

Abstract
We present GERMLINE, a robust algorithm for identifying segmental sharing indicative of recent common ancestry between pairs of individuals. Unlike methods with comparable objectives, GERMLINE scales linearly with the number of samples, enabling analysis of whole-genome data in large cohorts. Our approach is based on a dictionary of haplotypes that is used to efficiently discover short exact matches between individuals. We then expand these matches using dynamic programming to identify long, nearly identical segmental sharing that is indicative of relatedness. We use GERMLINE to comprehensively survey hidden relatedness both in the HapMap as well as in a densely typed island population of 3000 individuals. We verify that GERMLINE is in concordance with other methods when they can process the data, and also facilitates analysis of larger scale studies. We bolster these results by demonstrating novel applications of precise analysis of hidden relatedness for (1) identification and resolution of phasing errors and (2) exposing polymorphic deletions that are otherwise challenging to detect. This finding is supported by concordance of detected deletions with other evidence from independent databases and statistical analyses of fluorescence intensity not used by GERMLINE.
Recently, major advances in mapping complex traits have been made possible by genome-wide association studies in cohorts of purported unrelated individuals. The investigation of such large-scale genotype data brings renewed interest and potentially novel insights to an old question: Are individuals really unrelated and to what extent? While every pair of individuals is descended from the same person if you follow their genealogy infinitely into the past, we are particularly interested in recent common ancestry, occurring during the last few centuries. More specifically, we focus on relatives in the genetic sense, who not only share a recent progenitor, but also co-inherit some of that ancestor's genome. This portion of the genome is therefore identical by descent (IBD) in one haploid copy shared by contemporary progeny.
IBD between two individuals have been classically quantified (Malécot 1948) following Wright's inbreeding coefficient (Wright 1921), which prescribes probabilities of these individuals sharing two, one, or zero alleles by descent, averaged across the genome. Yet, such probabilities oversimplify coinheritance from a common ancestor: Even if the average chance of allele copies to be IBD between relatives is low, whenever an allele is co-inherited, a very long genomic segment around it is also likely to be shared. Formally, a pair of diploid kth generation descendents is IBD at a particular locus if the ancestral haplotype is copied and transmitted across 2k meioses. Each meiosis having a 0.5 probability of transmitting a copy, the probability of such a copy being inherited down both lineages is 21 − 2k—<1% for any k ≥ 4 (third half-cousins or less related). Despite being a low probability event, when it occurs, such sharing would imply a very long segment to be nearly identical across the two samples. Using the same example, segment length is expected to be d/2k, where d is the genetic distance, defined as 100 centimorgans (cM). This rate of change in IBD status along a pair of genomes facilitates computing the expected number of IBD segments genome-wide. Table 1 illustrates these statistics for a pair of individuals k generations apart. The practical implication is that even if IBD is rare, when it is present at a segment, the alleles it carries will provide unequivocal evidence for relatedness.
Table 1.
IBD between individuals of shared ancestry from a single source

aHuman genome length taken from Kong et al. (2004).
Modern association studies, with individuals by the thousands and marker counts running into the millions, bring forth fresh incentives for quantifying relatedness, alongside new opportunities and technical challenges. In terms of motivation, such studies impel kinship analysis as they hinge on the premise of independent, identically distributed observations of unrelated individuals sampled from the population, an assumption that is violated among related individuals whose alleles along some haploid segments of the genome are IBD. Related individuals bias not only association statistics in the regions of the genome shared between them, but may also taint analysis of population structure that affects results for the entire genome.
The magnitude of current data presents the opportunity to examine shared, inherited segments with more information and finer marker resolution for defining their boundaries (Hinrichs et al. 2005). Indeed, renewed interest in IBD (Thompson 2008) has led to the development of novel methods to quantify relatedness, by either genome-wide estimates (Mao and Xu 2005), segment-by-segment analysis (Hill and Hernandez-Sanchez 2007; Hill and Weir 2007), or both (Purcell et al. 2007). Applying such methods, the recently published Human Haplotype Map (The International HapMap Consortium 2005) reported the surprising discovery of abundant long haplotype segments shared between individuals purported to be unrelated (Frazer et al. 2007) coining them as hidden relatives.
Looking ahead, genotyped sets of individuals 100-fold larger than the HapMap represent more complete sampling of the general population than previously available, facilitating many more opportunities for any founder of the population to be observed as a co-ancestor of individuals in the cohort. In particular, a recent study of IBD among 35,000 Icelanders has demonstrated the relevance of using hidden relatedness in haplotype inference and rare variant discovery (Kong et al. 2008). Such research motivates analysis of IBD in large cohorts. However, as available sample size increases, previous methods cannot keep up with the current torrent of genotype data. Specifically, methods based on examining each pair of individuals require quadratic time (Purcell et al. 2007) and rapidly become impractical.
This study introduces an algorithm for linear-time discovery of segmental sharing and the corresponding implementation: GERMLINE (genetic error-tolerant regional matching with linear-time extension). Inspired by the quintessential matching algorithms for noisily homologous sequences (Altschul et al. 1990; Kent 2002) GERMLINE is based on a two-stage process: First, GERMLINE detects completely identical match-seeds of potentially shared segments by creating a dictionary (Ayers et al. 2006) of allele combination words across the population observed at different slices along the genome. The second stage involves extending these candidate matches to resolve likelihood of IBD by a dynamic programming algorithm along different slices. We developed the method for use with phased genotype data.
