pulmonary disorders and were not using prescribed or over the counter medications. Before the experiment, each subject gave informed written consent and visited the laboratory for familiarization with the techniques and procedures. All procedures conformed to the standards set by the Declaration of Helsinki. Subjects were requested to abstain from caffeinated beverages for 12 h and strenuous physical activity and alcohol for at least 24 h before the day of the experiment.
Measurements
All studies were performed at a constant room temperature between 23 and 24°C with external stimuli minimized. Heart rate (HR) was monitored using a lead II electrocardiogram (ECG). A catheter (0.47 mm i.d., 24 gauge) was placed in the brachial artery of the non-dominant arm for arterial blood samples and measurement of the arterial blood pressure (ABP) with a pressure transducer (DX-200, Nihon-Koden, Tokyo, Japan) positioned at the level of the right atrium in the mid-axillary line, fastened to the subject and connected to a pressure-monitoring system (RM-6000, Nihon-Koden). Arterial blood samples were obtained at rest and after reaching steady state in each experimental condition. Samples were immediately analysed for pH, Pa,CO2 and the partial pressure of arterial oxygen (Pa,O2) using a blood gas analyser (IL 1620, Instrumentation Laboratory, USA). The middle cerebral artery blood velocity (MCA V) was obtained by transcranial Doppler ultrasonography (WAKI, Atys Medical, St Genislaval, France). A 2 MHz Doppler probe was placed over the temporal window and fixed with an adjustable headband and adhesive ultrasonic gel (Tensive, Parker Laboratories, Orange, NJ, USA). The MCA V waveform was isonated at the same depth (5 cm from the skin surface of the temple window) in all subjects. Ventilatory responses were measured using an open-circuit apparatus. The subjects breathed through a face mask attached to a low-resistance one-way valve with a built-in hot-wire flow meter. The valve mechanism allowed subjects to inspire room air or a selected gas mixture from a 200 l Douglas bag containing 0.0, 3.5 or 5.0% CO2 in 40% O2 with nitrogen (N2) balance. These concentrations of CO2 administration were determined by previous studies (Ellingsen et al. 1987a,b). The respiratory CO2 sensitivity is close to constant within the range 0–5% CO2 in the inspired gas. We used three progressive CO2 stimulus points within the range 0–5% CO2 to identify respiratory chemoreflex in the model. The total instrumental dead space was 200 ml. Respiratory and metabolic data during the experiments were recorded by an automatic breath-by-breath respiratory gas-analysing system consisting of a differential pressure transducer, sampling tube, filter, suction pump and mass spectrometer (ARCO2000-MET, Arcosystem, Chiba, Japan). We digitized expired flow, CO2 and O2 concentrations, and derived tidal volume (VT), minute ventilation 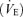 , end-tidal O2 (PET,O2) and end-tidal CO2 (PET,CO2). Flow signals were computed to single breath data, and matched to gas concentrations identified as single breaths using the peak PET,CO2, after accounting for the time delay in gas concentration measurements. The corresponding O2 uptake and CO2 output values for each breath were calculated from inspired–expired gas concentration differences, and by expired ventilation, with inspired ventilation being calculated by N2 correction. During each protocol, HR, ABP,
, end-tidal O2 (PET,O2) and end-tidal CO2 (PET,CO2). Flow signals were computed to single breath data, and matched to gas concentrations identified as single breaths using the peak PET,CO2, after accounting for the time delay in gas concentration measurements. The corresponding O2 uptake and CO2 output values for each breath were calculated from inspired–expired gas concentration differences, and by expired ventilation, with inspired ventilation being calculated by N2 correction. During each protocol, HR, ABP, 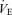 , PET,O2, PET,CO2 and MCA V were recorded continuously at 200 Hz.
, PET,O2, PET,CO2 and MCA V were recorded continuously at 200 Hz.
Experimental protocol
On the first day, each subject performed maximal cycle exercise for the measurement of maximal oxygen uptake 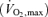 . In addition, with the exception of arterial blood gas sampling, each subject underwent the same experiment procedures as those used during the main experimental day to ensure familiarization with the experimental protocols.
. In addition, with the exception of arterial blood gas sampling, each subject underwent the same experiment procedures as those used during the main experimental day to ensure familiarization with the experimental protocols.
On the experimental day, subjects arrived at the laboratory at least 2 h after a light meal. Following instrumentation, the subjects rested in a comfortable chair. Five minutes of baseline data were recorded whilst the subjects breathed room air, wearing the face mask. To characterize the central controller and peripheral plant, subjects underwent two experimental procedures, which consisted of the 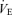 response to hypercapnia and the Pa,CO2 response to hypo- and hyperventilation, at rest and during exercise
response to hypercapnia and the Pa,CO2 response to hypo- and hyperventilation, at rest and during exercise 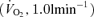 .
.
Exercise capacity
The 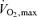 was assessed with an incremental protocol on a cycle ergometer (Corival1000SS, Lode, Groningen, the Netherlands). The workload was set at 20 W and was increased by 20 W every minute until the subject could no longer maintain the pedalling frequency at 60 r.p.m. despite strong verbal encouragement. The subjects breathed through a facemask attached to a volume transducer while gases were continuously sampled for analysis of fractional concentrations of O2, CO2 and N2. The respiratory gas analysis system was calibrated before each test using known standard gases.
was assessed with an incremental protocol on a cycle ergometer (Corival1000SS, Lode, Groningen, the Netherlands). The workload was set at 20 W and was increased by 20 W every minute until the subject could no longer maintain the pedalling frequency at 60 r.p.m. despite strong verbal encouragement. The subjects breathed through a facemask attached to a volume transducer while gases were continuously sampled for analysis of fractional concentrations of O2, CO2 and N2. The respiratory gas analysis system was calibrated before each test using known standard gases.
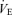 response to hypercapnia (CO2 administration)
response to hypercapnia (CO2 administration)
The 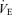 response to hypercapnia consisted of three trials (fraction of inspired CO2 (FI,CO2) 0.00, 0.035, 0.05), which was induced by rapidly changing the FI,CO2. Each FI,CO2 trial ran for 12 min at approximately 10–15 min intervals. This duration is long enough to permit CO2 to reach its new steady-state value at the central chemoreceptors (Honda et al. 1983; Poon & Greene, 1985; Pianosi et al. 1994; Teppema et al. 2000). During the interval periods, the subjects inspired room air. Each subject performed these three trials at rest and during exercise. The order of the trials was randomized for each subject. We performed all trials under the hyperoxic condition to abolish the O2-sensitive chemoreflex (Ohyabu et al. 1982; Robbins, 1988; Mohan & Duffin, 1997).
response to hypercapnia consisted of three trials (fraction of inspired CO2 (FI,CO2) 0.00, 0.035, 0.05), which was induced by rapidly changing the FI,CO2. Each FI,CO2 trial ran for 12 min at approximately 10–15 min intervals. This duration is long enough to permit CO2 to reach its new steady-state value at the central chemoreceptors (Honda et al. 1983; Poon & Greene, 1985; Pianosi et al. 1994; Teppema et al. 2000). During the interval periods, the subjects inspired room air. Each subject performed these three trials at rest and during exercise. The order of the trials was randomized for each subject. We performed all trials under the hyperoxic condition to abolish the O2-sensitive chemoreflex (Ohyabu et al. 1982; Robbins, 1988; Mohan & Duffin, 1997).
Pa,CO2 response to hypo- and hyperventilation (voluntary changes in respiration)
The Pa,CO2 response to ventilation consisted of three trials: two periods of hyperventilation and one period of hypoventilation. To avoid the possible effects of different breathing patterns on the 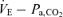 relationship, in the hyperventilation trials, both VT and breathing frequency were altered deliberately by matching the breathing pattern to that recorded during hypercapnia trials, whilst inhaling 0% CO2 in 40% O2 with N2 balance. In the hypoventilation trial,
relationship, in the hyperventilation trials, both VT and breathing frequency were altered deliberately by matching the breathing pattern to that recorded during hypercapnia trials, whilst inhaling 0% CO2 in 40% O2 with N2 balance. In the hypoventilation trial, 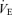 was set to 80% of
was set to 80% of 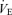 during the 0.00 FI,CO2 trial (i.e. during spontaneous breathing). The breathing pattern was estimated from the relationships between
during the 0.00 FI,CO2 trial (i.e. during spontaneous breathing). The breathing pattern was estimated from the relationships between 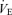 and VT in each subject. Each trial ran for 12 min with an interval of 10–15 min. Each subject performed these three trials at rest and during exercise, and the order of the trials was randomized.
and VT in each subject. Each trial ran for 12 min with an interval of 10–15 min. Each subject performed these three trials at rest and during exercise, and the order of the trials was randomized.