The study is structured as follows. We first present the GERMLINE analysis results in several varied populations; we analyze GERMLINE's efficiency and accuracy as compared with state-of-the-art segmental sharing applications in simulated and real data; we then explore the performance of GERMLINE in a large and unphased population; finally, we detail a novel application of IBD segments to identifying phasing error and structural variation. The algorithm is presented in detail in the Methods section: first, we introduce the hashing approach to identifying whole haplotype segment sharing; then, we extend this algorithm to analyze smaller slices and merge partially matching contiguous slices into long shared segments.
Results
Comparison with other methods
To determine the effectiveness of haplotype word matching in phase-known data, GERMLINE was used to identify IBD in the HapMap Phase II phased release. We compare the results with those of the PLINK whole-genome data analysis toolset (Purcell et al. 2007; S. Purcell, PLINK 1.00, http://pngu.mgh.harvard.edu/purcell/plink/), which can detect extended IBD with the “segmental sharing” runtime option. PLINK uses a hidden Markov Model (HMM) approach to estimate multipoint probabilities of IBD in pairs of individuals based on identity by state (IBS) sharing. We used both real and simulated data to compare accuracy and efficiency of the two algorithms.
We simulated instances of pairwise segmental sharing of varying length, planted on a background of unrelated samples, with realistic genotyping error. The average accuracy rates from both algorithms are presented in Table 2 as evaluated by three figures of merit: (1) Sensitivity—the percentage of the simulated IBD fragment that was detected (in single nucleotide polymorphisms [SNPs]); (2) False positive extension—the fraction of non-IBD markers flanking a true IBD segment that were falsely detected to be IBD, as a percentage of the true shared segment length; (3) False positive (nonflanking)—the remaining number of falsely detected non-IBD markers as a percentage of the true shared segment length. In all instances, GERMLINE has both a higher sensitivity than PLINK and a lower overall false-positive rate. Furthermore, because PLINK discovery depends significantly on allele frequencies in the cohort analyzed (randomly sampled, in this case), a number of the simulations lead to false discovery of completely nonflanking IBD; GERMLINE did not exhibit this behavior.
Table 2.
Sharing concordance between GERMLINE and PLINK (simulated data)

aIBD segments were planted onto the background genotype data of Kosrae individuals that are otherwise unrelated according to both methods, with the entire population as the cohort analyzed. A total of 25 pairs of parents were randomly selected from different trios without any 4-Mb-long IBD segment detectable by either of the compared methods. A total of 10 random regions of varying lengths were copied in turn from the untransmitted chromosome 15 haplotype of one parent to its counterpart with 1% simulated difference due to genotyping error. For PLINK, IBD detection was attempted within an analyzed cohort of 50 individuals—required for estimation of allele frequencies. For GERMLINE, no such cohort was needed.
bThe set of potential pairs of individuals who are unrelated, i.e., do not share a 2.5-cM segment, was too small for this analysis.
Table 3 shows runtime results for GERMLINE as well as the two comparison implementations using the HapMap cohorts (see Methods, Implementation section). We calculated two sets of results for the segmental sharing option: a default under which every available SNP was processed, and a pruned set that excluded highly linked SNPs as detailed in the PLINK documentation (S. Purcell, PLINK 1.00, http://pngu.mgh.harvard.edu/purcell/plink/). Analyzing all SNPs, GERMLINE runs more than 100-fold faster than PLINK. When PLINK is executed on each of the pruned datasets, consisting of 2.3%–4.9% of their respective originals, its run time (pruning time not included) was still slower by a factor of from 3.46 to 10.55.
Table 3.
Runtime comparison between GERMLINE and PLINK on HapMap cohorts

Minimum length of segments to be detected set to 1 Mb.
Examining discovery of shared segments longer than 2.5cM in these HapMap cohorts, Table 4 details the differences in segments found between the two algorithms. We observe high concordance between the methods for long segments, where in populations where trip data is available, the fraction of PLINK-identified segments not reported by GERMLINE is consistent with false-positive rate of HMM analysis. In particular, the three pairs of YRI individuals previously identified as closely related can serve as positive controls, and all displayed >90% concordance between methods. They represent only a small part of the detected set of related pairs: Most of the shared segments involve pairs of remotely related individuals. Without trio data, it appears GERMLINE has only limited power to detect IBD. For segments shorter than 5 cM we observed significantly more segments reported by GERMLINE: We further compare average statistics of detected segments to the results reported by the HapMap Project (Frazer et al. 2007), mirroring the parameters of their analysis by seeking segments over 1 Mb in length with at least 50 SNPs. Table 5 shows that in all populations, GERMLINE identified significantly more segments and maintained a higher total distance spanned at a near perfect IBS rate. This suggests the enrichment of reported segments by GERMLINE to reflect increased sensitivity rather than additional false positives. Furthermore, sensitivity to shorter IBD segments facilitates detection of breaks in IBD matches—breaks that have real biological meaning, as explained below. These effects further reduce the mean and maximum length of detected segments, while increasing accuracy.
Table 4.
Sharing concordance between GERMLINE and PLINK (HapMap cohorts)

Fraction of total PLINK-identified sharing also detected by GERMLINE.
Table 5.
Shared segment discovery in HapMap

aAverage across all shared segment pairs. Not available for HapMap relatedness study.
Application to whole-genome, whole-population data
We used GERMLINE to detect IBD in SNP array data from 3000 individuals, essentially the entire adult population of the Island of Kosrae, Micronesia (see Methods for dataset description). We first phased the entire population using the Beagle localized haplotype clustering tool (Browning and Browning 2007). We then applied GERMLINE to scan for shared segments over 10 cM in length according to a consensus database (Duffy 2006) of standard genetic maps (Lien et al. 2000; Kong et al. 2004).