During hypo- and hyperventilation trials, the inspired and expired volume curves were continuously displayed on a screen monitor. Visual and audio signals were constructed from the breathing pattern of the subjects during the hypercapnia trials. The target VT level was simultaneously displayed on the same screen monitor in each trial. The subjects were instructed to match their volume curve with the target VT level and to breathe according to the sound of the metronome. As a result, both the VT and breathing frequency, and thus 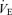 , were precisely controlled by the visual feedback.
, were precisely controlled by the visual feedback.
Since our preliminary measurements indicated that Pa,CO2 responses to 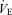 , and the
, and the 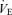 response to Pa,CO2 reached steady states within 8–12 min, we represented each response by averaging it in the last 2 min. The arterial blood sample (2.5 ml) was collected at minute 11.5 of each trial period. The measured values of operating points (OPs) in the subjects were defined to be the steady-state values for
response to Pa,CO2 reached steady states within 8–12 min, we represented each response by averaging it in the last 2 min. The arterial blood sample (2.5 ml) was collected at minute 11.5 of each trial period. The measured values of operating points (OPs) in the subjects were defined to be the steady-state values for 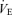 and Pa,CO2 that were obtained during the 0.00 FI,CO2 trial without visual feedback (i.e. during spontaneous breathing).
and Pa,CO2 that were obtained during the 0.00 FI,CO2 trial without visual feedback (i.e. during spontaneous breathing).
Data analysis
The cerebrovascular and ventilatory equilibrium diagram model is depicted in Fig. 1. The respiratory chemoreflex system consists of two subsystems, the central controller (I) and peripheral plant (II). These subsystems act as a feedback control system, which regulates the systemic CO2 level. In the brain, CBF (III) is influenced by systemic CO2 and regulates CO2 at the brain level, which then feeds back into the central controller.
Figure 1. Equilibrium diagram model.

A, systemically, partial pressure of arterial carbon dioxide (Pa,CO2) is controlled by the respiratory chemoreflex system which consists of two subsystems: the central controller (controlling element; I) and peripheral plant (controlled element; II). In the brain, cerebral CO2 is strongly regulated by change in cerebral blood flow (CBF; III). In addition, brain tissue CO2 influences the central chemoreflex, and can be characterized by observing CBF response to changes in CO2. B, in the central controller the input parameter is Pa,CO2, the output parameter is minute ventilation 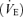 . To characterize the central controller, fraction of inspired CO2 (0, 3.5, 5% CO2 in 40% O2 with N2 balance) and the
. To characterize the central controller, fraction of inspired CO2 (0, 3.5, 5% CO2 in 40% O2 with N2 balance) and the 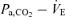 relationship were measured. The central controller can be characterized by observing changes in
relationship were measured. The central controller can be characterized by observing changes in 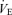 in response to changes in Pa,CO2. In the peripheral plant, input is
in response to changes in Pa,CO2. In the peripheral plant, input is 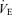 , and output is Pa,CO2. To characterize the peripheral plant,
, and output is Pa,CO2. To characterize the peripheral plant, 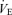 was altered by hyper- and hypoventilation using a visual feedback method, which made it possible to control both tidal volume and breathing frequency; the
was altered by hyper- and hypoventilation using a visual feedback method, which made it possible to control both tidal volume and breathing frequency; the 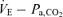 relationship was then quantified. Since both relationships share common variables, the resultant operating point of ventilatory response under the closed-loop condition is determined by intersection of the two relationships.
relationship was then quantified. Since both relationships share common variables, the resultant operating point of ventilatory response under the closed-loop condition is determined by intersection of the two relationships.
Central controller (I)
Change in Pa,CO2 (input) stimulates chemoreceptor activity and alters ventilation (output) via the central controller. To characterize the central controller, we used a protocol of CO2 administration (three levels), a conventional linear equation, 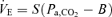 , and determined the slope S and intercept B using a least-squares regression method. The slope (S) also identifies the gain of the central controller (GC);
, and determined the slope S and intercept B using a least-squares regression method. The slope (S) also identifies the gain of the central controller (GC); 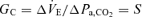 .
.
Peripheral plant (II)
The central controller-induced changes in ventilation (input) also alters Pa,CO2 (output). To characterize the peripheral plant, we used a protocol of voluntary changes in respiration (four levels), the modified metabolic hyperbola as 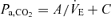 , and determined the values of A and C by the least-squares regression method. During hyper- and hypocapnia, the gain of the peripheral plant (GP) to operating point (OP) was calculated from the following equations under hyper- (
, and determined the values of A and C by the least-squares regression method. During hyper- and hypocapnia, the gain of the peripheral plant (GP) to operating point (OP) was calculated from the following equations under hyper- (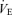 : −2 l min−1 from OP) and hypocapnia (
: −2 l min−1 from OP) and hypocapnia (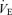 : +2 l min−1 from OP) conditions, respectively.
: +2 l min−1 from OP) conditions, respectively.
Total respiratory loop gain (I + II)
Total respiratory loop gain (GTR) to OP, hyper- and hypocapnia was calculated by the following equations.
Cerebral CO2 reactivity (III)
Pa,CO2 (input) alters CBF (output) via cerebral CO2 reactivity. To characterize cerebrovascular reactivity to CO2, we used protocols of CO2 administration and voluntary changes in respiration (six levels), an exponential function, %MCA 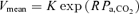 , and determined the values of K and R. The cerebral CO2 reactivity (GB) to OP, hyper- and hypocapnia was calculated from the following equations at the OP, and under hyper- (Pa,CO2: +5 mmHg from OP) and hypocapnic (Pa,CO2: −5 mmHg from OP) conditions, respectively.
, and determined the values of K and R. The cerebral CO2 reactivity (GB) to OP, hyper- and hypocapnia was calculated from the following equations at the OP, and under hyper- (Pa,CO2: +5 mmHg from OP) and hypocapnic (Pa,CO2: −5 mmHg from OP) conditions, respectively.
Statistical analysis
A paired t test was used to assess the differences in the steady-state haemodynamic variables between rest and exercise conditions. Two-way analysis (CO2 and exercise) of variance with repeated measures was used to assess the differences in the GP, GTR and GB between all conditions. A Student–Newman–Keul's test was employed post hoc when main effects were significant, i.e. P < 0.05. Data are expressed as mean ±s.e.m. and analyses were conducted using SigmaStat (Jandel Scientific Software, SPSS Inc., Chicago, IL, USA).
Results
Averaged O2 uptake during the cycling exercise was 32 ± 2%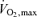 . This mild exercise increased HR and caused slight elevations in MAP (92 to 97 mmHg; P = 0.169) and MCA Vmean (51.4 to 53.3 cm s−1; P = 0.085, Table 1). During exercise, both
. This mild exercise increased HR and caused slight elevations in MAP (92 to 97 mmHg; P = 0.169) and MCA Vmean (51.4 to 53.3 cm s−1; P = 0.085, Table 1). During exercise, both 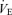 (P <0.001) and PET,CO2 (P = 0.004) were increased whilst pH and Pa,CO2 were unchanged.
(P <0.001) and PET,CO2 (P = 0.004) were increased whilst pH and Pa,CO2 were unchanged.
Table 1.