The available pedigree for the Kosrae samples brings forth multiple pairs of related individuals as positive controls. Simplified to a single summary statistic, the overall fraction of the genome shared by a related pair, their analysis (Fig. 1) provides additional validation for the GERMLINE method, agreeing with theoretical expectation (Table 1) for relatives up to four meioses apart. Distant relationships, however, show more sharing than expected, suggesting a background of hidden relatedness in these descendants of a small, isolated ancestral population.
Figure 1.

Expected and detected genome-wide sharing (Kosrae Cohort). (Blue, ▲) Expected genome-wide sharing; (green, ■) detected genome-wide sharing.
We further demonstrate the utility of segmental IBD analysis beyond the single statistic of genome-wide sharing. To this end, we compare and resolve relationships that are indiscriminate using genome-wide averages alone. Specifically, we juxtaposed pairs of individuals that are half-siblings (one shared parent) versus those that are related through a complete avuncular relationship. These two relationships are expected to have the same amount of overall sharing statistics in terms of both 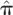 , the genome-wide proportion of IBD, as well as Z1, the overall probability of sharing one allele by IBD. In contrast, avuncular and half-sib pairs are expected to differ in segment length distribution. In accordance with expectation, Figure 2 presents a significant difference between such pairs of Kosraens in the average shared segment length, as identified by GERMLINE. No such difference is observed when comparing and Z1. With these measures alone, one cannot distinguish between avuncular and half-sib individuals, whereas shared segment analysis facilitates resolving these relationships by average segment length as a classifier.
, the genome-wide proportion of IBD, as well as Z1, the overall probability of sharing one allele by IBD. In contrast, avuncular and half-sib pairs are expected to differ in segment length distribution. In accordance with expectation, Figure 2 presents a significant difference between such pairs of Kosraens in the average shared segment length, as identified by GERMLINE. No such difference is observed when comparing and Z1. With these measures alone, one cannot distinguish between avuncular and half-sib individuals, whereas shared segment analysis facilitates resolving these relationships by average segment length as a classifier.
Figure 2.

PLINK metrics and share length for equally related pairs. Comparison of PLINK and Z1 values with GERMLINE share length for individuals of equal relationship coefficients. (Left) PLINK values; (middle) PLINK Z1 values; (right) GERMLINE share length (cM). Error bars, 99% CI.
We note that in our attempt to confirm these results on segmental sharing with the PLINK algorithm, it was only able to identify whole-chromosome sharing. With PLINK's focus at less-related cohorts, this tool may need specific tuning for resolving relatedness in the inbred Kosraean data set. Computationally, PLINK required 556 h to complete analysis of the eight shortest chromosomes, while GERMLINE processed the same data in 40.3 h (30.8 for Beagle phasing and 9.5 for GERMLINE analysis).
Segmental gaps
In identifying IBD on the HapMap data, we discovered a number of long shared segments, which were broken up by short regions (generally <100 SNPs) that contained unusually low IBS, referred to as “gaps.” We suspected these gaps to be indicative of phasing errors or structural variation. We demonstrate that, indeed, IBD gaps come in these two flavors, as manifested by their allelic makeup.
Each individual in the HapMap phased set is considered to have one transmitted (T) and one untransmitted (U) haplotype relative to its child. In regions of high heterozygosity, we identified gaps in which putative IBD switches in an individual from one haplotype to the other and then back at the end of the gap. This pattern may be explained by phasing inconsistency, which results in incorrectly oriented haplotypes at heterozygous sites, commonly referred to as “switch error” (Lin et al. 2002; Marchini et al. 2006). Table 6 provides an example of three contiguous segments on chromosome 18 between individuals NA06993 and NA07056 (CEU population) with IBS measurements taken for the two pairs of notable haplotypes. In the first region, the two individuals are in nearly complete IBS along their respective T haplotypes. In the subsequent 42 SNP gap, none of the 16 heterozygous positions continue the shared segment; rather, they match the T haplotype of NA06993 with the U haplotype of NA07056. In the remainder of the shared segment, the IBS switches back to the two transmitted chromosomes. This IBS switch back and forth is consistent with two closely spaced recombination sites during the NA07056 meiosis. However, the genotype data for the trio involving NA06993 reveals all three samples to be heterozygous at these 16 positions, implying that the phasing is completely computational and not constrained by Mendelian relationships; the lack of direct information makes such regions particularly prone to short phasing errors. Searching the two HapMap cohorts with known trio data for gaps in which IBD mismatches were contained to heterozygous sites and phasing was not based on familial information, we identified 58 such regions.
Table 6.
Potential phasing irregularity in HapMap

aSamples NA06993 and NA07056—CEU population.
bIdentity by state measured across heterozygous sites.
T, Transmitted; U, untransmitted.
An even more interesting class of gaps is characterized by regions of unusually low heterozygosity, suggestive of structural variation. A region that exhibits loss of heterozygosity may, in fact, represent incorrectly typed hemizygosity resulting from a segmental deletion along the otherwise shared haplotype. Such regions would also feature lower identity rates than expected by IBD, because SNP matching is effectively being counted on the haplotypes alternate to those that are actually IBD. We searched through the HapMap samples for gaps in long segmental sharing, which exhibited this characteristic of loss of heterozygosity as well as a high rate of IBS mismatches. Table 7 documents an example of two such regions between pairs of individuals in the CEU population. In the first region (NA12264 and NA12155), two shared segments >6000 SNPs in length straddle a 44-SNP gap that exhibits loss of heterozygosity and a decrease in IBS. Similarly, the second region (NA12717 and NA11840) contains two shared segments of >5000 SNPs in length straddling a 14-SNP gap that exhibits loss of heterozygosity and nearly complete lack of identity. The large size of shared segments essentially guarantees these regions to be IBD. This assumption, coupled with significant loss of heterozygosity in only one individual of the pair suggests that such gaps are de novo segmental deletions.