Ventilatory and haemodynamic variables at rest and during exercise
| Rest (R) | Exercise (E) | R versus E | |
|---|---|---|---|
| PET,CO2 (mmHg) | 38.2 ± 1.2 | 44.1 ± 1.7 | P = 0.004 |
| Pa,CO2 (mmHg) | 43.2 ± 1.7 | 44.6 ± 01.2 | P = 0.294 |
| Pa,O2 (mmHg) | 225 ± 17 | 244 ± 2 | P = 0.578 |
| pH | 7.40 ± 0.01 | 7.39 ± 0.01 | P = 0.668 |
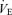 (l min−1) (l min−1) |
11.6 ± 0.5 | 26.0 ± 1.2 | P < 0.001 |
| VT (ml) | 1059 ± 330 | 1431 ± 163 | P = 0.156 |
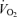 (ml min−1) (ml min−1) |
330 ± 63 | 942 ± 40 | P < 0.001 |
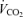 (ml min−1) (ml min−1) |
338 ± 24 | 849 ± 32 | P < 0.001 |
| MAP (mmHg) | 92 ± 1 | 97 ± 2 | P = 0.169 |
| HR (beats min−1) | 69 ± 5 | 98 ± 8 | P < 0.001 |
| MCA Vmean (cm s−1) | 51.4 ± 4.5 | 53.3 ± 4.2 | P = 0.085 |
Values are means ±s.e.m. PET,CO2, end-tidal carbon dioxide (CO2) tension; Pa,CO2, partial pressure of arterial CO2; Pa,O2, partial pressure of arterial O2; 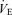 , minute ventilation; VT, tidal volume;
, minute ventilation; VT, tidal volume; 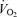 , oxygen uptake;
, oxygen uptake; 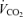 , CO2 uptake; MAP, mean arterial pressure; HR, heart rate; MCA Vmean, middle cerebral artery mean blood velocity.
, CO2 uptake; MAP, mean arterial pressure; HR, heart rate; MCA Vmean, middle cerebral artery mean blood velocity.
In the central controller, the regression line of the 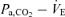 relation was reset to higher
relation was reset to higher 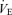 and Pa,CO2 during exercise (Fig. 2) without a change in gain (GC) (1.7 ± 0.3 to 1.8 ± 0.5 l min−1 mmHg−1, P = 0.837; Fig. 3). The functional curve of the peripheral plant also reset to higher
and Pa,CO2 during exercise (Fig. 2) without a change in gain (GC) (1.7 ± 0.3 to 1.8 ± 0.5 l min−1 mmHg−1, P = 0.837; Fig. 3). The functional curve of the peripheral plant also reset to higher 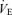 and Pa,CO2 during exercise (Fig. 2); however, the change in gain (GP) was different from that of GC, i.e. the GP at the OP, during both hypercapnia and hypocapnia, was decreased from rest to exercise; the change in GP was especially marked during hypercapnia (−4.1 ± 0.8 to −2.0 ± 0.2 mmHg l−1 min, P = 0.009; Fig. 3). Therefore, total respiratory loop gain (GTR) during hypercapnia decreased during exercise (−8.0 ± 2.3 to −3.5 ± 1.0 U, P = 0.019) despite no change in GTR at OP and under hypocapnia. If the change in Pa,CO2 is 2 mmHg,
and Pa,CO2 during exercise (Fig. 2); however, the change in gain (GP) was different from that of GC, i.e. the GP at the OP, during both hypercapnia and hypocapnia, was decreased from rest to exercise; the change in GP was especially marked during hypercapnia (−4.1 ± 0.8 to −2.0 ± 0.2 mmHg l−1 min, P = 0.009; Fig. 3). Therefore, total respiratory loop gain (GTR) during hypercapnia decreased during exercise (−8.0 ± 2.3 to −3.5 ± 1.0 U, P = 0.019) despite no change in GTR at OP and under hypocapnia. If the change in Pa,CO2 is 2 mmHg, 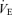 changes are 3.5 ± 0.7 and 3.6 ± 1.0 l min−1 at rest and during exercise, respectively. However, these similar
changes are 3.5 ± 0.7 and 3.6 ± 1.0 l min−1 at rest and during exercise, respectively. However, these similar 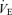 changes cause a different correction in Pa,CO2 between the hypo- and hypercapnia conditions. Under conditions of hypocapnia,
changes cause a different correction in Pa,CO2 between the hypo- and hypercapnia conditions. Under conditions of hypocapnia, 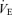 change similarly alters Pa,CO2 at rest (8 ± 2 mmHg) and during exercise (5 ± 2 mmHg); however, under conditions of hypercapnia, large differences in alterations in Pa,CO2 are observed between those at rest (16 ± 5 mmHg) and those during exercise (7 ± 2 mmHg).
change similarly alters Pa,CO2 at rest (8 ± 2 mmHg) and during exercise (5 ± 2 mmHg); however, under conditions of hypercapnia, large differences in alterations in Pa,CO2 are observed between those at rest (16 ± 5 mmHg) and those during exercise (7 ± 2 mmHg).
Figure 2. Characteristics of central controller (I; A) and peripheral plant (II; B) at rest and during exercise.

A (central controller), 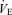 linearly increased with Pa,CO2 at rest and during exercise. The averaged regression lines were
linearly increased with Pa,CO2 at rest and during exercise. The averaged regression lines were 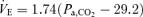 and
and 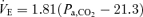 at rest and during exercise, respectively. B (peripheral plant), the peripheral plant was characterized by a modified metabolic hyperbola. The averaged fitted hyperbolae were
at rest and during exercise, respectively. B (peripheral plant), the peripheral plant was characterized by a modified metabolic hyperbola. The averaged fitted hyperbolae were 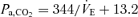 and
and 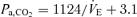 at rest and during exercise, respectively. Arrows denote operating points.
at rest and during exercise, respectively. Arrows denote operating points.
Figure 3. Group-averaged central controller gain (GC, A), peripheral plant gain (GP, B) and respiratory total loop gain (GTR, C) at rest and during exercise.

The central controller was characterized by a conventional linear equation, thus the gain under hypocapnic conditions was the same as that at the operating point (OP) and hypercapnia. The peripheral plant was characterized by a hyperbola therefore gains at OP, hypo- and hypercapnia were analysed. Values are means ±s.e.m.*P <0.05, different from rest; #P <0.05, different from operating point; $P <0.05, different from hypocapnia.
Hypercapnia resulted in an exponential elevation in MCA Vmean during exercise as well as at rest (Fig. 4). However, the functional curve of cerebral CO2 reactivity was not reset during exercise because of small changes in Pa,CO2 and MCA Vmean. In contrast to the ventilatory chemoreflex, all cerebrovascular reactivities (GB) to OP, hyper- and hypocapnia were increased during exercise despite unremarkable changes in both K (P = 0.662) and R (P = 0.286) of these curves (Table 2 and Fig. 5). The increases in GB were more marked in the hypercapnic condition (2.4 ± 0.2 to 2.8 ± 0.2 % mmHg−1, P = 0.025) compared to other conditions (OP, P = 0.049; hypocapnia, P = 0.086).
Figure 4. Characteristics of cerebrovascular CO2 reactivity (III) at rest and during exercise.

The cerebrovascular CO2 reactivity was characterized by an exponential function. The averaged fitted exponential equations were MCA 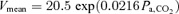 and MCA
and MCA 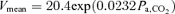 at rest and during exercise, respectively. Arrows denote operating points.
at rest and during exercise, respectively. Arrows denote operating points.
Table 2.
Characteristics of central controller, peripheral plant and cerebrovascular reactivity at rest and during exercise
| Rest (R) | Exercise (E) | R versus E | |
|---|---|---|---|
| Central controller (I) | |||
| S | 1.74 ± 0.32 | 1.81 ± 0.49 | P = 0.837 |
| B | 29.2 ± 8.51 | 21.28 ± 6.62 | P = 0.073 |
| Peripheral plant (II) | |||
| A | 344 ± 54 | 1124 ± 118 | P < 0.001 |
| C | 13.2 ± 3.0 | 3.1 ± 2.8 | P = 0.004 |
| Cerebrovascular CO2 reactivity (III) | |||
| K | 40.0 ± 2.5 | 38.9 ± 3.1 | P = 0.662 |
| R | 0.0216 ± 0.0013 | 0.0232 ± 0.0016 | P = 0.286 |
Values are means ±s.e.m. Central controller (I), 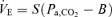 ; peripheral plant (II),
; peripheral plant (II), 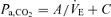 ; cerebrovascular reactivity to CO2 (III), MCA
; cerebrovascular reactivity to CO2 (III), MCA 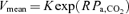 .
.
Figure 5. Group-averaged cerebrovascular CO2 reactivity (GB) at rest and during exercise.

The cerebrovascular CO2 reactivity was characterized by an exponential function therefore GB values at the operating point and under hypo- and hypercapnia were analysed. Values are means ±s.e.m.*P <0.05, different from rest; #P <0.05, different from operating point; $P <0.05, different from hypocapnia.
Discussion
The main finding of the present investigation was that, under conditions of hypercapnia and exercise, the total respiratory loop gain was markedly reduced. These changes in total loop gain occurred independently of the change in central controller gain because of a marked decrease in peripheral plant gain. Furthermore, cerebrovascular CO2 reactivity during each condition, especially during hypercapnia, was increased during exercise. These findings indicate that, despite an attenuated chemoreflex system controlling ventilation, elevations in cerebrovascular reactivity might help maintain CO2 homeostasis in the brain during exercise.
The respiratory chemoreflex
The respiratory chemoreflex is a powerful feedback control system which acts to maintain Pa,CO2 or pH remarkably constant; the tight regulation of pH is critical to maintain homeostatic function for all tissues (Chesler, 2003), especially neural activity. Exercise, which activates muscle metabolism and produces CO2, causes hyperpnoea via the ventilatory chemoreflex. The exercise-induced hyperpnoea was reflected in elevations in 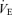 from 12 to 26 l min−1 (P <0.001). The mechanism(s) subserving ventilatory control during exercise remain controversial (Ward, 2007); however, traditionally these mechanisms are proposed to include elements of proportional feedback, central and carotid chemosensory, and feedforward systems, central command and muscle reflex (Dempsey et al. 2006; Waldrop & Iwamoto, 2006; Ward, 2007). Acute hypoxia causes hyperventilation and alkalosis at the medullary chemoreceptors, which reduce their drive (Crawford & Severinghaus, 1978). In addition, the change of this cerebrospinal fluid alkalosis modifies the respiratory control (Severinghaus et al. 1963). Although the mechanism of exercise-induced hyperpnoea is different from that associated with hyperventilation at high altitude, it seems that exercise-induced hyperpnoea alters central chemoreflex. The role of the ventilatory chemoreflex in the regulation of exercise hyperpnoea has been extensively investigated (Cunningham, 1987). The ventilatory sensitivity to hypoxia is increased from rest during exercise (Bhattacharyya et al. 1968); however, the effect of exercise on the respiratory chemoreflex remains controversial. For example, Asmussen & Nielsen (1957) demonstrated that the ventilatory–PCO2 relationship line shifted to the left without a change in its sensitivity. In contrast, Poon & Greene (1985) showed that the slope of the ventilatory–PCO2 relationship was increased by exercise. In addition, the ‘chemoreflex response’ is not dictated by the level of chemical drive. Such an integrative response involves a dynamic interaction between the respiratory controller and the chemical drive, and is influenced by respiratory mechanical constraints (Poon et al., 2007). Under a closed-loop condition (Fig. 1), ventilatory output is determined by chemical and metabolic drives, although this ventilatory loading alters these drives in the lung system which feeds back to ventilatory output. However, previous studies have failed to consider the importance of metabolic changes due to the work of breathing (Miyamoto et al. 2004). We have used a new equilibrium diagram model (Miyamoto et al. 2004) to resolve this question. The previous studies by Severinghaus et al. (Severinghaus et al. 1963; Severinghaus & Carcelen, 1964; Crawford & Severinghaus, 1978) demonstrated the regulation of cerebrospinal fluid pH during the hyperventilation associated with high altitude by using a similar model for ventilatory control. The respiratory model of the present study has a limitation in identifying the regulation of cerebrospinal fluid pH or the interaction between central and peripheral chemoreflexes. In the present study, the equilibrium diagram model demonstrates the effect of CO2 change on respiratory control or the effect of respiratory change on cerebral CO2 haemodynamics during exercise. Compared with the model used in previous work (Severinghaus et al. 1963; Severinghaus & Carcelen, 1964; Crawford & Severinghaus, 1978), our model gave similar information about respiratory control during conditions of exercise rather than at high altitude.
from 12 to 26 l min−1 (P <0.001). The mechanism(s) subserving ventilatory control during exercise remain controversial (Ward, 2007); however, traditionally these mechanisms are proposed to include elements of proportional feedback, central and carotid chemosensory, and feedforward systems, central command and muscle reflex (Dempsey et al. 2006; Waldrop & Iwamoto, 2006; Ward, 2007). Acute hypoxia causes hyperventilation and alkalosis at the medullary chemoreceptors, which reduce their drive (Crawford & Severinghaus, 1978). In addition, the change of this cerebrospinal fluid alkalosis modifies the respiratory control (Severinghaus et al. 1963). Although the mechanism of exercise-induced hyperpnoea is different from that associated with hyperventilation at high altitude, it seems that exercise-induced hyperpnoea alters central chemoreflex. The role of the ventilatory chemoreflex in the regulation of exercise hyperpnoea has been extensively investigated (Cunningham, 1987). The ventilatory sensitivity to hypoxia is increased from rest during exercise (Bhattacharyya et al. 1968); however, the effect of exercise on the respiratory chemoreflex remains controversial. For example, Asmussen & Nielsen (1957) demonstrated that the ventilatory–PCO2 relationship line shifted to the left without a change in its sensitivity. In contrast, Poon & Greene (1985) showed that the slope of the ventilatory–PCO2 relationship was increased by exercise. In addition, the ‘chemoreflex response’ is not dictated by the level of chemical drive. Such an integrative response involves a dynamic interaction between the respiratory controller and the chemical drive, and is influenced by respiratory mechanical constraints (Poon et al., 2007). Under a closed-loop condition (Fig. 1), ventilatory output is determined by chemical and metabolic drives, although this ventilatory loading alters these drives in the lung system which feeds back to ventilatory output. However, previous studies have failed to consider the importance of metabolic changes due to the work of breathing (Miyamoto et al. 2004). We have used a new equilibrium diagram model (Miyamoto et al. 2004) to resolve this question. The previous studies by Severinghaus et al. (Severinghaus et al. 1963; Severinghaus & Carcelen, 1964; Crawford & Severinghaus, 1978) demonstrated the regulation of cerebrospinal fluid pH during the hyperventilation associated with high altitude by using a similar model for ventilatory control. The respiratory model of the present study has a limitation in identifying the regulation of cerebrospinal fluid pH or the interaction between central and peripheral chemoreflexes. In the present study, the equilibrium diagram model demonstrates the effect of CO2 change on respiratory control or the effect of respiratory change on cerebral CO2 haemodynamics during exercise. Compared with the model used in previous work (Severinghaus et al. 1963; Severinghaus & Carcelen, 1964; Crawford & Severinghaus, 1978), our model gave similar information about respiratory control during conditions of exercise rather than at high altitude.
The regression line of the central controller was shifted rightward and upward around the higher operating point of 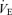 and Pa,CO2 during exercise (Fig. 2). Both neural and humoral mechanisms may be involved in the ventilatory chemoreflex responses of the central controller associated with exercise. During exercise, increases in peripheral chemoreflex hypoxic sensitivity can be related to lactic acidosis (Asmussen & Nielsen, 1958; Wasserman et al. 1975), circulating catecholamines (Cunningham et al. 1963) and potassium (Linton et al. 1984; Qayyum et al. 1994). However, the sensitivity of the central controller (GC) was unchanged during exercise (P = 0.837, Fig. 3). This finding may be related to the small changes in lactate, catecholamines and potassium concentrations during such a light exercise workload (32%
and Pa,CO2 during exercise (Fig. 2). Both neural and humoral mechanisms may be involved in the ventilatory chemoreflex responses of the central controller associated with exercise. During exercise, increases in peripheral chemoreflex hypoxic sensitivity can be related to lactic acidosis (Asmussen & Nielsen, 1958; Wasserman et al. 1975), circulating catecholamines (Cunningham et al. 1963) and potassium (Linton et al. 1984; Qayyum et al. 1994). However, the sensitivity of the central controller (GC) was unchanged during exercise (P = 0.837, Fig. 3). This finding may be related to the small changes in lactate, catecholamines and potassium concentrations during such a light exercise workload (32%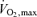 ), or a differential influence of CO2 on peripheral chemoreflex activity as opposed to hypoxia. Moreover, because oscillations in pH increase during exercise (Band et al. 1980), these changes might also modify the results of respiratory control during exercise as identified in the present study.