Table 7.
Potential deletion regions in HapMap

aCEU sample NA12264.
bCEU sample NA12155.
cAverage across both involved samples.
dCEU sample NA12717.
eCEU sample NA11840.
To validate these supposed deletions, we searched for overlapping deletion regions in the Database of Genomic Variants (Iafrate et al. 2004), a shared database of structural variation identified in a number of studies, including experimental examination of structural variants in the HapMap cell lines (Redon et al. 2006). Because identification of such gaps by GERMLINE is explicitly dependent on the presence of IBD, we targeted reported gaps in HapMap that overlapped with IBD regions identified by GERMLINE. Table 8 shows these results, split up by the validation source: experimental (Perry et al. 2008) and computational (Pinto et al. 2007; Wang et al. 2007). The “verified gaps” column shows the number of gaps identified by GERMLINE that overlapped with those in the dbGV reference, while the “false negatives” column shows the number of IBD regions that spanned across a reported deletion in the dbGV. We note that IBD can only detect deletions along the one haplotype that is shared; therefore, power to detect a deletion is bounded by 50%. Overall, of the reported deletions present in an IBD region, 42% were picked out by GERMLINE as gaps.
Table 8.
Verification of gaps in HapMap with deletions in the database of genomic variants

We further explored the discovery of segmental deletions in the Kosraean population data where IBD is more prevalent, and we observe a larger number of gaps. We used a binomial score to rank potential deletions in homozygous gaps based on the number of mismatching SNPs and the rate of mismatch in the flanking shared segments, measured across all shared segments with the suspected gap (see Methods, Gap Likelihood Scoring section). Figure 3 shows an example of such a gap, plotting the normalized fluorescence intensity measures across a 2-Mb region containing the putative deletion. This region clearly coincides with a significant decrease in intensity values, supporting the hypothesis. We attempted to validate the 200 most statistically significant gaps with three means of verification: (1) the Affymetrix Copy Number Analysis Tool (CNAT) (Huang et al. 2004), which processes fluorescence intensity in an HMM-based algorithm to identify blocks of structural variation common to many individuals, (2) examining deviations from the average in normalized fluorescence intensity values, which can help identify deletions that are too short or uncommon for CNAT, and (3) overlap with deletions reported in the Database of Genomic Variants (dbGV). Figure 4 reports validation by these criteria: 21 segments were identified by CNAT, 65 showed significant deviations in intensity (Intensity), and 124 were verified in the dbGV. Focusing specifically on CNAT, we ran a similar concordance analysis to that in Table 8—matching deletions identified by CNAT with GERMLINE IBD regions and gaps and counting unique deletions rather than individual segments. We found that of the CNAT deletions identified in an IBD region, 9% were picked out by GERMLINE as gaps. However, one-third of the SNPs CNAT implicated in deletions did not pass quality control, and 92.7% of the called deletions contained at least one QC-failed SNP, making these deletion calls suspected as false positives, while also hiding these SNPs from GERMLINE analysis of SNPs passing QC.
Figure 3.

Candidate deletion fluorescence intensity. (Black line) Population average; (open circles) individuals identified as having the deletion; (blue bar at bottom) deletion identified by GERMLINE.
Figure 4.

Verified candidate deletion regions (top 200). (dbGV) Identified in Database of Genomic Variants; (CNAT) verified by Affymetrix Copy Number Analysis Tool; (Intensity) verified as deviation from population average intensity.
Discussion
We presented GERMLINE, a method for genome-wide discovery of IBD segments shared within large populations. We introduced a linear-time time algorithm for identifying short identical genomic “slices” between pairs of individuals, and then extending the boundaries of these slices to discover long shared segments representative of IBD. With efficiency in mind, the program is specifically intended for analyzing large and complex datasets.
A potential limitation of such an exact hashing algorithm can be susceptible to false negatives if GERMLINE fails to find an exact match to use as a seed. Such a scenario would occur if mismatches are distributed evenly in every slice across the match. We have taken steps to alleviate this limitation at the cost of decreased efficiency by implementing an optional feature of a sliding overlapping window; however, the accuracy increase was slight (data not shown) and was not used to obtain the results presented in this study. Overall, simulated experiments showed GERMLINE to accurately detect IBD with a realistic error rate, even for segments as short as 5 Mb. Additional limitations of GERMLINE presently include the use of user-defined, constant thresholds for word length and mismatch rate, rather than a model-based approach that may be even more robust.
Real data established the stability and accuracy of GERMLINE in the trio-based populations of the HapMap, alongside advantages in runtime and scalability. In simulated data, GERMLINE was highly accurate across varying lengths of IBD, particularly short segments, and identified few spurious fragments. With real data, the concordance between GERMLINE and another method (PLINK) is reduced for shorted fragments with non-trio data. Nevertheless, overall statistics compared with results of the International HapMap show that GERMLINE consistently finds more segmental sharing while maintaining nearly identical identity by state. In particular, GERMLINE identified a larger number of short, shared segments—a self-described weak point of previous methods (Frazer et al. 2007).
The accuracy and efficiency of GERMLINE on phased data from HapMap motivated IBD analysis of genotype data from the significantly larger and more densely related population of Kosrae, with an exceptional challenge due to extensive inbreeding. Our results mirrored expectations for close relatives, and further resolved relationships with statistically indistinguishable totals of genome-wide sharing by revealing their divergence in the average length of GERMLINE-identified segments.