), or a differential influence of CO2 on peripheral chemoreflex activity as opposed to hypoxia. Moreover, because oscillations in pH increase during exercise (Band et al. 1980), these changes might also modify the results of respiratory control during exercise as identified in the present study.
The lung system (peripheral plant) is an important subsystem of respiratory chemoreflex, because it is an effector to change CO2 systemically via an alteration in ventilation (Miyamoto et al. 2004). The sensitivity of peripheral plant is non-linear and is changed by ventilation (Fig. 2). At rest, GP was increased due to a decrease in ventilation, suggesting that the peripheral plant is more effective in controlling CO2 at low 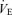 levels. This effect of ventilatory loading is particularly acute during hyperventilation as the respiratory apparatus is subject to increasing mechanical limitations, i.e. dead space (Poon et al. 2007). Therefore, considering the multiple effects of these subsystems, the central controller is generally more pronounced at low
levels. This effect of ventilatory loading is particularly acute during hyperventilation as the respiratory apparatus is subject to increasing mechanical limitations, i.e. dead space (Poon et al. 2007). Therefore, considering the multiple effects of these subsystems, the central controller is generally more pronounced at low 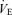 than high
than high 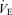 levels (Clark et al. 1980; Poon, 1989a,b). The respiratory total loop gain (GTR) was much higher under the hypercapnic condition caused by hypoventilation compared with normal (P <0.001) and hypocapnic (P <0.001) conditions at rest (Fig. 3).
levels (Clark et al. 1980; Poon, 1989a,b). The respiratory total loop gain (GTR) was much higher under the hypercapnic condition caused by hypoventilation compared with normal (P <0.001) and hypocapnic (P <0.001) conditions at rest (Fig. 3).
During exercise the functional curve of the peripheral plant also reset rightward and upward around the higher 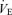 and Pa,CO2 (Fig. 2). However, the change in GP was different from that of GC. From rest to exercise, during hypercapnia and hypocapnia, there was a decrease in GP at the operating point (OP). Importantly, the change in GP at hypercapnia was larger (−51%) compared with that at other conditions (OP, −39% and hypocapnia −27%; Fig. 3). The sensitivity of the peripheral plant is non-linear and was decreased exponentially during elevations in ventilation. Therefore, these exercise-induced GP reductions were related such that the OP moved rightward on the functional curve of the peripheral plant during exercise compared with rest (Fig. 2). The rightward shift of the exercise OP was determined by the resetting of central controller (rightward and upward shift), indicating that the mechanism of the change in GP during exercise depends on the interaction with alteration in the central controller. As a consequence, total respiratory loop gain (GTR) at hypercapnia decreased during exercise despite no changes in GTR at the OP and under the hypocapnic condition. These findings suggest that the respiratory chemoreflex was attenuated during exercise under the hypercapnic condition despite no change in the sensitivity of the central controller. The interaction between the central controller and the plant was non-linear. Moreover, these results were not consistent with the traditional chemoreflex feedback model, which ignores the mechanical plant. The ventilatory response to chemical or exercise inputs is also potentiated by increases in physiological dead space or shunt (Poon et al. 2007). In addition, congestive heart failure patients with increased physiological dead space are reported to have an augmented
and Pa,CO2 (Fig. 2). However, the change in GP was different from that of GC. From rest to exercise, during hypercapnia and hypocapnia, there was a decrease in GP at the operating point (OP). Importantly, the change in GP at hypercapnia was larger (−51%) compared with that at other conditions (OP, −39% and hypocapnia −27%; Fig. 3). The sensitivity of the peripheral plant is non-linear and was decreased exponentially during elevations in ventilation. Therefore, these exercise-induced GP reductions were related such that the OP moved rightward on the functional curve of the peripheral plant during exercise compared with rest (Fig. 2). The rightward shift of the exercise OP was determined by the resetting of central controller (rightward and upward shift), indicating that the mechanism of the change in GP during exercise depends on the interaction with alteration in the central controller. As a consequence, total respiratory loop gain (GTR) at hypercapnia decreased during exercise despite no changes in GTR at the OP and under the hypocapnic condition. These findings suggest that the respiratory chemoreflex was attenuated during exercise under the hypercapnic condition despite no change in the sensitivity of the central controller. The interaction between the central controller and the plant was non-linear. Moreover, these results were not consistent with the traditional chemoreflex feedback model, which ignores the mechanical plant. The ventilatory response to chemical or exercise inputs is also potentiated by increases in physiological dead space or shunt (Poon et al. 2007). In addition, congestive heart failure patients with increased physiological dead space are reported to have an augmented 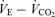 sensitivity (Wasserman et al. 1997). Therefore, an interaction with the attenuation in peripheral plant gain may be another mechanism underlining the lack of change in the controller gain during exercise.
sensitivity (Wasserman et al. 1997). Therefore, an interaction with the attenuation in peripheral plant gain may be another mechanism underlining the lack of change in the controller gain during exercise.
Cerebrovascular CO2 reactivity
At rest, hypercapnic cerebral CO2 reactivity was greater than the hypocapnic reactivity (Fig. 5) because of the increase in CO2 exponentially elevated MCA Vmean when a wider range of CO2 challenge was applied (Rasmussen et al. 2006). Animal studies indicate that the mechanisms underlying the normal greater reactivity to hypercapnia compared with hypocapnia may be related to a greater influence of vasodilator mediators on intracranial vascular tone compared with vasoconstrictive mediators (Toda & Okamura, 1998). During exercise, cerebral CO2 reactivity (GB) to the OP, in both the hyper- and hypocapnia conditions, was increased. Enhanced cerebral CO2 reactivity at OP with exercise has been reported (Rasmussen et al. 2006). Our new finding is that the increase in GB during the hypercapnic condition was much larger compared with other conditions at rest and during exercise. Moreover, cerebral CO2 reactivity (GB) to OP (P = 0.049) and hypercapnia (P = 0.025) was increased during exercise while GB to hypocapnia was unchanged (P = 0.086, Fig. 5). This enhanced cerebral CO2 reactivity during exercise may relate to interactions with the central controller; however, the mechanism(s) underpinning such changes remain unclear.
The role of autonomic neural control of the cerebral circulation is controversial and, despite rich sympathetic nerve innervation of the cerebral arteries (Nielsen & Owman, 1967; Nelson & Rennels, 1970; Edvinsson, 1975), the traditional thinking is that changes in sympathetic tone appear to have a limited effect on CBF. In contrast, Meadows et al. (2003) found that sleep decreased cerebral CO2 reactivity, suggesting that the level of cerebral activation influences the cerebrovascular reactivity to CO2. In addition, sympathetic nervous activation attenuates the CO2-induced increase in CBF at rest (Jordan et al. 2000). Therefore, exercise-induced physiological changes (e.g. autonomic neural control) may also modify the cerebral CO2 reactivity. However, these findings contrast with a study which reported that sympatho-excitation induced with lower body negative pressure did not alter the cerebral CO2 reactivity (LeMarbre et al. 2003). Collectively, the mechanisms underlying heightened sympathetic nerve activity during exercise on the regulation of CBF remain unclear.
Cerebral autoregulation is well maintained during mild and moderate dynamic exercise (Brys et al. 2003; Ogoh et al. 2005a,b, 2007), suggesting that CBF regulation is not influenced by ABP during exercise. However, at rest in the supine position, Aaslid et al. (1989) have reported that cerebral autoregulation is also affected by the basal vascular tone and it is attenuated by hypercapnia. Via sympathoexcitation, arterial blood pressure increases with CO2 administration (Ainslie et al. 2005). Thus, because of an attenuation in normal cerebral autoregulation under hypercapnic conditions, CBF may be influenced by an increased ABP with CO2 administration and this phenomenon may be further altered by exercise. During exercise the additional CO2-induced elevations in blood pressure and a lowered cerebral autoregulation might explain the exponential change in cerebral CO2 reactivity during hypercapnia.
The interaction between total respiratory chemoreflex and cerebrovascular reactivity
An increase in cerebrovascular CO2 reactivity compensated an attenuated respiratory chemoreflex system during steady-state exercise, especially under the hypercapnic condition. Although the interaction between systemic and cerebral CO2 controlling mechanisms during exercise remains unknown, previous investigations (Chapman et al. 1979; Dempsey, 2005; Xie et al. 2005, 2006; Ainslie et al. 2007; Peebles et al. 2007) indicate that cerebral CO2 reactivity is linked with the ventilatory response to CO2. Changes in cerebrovascular CO2 reactivity affect the stability of the ventilatory responsiveness to CO2 via alterations in the degree of washout in central chemoreceptor hydrogen [H+]; these changes have been documented in a range of physiological (Xie et al. 2006; Ainslie et al. 2007) and pathophysiological disorders (Xie et al. 2005). Peebles et al. (2007) reported that hypercapnic cerebral CO2 reactivity was inversely related to the increase in ventilatory change and suggested that a reduced cerebral CO2 reactivity resulted in less central CO2 washout and greater ventilatory stimulus. However, our findings indicate that the relationship between the two systems during exercise cannot be explained only by these mechanisms, because the central controller gain was unchanged during exercise despite an enhanced cerebral CO2 reactivity. This dissociation may depend on a peripheral chemoreflex distribution to the central controller or CBF distributions to central chemoreflex that are different between rest and exercise. These findings highlight the interdependence of total respiratory chemoreflex to many other variables through a complex and probably non-linear relationship.
Technological considerations
PET,CO2 measurement has been used as an estimate of Pa,CO2. The difference between Pa,CO2 and PET,CO2 is influenced by metabolic CO2 production and tidal volume and the relationship between these two variables is not altered by breathing frequency and exercise (Jones et al. 1979). In addition, the estimated different cerebral CO2 reactivity between rest and exercise from Pa,CO2 was the same as that from PET,CO2 (Rasmussen et al. 2006). However, PET,CO2 is higher than Pa,CO2 when metabolic CO2 production and 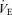 are increased (Jones et al. 1979). Peebles et al. (2007) demonstrated that cerebrovascular CO2 reactivity is underestimated by PET,CO2 when compared with Pa,CO2; therefore, we have made the calculations of central controller, peripheral plant and cerebral CO2 reactivity using Pa,CO2. Another important consideration is that PCO2 from the internal jugular vein is likely to be a closer index of brain tissue PCO2 than Pa,CO2 (Xie et al. 2006) and the medullary central chemoreceptors are not stimulated directly by Pa,CO2; rather, they are stimulated by [H+] via alterations in brain tissue CO2 tension (Peebles et al. 2007). Whilst studies have ‘corrected’ ventilatory reactivity against brain tissue PCO2 (Xie et al. 2006), these experiments were conducted at rest (Fencl, 1986; Peebles et al. 2007). Because exercise would modify the relationship between Pa,CO2 and internal jugular vein PCO2 as an index of brain tissue PCO2, we decided not to ‘correct’ ventilatory reactivity against brain tissue PCO2. Another potential limitation of estimating MCA V using transcranial Doppler ultrasonography is that changes in the diameter of the isonated vessels could modulate MCA V independently of flow. However, the MCA diameter appears to remain relatively constant in humans under several conditions (Giller et al. 1993; Schreiber et al. 2000; Serrador et al. 2000). In addition, the changes in MCA Vmean during submaximal dynamic exercise appear to be similar to the changes in CBF determined by other techniques, i.e. internal carotid artery blood flow (Hellström et al. 1996) and the 133Xe clearance technique (Jørgensen et al. 1992a,b). It should be noted, however, that the functional anatomy of the arteries supplying the brain varies between individuals. In addition, the proportion of total flow in any single vessel may not be constant, and flow redistribution between major cerebral vessels may occur. Therefore, accurate measurement of global CBF cannot be assured unless simultaneous flow in all major vessels is measured. Third, the change in MCA Vmean in relation to a ‘central controller’versus a ‘peripheral plant’ could not be evaluated in the model of the present study. Each mechanism was identified separately at rest and during exercise. Thus, its relationship remains unclear at rest and during exercise and further studies incorporating linear and non-linear models are needed to provide further insight.
are increased (Jones et al. 1979). Peebles et al. (2007) demonstrated that cerebrovascular CO2 reactivity is underestimated by PET,CO2 when compared with Pa,CO2; therefore, we have made the calculations of central controller, peripheral plant and cerebral CO2 reactivity using Pa,CO2. Another important consideration is that PCO2 from the internal jugular vein is likely to be a closer index of brain tissue PCO2 than Pa,CO2 (Xie et al. 2006) and the medullary central chemoreceptors are not stimulated directly by Pa,CO2; rather, they are stimulated by [H+] via alterations in brain tissue CO2 tension (Peebles et al. 2007). Whilst studies have ‘corrected’ ventilatory reactivity against brain tissue PCO2 (Xie et al. 2006), these experiments were conducted at rest (Fencl, 1986; Peebles et al. 2007). Because exercise would modify the relationship between Pa,CO2 and internal jugular vein PCO2 as an index of brain tissue PCO2, we decided not to ‘correct’ ventilatory reactivity against brain tissue PCO2. Another potential limitation of estimating MCA V using transcranial Doppler ultrasonography is that changes in the diameter of the isonated vessels could modulate MCA V independently of flow. However, the MCA diameter appears to remain relatively constant in humans under several conditions (Giller et al. 1993; Schreiber et al. 2000; Serrador et al. 2000). In addition, the changes in MCA Vmean during submaximal dynamic exercise appear to be similar to the changes in CBF determined by other techniques, i.e. internal carotid artery blood flow (Hellström et al. 1996) and the 133Xe clearance technique (Jørgensen et al. 1992a,b). It should be noted, however, that the functional anatomy of the arteries supplying the brain varies between individuals. In addition, the proportion of total flow in any single vessel may not be constant, and flow redistribution between major cerebral vessels may occur. Therefore, accurate measurement of global CBF cannot be assured unless simultaneous flow in all major vessels is measured. Third, the change in MCA Vmean in relation to a ‘central controller’versus a ‘peripheral plant’ could not be evaluated in the model of the present study. Each mechanism was identified separately at rest and during exercise. Thus, its relationship remains unclear at rest and during exercise and further studies incorporating linear and non-linear models are needed to provide further insight.
Acknowledgments
The authors appreciate the time and effort expended by all the volunteer subjects. This study was supported in part by a Grant-in-Aid for Scientific Research (No. 19500574) from the Japanese Ministry of Education, Culture, Sports, Science and Technology, a grant from the Descente and Ishimoto Memorial Foundation for the Promotion of Sports Science and a Grant from the Kouzuki Foundation for sports and education.