A novel result of our IBD analysis in various populations was the identification of short “gaps” in long IBD segments. We hypothesized that these gaps were indicative of phasing error or structural variation, and drew support for these conclusions from independent data sets. Specifically, putative phasing errors were consistently found to be unconstrained by Mendelian segregation, thus prone to errors by computational phasing methods. This highlights the potential use of genome-wide, fine IBD structure for phasing, as recently proposed for a per-locus method (Kong et al. 2008). Putative deletions significantly overlapped cataloged deletion variants as well as structural variants discovered in our raw data using analysis of fluorescence intensity. Power to detect deletions using GERMLINE remains limited to sufficiently long structural events within IBD segments whose SNPs pass QC, but enjoys independence from probe-level image data. This original strategy of detecting polymorphic deletions using a GERMLINE-enabled fine-scale map of IBD can therefore complement existing tools for the hotly debated association analysis of microdeletions (Kumar et al. 2008; Weiss et al. 2008).
Recently, IBD has been reported in Icelanders (Kong et al. 2008) based on statistic arguments and pairwise analysis of samples. These features of the report are shared by PLINK's IBD analysis, and are therefore expected to limit both resolution of segment detection and computational efficiency of genome-wide, population-wide scanning for IBD. Our own implementation of this method demonstrated it would take 48 CPU days to process the Kosrae data. Indeed, Kong et al. (2008) report IBD only for a handful of regions, and focus on course analysis of >10 Mb segments. Such work only increases motivation for a tool like GERMLINE, which allows resolving short segments genome-wide, and scales better with population size.
Looking ahead, as genotyping data volume continues to increase, hidden relatedness will become ubiquitous. With GERMLINE, we have overcome the computational barrier of pairwise sample analysis and can now scale the analysis linearly with the sample size. Understanding such shared genomic segments has previously been shown to add statistical power to heritable trait association mapping (Almasy and Blangero 1998; Dodds et al. 2007; Meuwissen and Goddard 2007) as well as gene detection in the presence of pedigree data (Thomas et al. 2008). Furthermore, recent IBD research in a large population has demonstrated its effectiveness for haplotype inference and the tracking of known structural variation in specific regions (Kong et al. 2008). Nevertheless, these methods still require all-pairs analysis and have therefore been significantly restricted to particular regions and conservative rule-based thresholds. The GERMLINE algorithm is a robust framework for identifying sharing in even larger cohorts—precisely where IBD presence has the most significant impact.
Methods
Haplotype IBD matching
We devise a search for IBD that is based on directly matching portions of haplotypes between individual samples. Such a search is naturally simpler in the hypothetical case when shared segments are identical throughout the entirety of the haplotype considered, allele calls are error free, and the phase of input sequences is known. This simple case facilitates direct matching of haplotypes to one another, and we first present GERMLINE, our methodology for efficient IBD detection, in such a demonstrative scenario (see Detection of IBD Along the Entire Haplotype Copy section, below). Subsequently, we introduce realistic complexities of segment-limited matching (see Identical Matching Across Subsets of H section, below) and data errors (see Genotyping Error section, below). While our implementation includes the handling of unphased data, description is omitted for brevity, as results presented in this manuscript use an existing software tool for phasing (Browning and Browning 2007).
Detection of IBD along the entire haplotype copy
We first consider a search for pairs of haplotypes that are identical throughout the input data, a set of observed haplotypes for n individuals and s SNPs along a genomic segment of interest. Formally, the input is a 2n × s matrix H with rows corresponding to haplotypes and columns to SNPs. The output is a set of pairs of identical rows of H. Haplotype calls are represented by a binary alphabet corresponding to the alleles. A matrix entry H[i,j] is 1 if haplotype i carries the minor allele of SNP j, and 0 otherwise. Each row of H can therefore be regarded as a binary vector. When two haplotypes (i,i′) are IBD, the corresponding rows (i,i′) will be identical in H. The goal of the algorithm is therefore to accept a matrix H and output a set L of IBD shared segments (i,i′′). Due to errors and noise that are likely to be present in the input, we distinguish between the observed H and its underlying counterpart, the matrix Hreal of true haplotypes, without errors or missing data. We defer handling of errors to the Genotyping Error section below, and in this section we define the algorithm using H.
We first identify matches across the s SNPs in matrix H by relying on a dictionary of haplotypes: the set D of size no larger than 2n consisting of nonredundant rows from H. D is implemented as a hash-table data structure with constant-time insertion and lookup: The key is a binary vector of length s, and the value is a set of individual haplotypes having identical rows. Once D is constructed, each pair of rows indexed by the same key in D is a match. The set M(H) of all such matches can be obtained by traversing all keys in D, and all pairs of rows per key as in match (Algorithm 1).
 |
Identical matching across subsets of H
When considering a large fraction of a chromosome, true IBD may not span all of the available SNPs. Therefore, in a whole-genome or whole-chromosome context we are interested in detecting partial matches, or pairs of individuals that share a common recent ancestor only along a segment of a chromosome. As such, we establish a defined threshold on the minimum length for an IBD shared segment. The choice of LIBD corresponds to the expected segment length for the most distantly related individuals we aim at detecting (see Table 1). Formally, we define the length L(j,j′) of an interval between columns j and j′ as the genetic distance between the corresponding genes. A pair of haplotypes (i,i′) is defined to be sharing a segment in a SNP region [j .. j′] if their included SNPs are identical and L(j,j′) exceeds LIBD. The problem is now to find, given H, a set of quartets (i,i′,j,j′) such that (i,i′) shares the segment [j .. j′]. We now propose a divide and conquer strategy for using match to discover such shared segments. Our goal is to identify pairs of long identical segments that are shorter than the length of H. We can approximate this by dividing the columns of H into equal vertical intervals, or slices, and finding pairs of individuals that match along contiguous slices. We thus distinguish between a haplotype, which is an entire row of H and a word that represents the part of a haplotype that intersects a slice of H. A match between two individuals along several slices can be considered extended if the words of these individuals also match in the succeeding slice, otherwise the match terminates. A shared segment can thus be redefined as belonging to a pair of individuals that extend across several word pairs, and will represent an IBD segment rounded to the nearest slice break.
Formally, our algorithm accepts as input H and iteratively uses match to generate a set M′ of all shared segments in H. We vertically slice H into nonoverlapping, equal width submatrices of δ columns, with each slice denoted as Hk. The algorithm is a dynamic program that scans slices along the chromosome and maintains sets of the terminated and extendable matches within the current slice. To avoid redundant conversion from SNPs to slices, we will henceforth refer to a match (i,i′; j . j′) as (i,i′; m . m′) such that j = mδ and j′ = (m′ + 1)δ − 1. At each slice k = 0→(s/δ) − 1, we compute independent Mk = match(Hk), the set of identical matches at k. As detailed in EXTEND (Algorithm 2), we extend complementary matches in neighboring slices by examining Mk and generating a corresponding set Mk′ that contains all extended matches; naturally, each extended match contains m, the start of the match, in the range of from 0 to k-1 and m′, the end of the match, equal to k. In the first slice, define M0′ = M0; subsequently, initiate Mk′ = Mk, and search through all matches Mk′ for extendable matches from Mk-1′. Where they exist, we updated the starting position for matches in slice k to be that of the match in slice k − 1. Similarly, terminated matches present in Mk-1′ but not Mk, are either discarded or added as IBD to M′, depending on their length. Upon completing the final iteration, all matches in M(s/δ) − 1 are discarded or added to M′ in the same manner.
Genotyping error
Up until this point, we have ignored the effect genotyping errors may have on identifying matches. While modern genotyping platforms achieve accuracy levels >99% (Paynter et al. 2006), across many slices the chance of an error becomes non-negligible, even in a single sample. Across thousands of samples and along densely typed, complete chromosomes, the presence of errors approaches certainty. For IBD matching, random errors are unlikely to produce false positives. We are, however concerned about error-induced false negatives. Intuitively, a true IBD match would be present in several consecutive slices and may be detected by matching in any of them. Assuming random, uniform, and independent error rate ɛ per SNP, the number of mismatching allele calls between a pair of IBD haplotype intervals of length δ SNPs is Poisson distributed with parameter λ = 2δɛ. Across an IBD segment of length sIBD SNPs [where L(sIBD) = LIBD], the expected number of matching slices can be modeled as
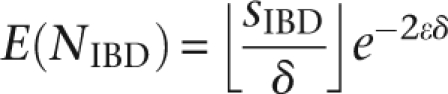 |
and typically being flanked by nearly identical slices. Specifically, the chance of these words to be identical is (1 − ɛ)2δ, with the length of a complete observed match in an IBD region thus geometrically distributed.
Nearly identical matching
In practice, with δ such that E(NIBD) >1, long IBD segments will likely have identical matches therein. The entire segment is identified by merging nearly identical flanking matches. ɛ determines the allowed mismatches in otherwise identical intervals. Specifically, conservatively assuming ɛ = 0.01; LIBD = 2,000 SNPs; δ = 100 SNPs; E(NIBD) = 2.7, allowing one mismatching bit/slice. The haplotypes corresponding to such slices are defined as being nearly identical. In MERGE-PARTIAL (Algorithm 3), we modify EXTEND to detect nearly identical matches by post-processing L′ to include nearly identical matches.
 |
All of the modules integrate into a complete procedure with input matrix H and output set L of all contiguous identical or nearly identical matches in H, based on predefined length and mismatch thresholds. As described in Algorithm 4, H is divided into slices that are analyzed sequentially by MATCH, MERGE, and MERGE-PARTIAL. Unextendable matches from previous slices are discarded or output as L′. This procedure deals also with missing data.
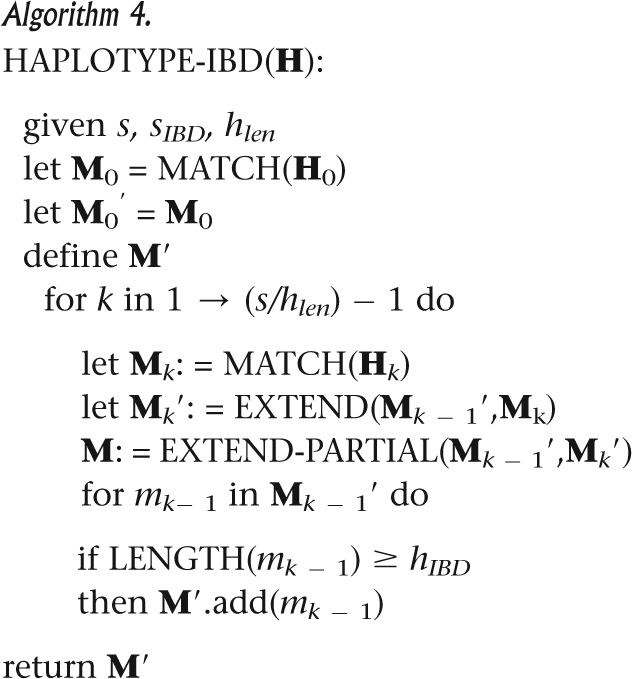 |
Algorithm complexity
Computationally, the algorithm has a significant gap between worst-case and typical scenarios. Specifically, the time complexity is highly dependent on expected number of matches, which is determined by the underlying population structure. In general, if the average number of matches per word is m, the complete computation requires O(sn) to build the dictionary and O(sm) to attempt extension on all matches. In the worst case, where L is formed by the Cartesian square of the 2n haplotypes, m = 4n2 and GERMLINE is comparable to pairwise exhaustive search. In practice, if we make a naïve assumption of independence of sites, the expected number of matches occurring at a slice at random is
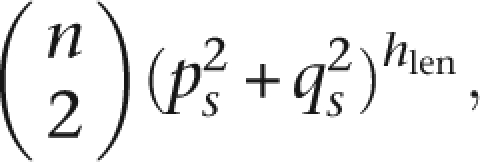 |
where ps and qs are the allele frequencies. A more realistic assumption would acknowledge local LD within each segment. If we denote a set of population haplotype frequencies f of size fn, the expected number of matches occurring at a word is
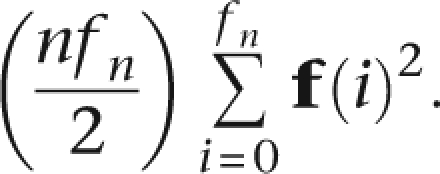 |
Even in large samples sizes, this factor is low enough where overall complexity approaches O(sn).