References
- Aaslid R, Lindegaard KF, Sorteberg W, Nornes H. Cerebral autoregulation dynamics in humans. Stroke. 1989;20:45–52. doi: 10.1161/01.str.20.1.45. [DOI] [PubMed] [Google Scholar]
- Ainslie PN, Ashmead JC, Ide K, Morgan BJ, Poulin MJ. Differential responses to CO2 and sympathetic stimulation in the cerebral and femoral circulations in humans. J Physiol. 2005;566:613–624. doi: 10.1113/jphysiol.2005.087320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ainslie PN, Murrell C, Peebles K, Swart M, Skinner MA, Williams MJ, Taylor RD. Early morning impairment in cerebral autoregulation and cerebrovascular CO2 reactivity in healthy humans: relation to endothelial function. Exp Physiol. 2007;92:769–777. doi: 10.1113/expphysiol.2006.036814. [DOI] [PubMed] [Google Scholar]
- Asmussen E, Nielsen M. Ventilatory response to CO2 during work at normal and at low oxygen tensions. Acta Physiol Scand. 1957;39:27–35. doi: 10.1111/j.1748-1716.1957.tb01406.x. [DOI] [PubMed] [Google Scholar]
- Asmussen E, Nielsen M. Pulmonary ventilation and effect of oxygen breathing in heavy exercise. Acta Physiol Scand. 1958;43:365–378. doi: 10.1111/j.1748-1716.1958.tb01601.x. [DOI] [PubMed] [Google Scholar]
- Band DM, Wolff CB, Ward J, Cochrane GM, Prior J. Respiratory oscillations in arterial carbon dioxide tension as a control signal in exercise. Nature. 1980;283:84–85. doi: 10.1038/283084a0. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya NK, Cunningham DJ, Goode RC, Howson MG, Lloyd BB. The effects of hypoxia and light exercise on the respiratory response to CO2 in man. J Physiol. 1968;194:14P–16P. [PubMed] [Google Scholar]
- Brys M, Brown CM, Marthol H, Franta R, Hilz MJ. Dynamic cerebral autoregulation remains stable during physical challenge in healthy persons. Am J Physiol Heart Circ Physiol. 2003;285:H1048–H1054. doi: 10.1152/ajpheart.00062.2003. [DOI] [PubMed] [Google Scholar]
- Chapman RW, Santiago TV, Edelman NH. Effects of graded reduction of brain blood flow on chemical control of breathing. J Appl Physiol. 1979;47:1289–1294. doi: 10.1152/jappl.1979.47.6.1289. [DOI] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Clark JM, Sinclair RD, Lenox JB. Chemical and nonchemical components of ventilation during hypercapnic exercise in man. J Appl Physiol. 1980;48:1065–1076. doi: 10.1152/jappl.1980.48.6.1065. [DOI] [PubMed] [Google Scholar]
- Crawford RD, Severinghaus JW. CSF pH and ventilatory acclimatization to altitude. J Appl Physiol. 1978;45:275–283. doi: 10.1152/jappl.1978.45.2.275. [DOI] [PubMed] [Google Scholar]
- Cunningham DJ. Studies on arterial chemoreceptors in man. J Physiol. 1987;384:1–26. doi: 10.1113/jphysiol.1987.sp016440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham DJ, Hey EN, Patrick JM, Lloyd BB. The effect of noradrenaline infusion on the relation between pulmonary ventilation and the alveolar PO2 and PCO2 in man. Ann N Y Acad Sci. 1963;109:756–771. doi: 10.1111/j.1749-6632.1963.tb13504.x. [DOI] [PubMed] [Google Scholar]
- Dempsey JA. Crossing the apnoeic threshold: causes and consequences. Exp Physiol. 2005;90:13–24. doi: 10.1113/expphysiol.2004.028985. [DOI] [PubMed] [Google Scholar]
- Dempsey JA, Romer L, Rodman J, Miller J, Smith C. Consequences of exercise-induced respiratory muscle work. Respir Physiol Neurobiol. 2006;151:242–250. doi: 10.1016/j.resp.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Edvinsson L. Neurogenic mechanisms in the cerebrovascular bed. Acta Physiol Scand Suppl. 1975;427:1–35. Autonomic nerves, amine receptors and their effects on cerebral blood flow. [PubMed] [Google Scholar]
- Ellingsen I, Liestol K, Sydnes G, Hauge A, Nicolaysen G. Arterial PCO2 and lung ventilation in man exposed to 1–5% CO2 in the inspired gas. Acta Physiol Scand. 1987a;129:269–276. doi: 10.1111/j.1748-1716.1987.tb08069.x. [DOI] [PubMed] [Google Scholar]
- Ellingsen I, Sydnes G, Hauge A, Zwart JA, Liestol K, Nicolaysen G. CO2 sensitivity in humans breathing 1 or 2% CO2 in air. Acta Physiol Scand. 1987b;129:195–202. doi: 10.1111/j.1748-1716.1987.tb08059.x. [DOI] [PubMed] [Google Scholar]
- Fencl V. Acid-base balance in cerebral fluids. In: Fishman AP, Cherniack NS, Widdicombe JG, Geiger SR, editors. Handbook of Physiology, section 3, The Respiratory System, vol. 2, Control of Breathing. Bethesda, MD, USA: American Physiology Society; 1986. pp. 115–140. [Google Scholar]
- Giller CA, Bowman G, Dyer H, Mootz L, Krippner W. Cerebral arterial diameters during changes in blood pressure and carbon dioxide during craniotomy. Neurosurgery. 1993;32:737–741. discussion 741–742. [PubMed] [Google Scholar]
- Hellström G, Fischer-Colbrie W, Wahlgren NG, Jogestrand T. Carotid artery blood flow and middle cerebral artery blood flow velocity during physical exercise. J Appl Physiol. 1996;81:413–418. doi: 10.1152/jappl.1996.81.1.413. [DOI] [PubMed] [Google Scholar]
- Honda Y, Hayashi F, Yoshida A, Ohyabu Y, Nishibayashi Y, Kimura H. Overall ‘gain’ of the respiratory control system in normoxic humans awake and asleep. J Appl Physiol. 1983;55:1530–1535. doi: 10.1152/jappl.1983.55.5.1530. [DOI] [PubMed] [Google Scholar]
- Jones NL, Robertson DG, Kane JW. Difference between end-tidal and arterial PCO2 in exercise. J Appl Physiol. 1979;47:954–960. doi: 10.1152/jappl.1979.47.5.954. [DOI] [PubMed] [Google Scholar]
- Jordan J, Shannon JR, Diedrich A, Black B, Costa F, Robertson D, Biaggioni I. Interaction of carbon dioxide and sympathetic nervous system activity in the regulation of cerebral perfusion in humans. Hypertension. 2000;36:383–388. doi: 10.1161/01.hyp.36.3.383. [DOI] [PubMed] [Google Scholar]
- Jørgensen LG, Perko M, Hanel B, Schroeder TV, Secher NH. Middle cerebral artery flow velocity and blood flow during exercise and muscle ischemia in humans. J Appl Physiol. 1992a;72:1123–1132. doi: 10.1152/jappl.1992.72.3.1123. [DOI] [PubMed] [Google Scholar]
- Jørgensen LG, Perko G, Secher NH. Regional cerebral artery mean flow velocity and blood flow during dynamic exercise in humans. J Appl Physiol. 1992b;73:1825–1830. doi: 10.1152/jappl.1992.73.5.1825. [DOI] [PubMed] [Google Scholar]
- LeMarbre G, Stauber S, Khayat RN, Puleo DS, Skatrud JB, Morgan BJ. Baroreflex-induced sympathetic activation does not alter cerebrovascular CO2 responsiveness in humans. J Physiol. 2003;551:609–616. doi: 10.1113/jphysiol.2003.046987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linton RA, Lim M, Wolff CB, Wilmshurst P, Band DM. Arterial plasma potassium measured continuously during exercise in man. Clin Sci (Lond) 1984;67:427–431. doi: 10.1042/cs0670427. [DOI] [PubMed] [Google Scholar]
- Markwalder TM, Grolimund P, Seiler RW, Roth F, Aaslid R. Dependency of blood flow velocity in the middle cerebral artery on end-tidal carbon dioxide partial pressure – a transcranial ultrasound Doppler study. J Cereb Blood Flow Metab. 1984;4:368–372. doi: 10.1038/jcbfm.1984.54. [DOI] [PubMed] [Google Scholar]
- Meadows GE, Dunroy HM, Morrell MJ, Corfield DR. Hypercapnic cerebral vascular reactivity is decreased, in humans, during sleep compared with wakefulness. J Appl Physiol. 2003;94:2197–2202. doi: 10.1152/japplphysiol.00606.2002. [DOI] [PubMed] [Google Scholar]