Gap likelihood scoring
To prioritize the segmental gaps that were most likely to be representative of a deletion, we developed a scoring function based on the number of mismatching SNPs and levels of homozygosity in the gap. From per-gap data we estimated the probability p for an independent SNP to have a mismatch. We registered the number of mismatches n and the total number of SNPs k occurring in each respective pair showing a particular gap. The binomial distribution term 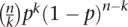 is therefore a likelihood score for each gap being due to mismatches consistent with the local rate of errors. Furthermore, a deletion is expected to be typed as completely homozygous, and we filtered the IBD-mismatching set for nearly complete loss of heterozygosity.
is therefore a likelihood score for each gap being due to mismatches consistent with the local rate of errors. Furthermore, a deletion is expected to be typed as completely homozygous, and we filtered the IBD-mismatching set for nearly complete loss of heterozygosity.
 |
Implementation
GERMLINE was implemented in C++ and made available at http://www.cs.columbia.edu/~itsik/Software.htm. All experiments were conducted on a Linux node of 2 × 2.4 GHz Xeon CPUs with 2 GB of memory.
Data
Pacific islanders from Kosrae, Federated States of Micronesia
Data included 3000 individuals genotyped for 600,000 SNPs at Affymetrix and Rockefeller University (Shmulewitz et al. 2006; Lowe et al. 2009). Post-QC we considered 2906 individuals and 429,925 autosomal markers, with partially known pedigrees.
International HapMap Project
HapMap Phase II release 21 phased data (Table 3) was used (The International HapMap Consortium 2005). These 30 trio parent pairs from the CEU and YRI populations, and 45 unrelated individuals from the JPT and CHB populations were originally passed using PHASE (Stephens et al. 2001). An average pair of individuals shares ∼0.5% of their genome through recent IBD (Frazer et al. 2007).
Acknowledgments
We thank the anonymous reviewers of this manuscript for their support and insightful comments. A.G. was supported by NIH grant 5 U54 CA121852 and the NSF Graduate Research Fellowship; I.P. was supported by NIH grant 5 U54 CA121852 and NSF grant NOA CCF-0829882; J.K.L. was supported in part by an NIH/NIDDK grant (5R01 DK60089) and by a Massachusetts Biomedical Research Corporation Tosteson Postdoctoral Fellowship. This work was further supported by grants from the Starr Foundation and Howard Hughes Medical Institute.
Footnotes
[Supplemental material is available online at www.genome.org. GERMLINE is freely available at http://www.cs.columbia.edu/~gusev/germline/.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.081398.108.
References
- Almasy L., Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 1998;62:1198–1211. doi: 10.1086/301844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Ayers K.L., Sabatti C., Lange K. Reconstructing ancestral haplotypes with a dictionary model. J. Comput. Biol. 2006;13:767–785. doi: 10.1089/cmb.2006.13.767. [DOI] [PubMed] [Google Scholar]
- Browning S.R., Browning B.L. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. Am. J. Hum. Genet. 2007;81:1084–1097. doi: 10.1086/521987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds K.G., Amer P.R., Auvray B. Using genetic markers in unpedigreed populations to detect a heritable trait. J. Zhejiang Univ. Sci. B. 2007;8:782–786. doi: 10.1631/jzus.2007.B0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy D.L. An integrated genetic map for linkage analysis. Behav. Genet. 2006;36:4–6. doi: 10.1007/s10519-005-9015-x. [DOI] [PubMed] [Google Scholar]
- Frazer K.A., Ballinger D.G., Cox D.R., Hinds D.A., Stuve L.L., Gibbs R.A., Belmont J.W., Boudreau A., Hardenbol P., Leal S.M., et al. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–861. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W.G., Hernandez-Sanchez J. Prediction of multilocus identity-by-descent. Genetics. 2007;176:2307–2315. doi: 10.1534/genetics.107.074344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill W.G., Weir B.S. Prediction of multi-locus inbreeding coefficients and relation to linkage disequilibrium in random mating populations. Theor. Popul. Biol. 2007;72:179–185. doi: 10.1016/j.tpb.2006.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs A.L., Bertelsen S., Bierut L.J., Dunn G., Jin C.H., Kauwe J.S., Suarez B.K. Multipoint identity-by-descent computations for single-point polymorphism and microsatellite maps. BMC Genet. 2005;6:S34. doi: 10.1186/1471-2156-6-S1-S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Wei W., Zhang J., Liu G., Bignell G.R., Stratton M.R., Futreal P.A., Wooster R., Jones K.W., Shapero M.H. Whole genome DNA copy number changes identified by high density oligonucleotide arrays. Hum. Genomics. 2004;1:287–299. doi: 10.1186/1479-7364-1-4-287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate A.J., Feuk L., Rivera M.N., Listewnik M.L., Donahoe P.K., Qi Y., Scherer S.W., Lee C. Detection of large-scale variation in the human genome. Nat. Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- The International HapMap Consortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W.J. BLAT—The BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X., Murphy K., Raj T., He C., White P.S., Matise T.C. A combined linkage-physical map of the human genome. Am. J. Hum. Genet. 2004;75:1143–1148. doi: 10.1086/426405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A., Masson G., Frigge M.L., Gylfason A., Zusmanovich P., Thorleifsson G., Olason P.I., Ingason A., Steinberg S., Rafnar T., et al. Detection of sharing by descent, long-range phasing and haplotype imputation. Nat. Genet. 2008;40:1068–1075. doi: 10.1038/ng.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R.A., KaraMohamed S., Sudi J., Conrad D.F., Brune C., Badner J.A., Gilliam T.C., Nowak N.J., Cook E.H., Jr, Dobyns W.B., et al. Recurrent 16p11.2 microdeletions in autism. Hum. Mol. Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Lien S., Szyda J., Schechinger B., Rappold G., Arnheim N. Evidence for heterogeneity in recombination in the human pseudoautosomal region: High resolution analysis by sperm typing and radiation-hybrid mapping. Am. J. Hum. Genet. 2000;66:557–566. doi: 10.1086/302754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Cutler D.J., Zwick M.E., Chakravarti A. Haplotype inference in random population samples. Am. J. Hum. Genet. 2002;71:1129–1137. doi: 10.1086/344347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J.K., Maller J.B., Pe'er I., Neale B.M., Salit J., Kenny E.E., Shea J.L., Burkhardt R., Ji W., Noel M., et al. Genome-wide association studies in an isolated founder population from the Pacific island of Kosrae. PLoS Genet. 2009 doi: 10.1371/journal.pgen.1000365. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malécot G. Les mathématiques de l'hérédité. Masson; Paris, France: 1948. [Google Scholar]
- Mao Y., Xu S. A Monte Carlo algorithm for computing the IBD matrices using incomplete marker information. Heredity. 2005;94:305–315. doi: 10.1038/sj.hdy.6800564. [DOI] [PubMed] [Google Scholar]
- Marchini J., Cutler D., Patterson N., Stephens M., Eskin E., Halperin E., Lin S., Qin Z.S., Munro H.M., Abecasis G.R., et al. A comparison of phasing algorithms for trios and unrelated individuals. Am. J. Hum. Genet. 2006;78:437–450. doi: 10.1086/500808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen T.H., Goddard M.E. Multipoint identity-by-descent prediction using dense markers to map quantitative trait loci and estimate effective population size. Genetics. 2007;176:2551–2560. doi: 10.1534/genetics.107.070953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paynter R.A., Skibola D.R., Skibola C.F., Buffler P.A., Wiemels J.L., Smith M.T. Accuracy of multiplexed Illumina platform-based single-nucleotide polymorphism genotyping compared between genomic and whole genome amplified DNA collected from multiple sources. Cancer Epidemiol. Biomarkers Prev. 2006;15:2533–2536. doi: 10.1158/1055-9965.EPI-06-0219. [DOI] [PubMed] [Google Scholar]
- Perry G.H., Ben-Dor A., Tsalenko A., Sampas N., Rodriguez-Revenga L., Tran C.W., Scheffer A., Steinfeld I., Tsang P., Yamada N.A., et al. The fine-scale and complex architecture of human copy-number variation. Am. J. Hum. Genet. 2008;82:685–695. doi: 10.1016/j.ajhg.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D., Marshall C., Feuk L., Scherer S.W. Copy-number variation in control population cohorts. Hum. Mol. Genet. 2007;16:R168–R173. doi: 10.1093/hmg/ddm241. [DOI] [PubMed] [Google Scholar]
- Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R., Ishikawa S., Fitch K.R., Feuk L., Perry G.H., Andrews T.D., Fiegler H., Shapero M.H., Carson A.R., Chen W., et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmulewitz D., Heath S.C., Blundell M.L., Han Z., Sharma R., Salit J., Auerbach S.B., Signorini S., Breslow J.L., Stoffel M., et al. Linkage analysis of quantitative traits for obesity, diabetes, hypertension, and dyslipidemia on the island of Kosrae, Federated States of Micronesia. Proc. Natl. Acad. Sci. 2006;103:3502–3509. doi: 10.1073/pnas.0510156103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens M., Smith N.J., Donnelly P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001;68:978–989. doi: 10.1086/319501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A., Camp N.J., Farnham J.M., Allen-Brady K., Cannon-Albright L.A. Shared genomic segment analysis. Mapping disease predisposition genes in extended pedigrees using SNP genotype assays. Ann. Hum. Genet. 2008;72:279–287. doi: 10.1111/j.1469-1809.2007.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson E.A. The IBD process along four chromosomes. Theor. Popul. Biol. 2008;73:369–373. doi: 10.1016/j.tpb.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Li M., Hadley D., Liu R., Glessner J., Grant S.F., Hakonarson H., Bucan M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007;17:1665–1674. doi: 10.1101/gr.6861907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss L.A., Shen Y., Korn J.M., Arking D.E., Miller D.T., Fossdal R., Saemundsen E., Stefansson H., Ferreira M.A., Green T., et al. Association between microdeletion and microduplication at 16p11.2 and autism. N. Engl. J. Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
- Wright S. Systems of mating. I. The biometric relations between parent and offspring. Genetics. 1921;6:111–123. doi: 10.1093/genetics/6.2.111. [DOI] [PMC free article] [PubMed] [Google Scholar]