- Miyamoto T, Inagaki M, Takaki H, Kawada T, Yanagiya Y, Sugimachi M, Sunagawa K. Integrated characterization of the human chemoreflex system controlling ventilation, using an equilibrium diagram. Eur J Appl Physiol. 2004;93:340–346. doi: 10.1007/s00421-004-1219-x. [DOI] [PubMed] [Google Scholar]
- Mohan R, Duffin J. The effect of hypoxia on the ventilatory response to carbon dioxide in man. Respir Physiol. 1997;108:101–115. doi: 10.1016/s0034-5687(97)00024-8. [DOI] [PubMed] [Google Scholar]
- Nelson E, Rennels M. Innervation of intracranial arteries. Brain. 1970;93:475–490. doi: 10.1093/brain/93.3.475. [DOI] [PubMed] [Google Scholar]
- Nielsen KC, Owman C. Adrenergic innervation of pial arteries related to the circle of Willis in the cat. Brain Res. 1967;6:773–776. doi: 10.1016/0006-8993(67)90134-5. [DOI] [PubMed] [Google Scholar]
- Ogoh S, Brothers RM, Barnes Q, Eubank WL, Hawkins MN, Purkayastha S, O-Yurvati A, Raven PB. The effect of changes in cardiac output on middle cerebral artery mean blood velocity at rest and during exercise. J Physiol. 2005a;569:697–704. doi: 10.1113/jphysiol.2005.095836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogoh S, Dalsgaard MK, Secher NH, Raven PB. Dynamic blood pressure control and middle cerebral artery mean blood velocity variability at rest and during exercise in humans. Acta Physiol (Oxf) 2007;191:3–14. doi: 10.1111/j.1748-1716.2007.01708.x. [DOI] [PubMed] [Google Scholar]
- Ogoh S, Fadel PJ, Zhang R, Selmer C, Jans O, Secher NH, Raven PB. Middle cerebral artery flow velocity and pulse pressure during dynamic exercise in humans. Am J Physiol Heart Circ Physiol. 2005b;288:H1526–H1531. doi: 10.1152/ajpheart.00979.2004. [DOI] [PubMed] [Google Scholar]
- Ohyabu Y, Yoshida A, Hayashi F, Honda Y. Ventilatory response to CO2 after brief stimulations of the peripheral chemoreceptors in man. Jpn J Physiol. 1982;32:627–636. doi: 10.2170/jjphysiol.32.627. [DOI] [PubMed] [Google Scholar]
- Peebles K, Celi L, McGrattan K, Murrell C, Thomas K, Ainslie PN. Human cerebrovascular and ventilatory CO2 reactivity to end-tidal, arterial and internal jugular vein PCO2. J Physiol. 2007;584:347–357. doi: 10.1113/jphysiol.2007.137075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pianosi P, Grondin D, Desmond K, Coates AL, Aranda JV. Effect of caffeine on the ventilatory response to inhaled carbon dioxide. Respir Physiol. 1994;95:311–320. doi: 10.1016/0034-5687(94)90093-0. [DOI] [PubMed] [Google Scholar]
- Poon CS. Effects of inspiratory elastic load on respiratory control in hypercapnia and exercise. J Appl Physiol. 1989a;66:2400–2406. doi: 10.1152/jappl.1989.66.5.2400. [DOI] [PubMed] [Google Scholar]
- Poon CS. Effects of inspiratory resistive load on respiratory control in hypercapnia and exercise. J Appl Physiol. 1989b;66:2391–2399. doi: 10.1152/jappl.1989.66.5.2391. [DOI] [PubMed] [Google Scholar]
- Poon CS, Greene JG. Control of exercise hyperpnea during hypercapnia in humans. J Appl Physiol. 1985;59:792–797. doi: 10.1152/jappl.1985.59.3.792. [DOI] [PubMed] [Google Scholar]
- Poon CS, Tin C, Yu Y. Homeostasis of exercise hyperpnea and optimal sensorimotor integration: the internal model paradigm. Respir Physiol Neurobiol. 2007;159:1–13. doi: 10.1016/j.resp.2007.02.020. discussion 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qayyum MS, Barlow CW, O'Connor DF, Paterson DJ, Robbins PA. Effect of raised potassium on ventilation in euoxia, hypoxia and hyperoxia at rest and during light exercise in man. J Physiol. 1994;476:365–372. doi: 10.1113/jphysiol.1994.sp020138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen P, Stie H, Nielsen B, Nybo L. Enhanced cerebral CO2 reactivity during strenuous exercise in man. Eur J Appl Physiol. 2006;96:299–304. doi: 10.1007/s00421-005-0079-3. [DOI] [PubMed] [Google Scholar]
- Robbins PA. Evidence for interaction between the contributions to ventilation from the central and peripheral chemoreceptors in man. J Physiol. 1988;401:503–518. doi: 10.1113/jphysiol.1988.sp017175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SJ, Gottschalk S, Weih M, Villringer A, Valdueza JM. Assessment of blood flow velocity and diameter of the middle cerebral artery during the acetazolamide provocation test by use of transcranial Doppler sonography and MR imaging. AJNR Am J Neuroradiol. 2000;21:1207–1211. [PMC free article] [PubMed] [Google Scholar]
- Serrador JM, Picot PA, Rutt BK, Shoemaker JK, Bondar RL. MRI measures of middle cerebral artery diameter in conscious humans during simulated orthostasis. Stroke. 2000;31:1672–1678. doi: 10.1161/01.str.31.7.1672. [DOI] [PubMed] [Google Scholar]
- Severinghaus JW, Carcelen A. Cerebrospinal fluid in man native to high altitude. J Appl Physiol. 1964;19:319–321. doi: 10.1152/jappl.1964.19.2.319. [DOI] [PubMed] [Google Scholar]
- Severinghaus JW, Mitchell RA, Richardson BW, Singer MM. Respiratory control at high altitude suggesting active transport regulation of CSF pH. J Appl Physiol. 1963;18:1155–1166. doi: 10.1152/jappl.1963.18.6.1155. [DOI] [PubMed] [Google Scholar]
- Teppema L, Sarton E, Dahan A, Olievier CN. The neuronal nitric oxide synthase inhibitor 7-nitroindazole (7-NI) and morphine act independently on the control of breathing. Br J Anaesth. 2000;84:190–196. doi: 10.1093/oxfordjournals.bja.a013402. [DOI] [PubMed] [Google Scholar]
- Toda N, Okamura T. Cerebral vasodilators. Jpn J Pharmacol. 1998;76:349–367. doi: 10.1254/jjp.76.349. [DOI] [PubMed] [Google Scholar]
- Waldrop TG, Iwamoto GA. Point: supraspinal locomotor centers do contribute significantly to the hyperpnea of dynamic exercise. J Appl Physiol. 2006;100:1077–1079. doi: 10.1152/japplphysiol.01528.2005. [DOI] [PubMed] [Google Scholar]
- Ward SA. Muscle-energetic and cardio-pulmonary determinants of exercise tolerance in humans: Muscle-energetic and cardio-pulmonary determinants of exercise tolerance in humans. Exp Physiol. 2007;92:321–322. doi: 10.1113/expphysiol.2006.034389. [DOI] [PubMed] [Google Scholar]
- Wasserman K, Whipp BJ, Koyal SN, Cleary MG. Effect of carotid body resection on ventilatory and acid-base control during exercise. J Appl Physiol. 1975;39:354–358. doi: 10.1152/jappl.1975.39.3.354. [DOI] [PubMed] [Google Scholar]
- Wasserman K, Zhang YY, Gitt A, Belardinelli R, Koike A, Lubarsky L, Agostoni PG. Lung function and exercise gas exchange in chronic heart failure. Circulation. 1997;96:2221–2227. doi: 10.1161/01.cir.96.7.2221. [DOI] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Khayat R, Dempsey JA, Morgan B, Russell D. Cerebrovascular response to carbon dioxide in patients with congestive heart failure. Am J Respir Crit Care Med. 2005;172:371–378. doi: 10.1164/rccm.200406-807OC. [DOI] [PubMed] [Google Scholar]
- Xie A, Skatrud JB, Morgan B, Chenuel B, Khayat R, Reichmuth K, Lin J, Dempsey JA. Influence of cerebrovascular function on the hypercapnic ventilatory response in healthy humans. J Physiol. 2006;577:319–329. doi: 10.1113/jphysiol.2006.110627. [DOI] [PMC free article] [PubMed] [Google Scholar]