

Abstract
Arterial calcification is a phenotype of vascular repair in atherosclerosis, diabetes, hyperphosphatemic renal failure, and aging. Arterial calcification is modulated by transition of arterial smooth muscle cells (SMCs) from contractile to chondro–osseous differentiation programmed in response to increases in Pi, bone morphogenetic protein-2, and certain other stimuli. Transglutaminase (TG)2 release modulates tissue repair, partly by transamidation-catalyzed covalent crosslinking of extracellular matrix substrates. TG2 regulates cultured SMC differentiation, resistance artery remodeling to vasoconstriction, and atherosclerotic lesion size. Here, TG2 expression was required for the majority of TG activity in mouse and human aortic SMCs. TG2−/− SMCs lost the capacity for Pi donor–induced formation of multicellular bone-like nodules and for increased expression of the type III sodium–dependent Pi cotransporter Pit-1 and certain osteoblast and chondrocyte genes (tissue-nonspecific alkaline phosphatase, the osteoblast master transcription factor runx2, and chondrocyte-restricted aggrecan), and for Pi donor– and bone morphogenetic protein-2–induced calcification. Uniquely in TG2−/− SMCs, Pi donor treatment increased expression of the physiological SMC chondro–osseous differentiation and calcification inhibitors osteoprotegerin, matrix Gla protein, and osteopontin. Conversely, TG2−/− SMCs, unlike wild-type SMCs, failed to maintain contractile differentiation on laminin. Exogenous catalytically active TG2 augmented calcification by TG2−/− SMC in response to Pi donor treatment. TG2 expression also drove Pi-stimulated calcification of mouse aortic ring organ cultures, which was suppressed by the TG2 catalytic site-specific inhibitor Boc-DON-Gln-Ile-Val-OMe (10 μmol/L). Our results suggest that TG2 release in injured arteries is critical for programming chondro–osseous SMC differentiation and calcification in response to increased Pi and bone morphogenetic protein-2.
Keywords: smooth muscle cells, atherosclerosis, osteopontin, matrix Gla protein, osteoprotegerin, laminin
Arterial smooth muscle cells (SMCs) mediate complex vascular repair and remodeling processes.1 SMCs are phenotypically plastic and are drawn out of a contractile differentiation by stimuli, including biomechanical forces, changes in vascular tone, extracellular matrix modifications, thrombotic factors, and inflammatory and growth factors exemplified by platelet-derived growth factor (PDGF).2 The transition of contractile to synthetic SMCs modulates repair of arterial injuries via the capacity to migrate, proliferate, remodel the extracellular matrix, modulate inflammation, and promote thrombosis.3
Normal artery SMC populations also contain cells that undergo phenotypic transition to calcifying osteoblastic and chondrocytic cells,4–8 a potentiality shared in the diseased artery by pericytes, adventitial myofibroblasts, and vascular stem cells.5,9,10–12 Pi generation and sodium-dependent Pi uptake, bone morphogenetic protein (BMP)-2 and Wnt signaling, and certain oxidized proatherogenic lipids are recognized to drive intraarterial chondro–osseous differentiation and calcification.4–12 SMC generation by NPP1 and transport by the murine progressive ankylosis gene (ANK) of the basic calcium phosphate crystal growth and chondrogenesis inhibitor PPi help physiologically hold SMC chondro–osseous differentiation and artery calcification in check.8,13 In this light, hydrolysis of PPi to Pi is a major activity of tissue-nonspecific alkaline phosphatase (TNAP), and TNAP expression is linked to SMC chondro–osseous differentiation that promotes calcification.8 Arterial calcification also is physiologically limited by SMC expression of the potent BMP-2 inhibitor (MGP),14 the potent basic calcium phosphate crystal growth inhibitor and mineral resorption promoter osteopontin (OPN),6,14 and osteoprotegerin (OPG), an inhibitor of RANKL and TRAIL signaling and potential modulator of the bone–vascular axis.15,16
Recently, the multifunctional protein transglutaminase (TG)2 has been implicated as a regulator of calcification by chondrocytes and osteoblasts.17–19 TGs, by calcium-dependent transamidation (EC2.3.2.13), covalently crosslink a broad array of substrate proteins with available glutamine and lysine residues (such as collagen I, fibronectin, laminin, and OPN).20 Producing protease-resistant isopeptide bonds, TGs directly mediate stabilization of extracellular matrices.20 TG2 also mediates cultured chondrocyte maturation to terminal hypertrophic differentiation and the capacity of chondrocytes to calcify the matrix in response to retinoic acid and certain inflammatory cytokines.17,18,21 TG2, although lacking signal peptide, is released by cells.18,20 Exogenous nanomolar TG2 is sufficient to promote chondrocyte hypertrophy,18 and direct effects of extracellular TG2 on cell differentiation have been linked to consequences of TG2-catalyzed pericellular matrix protein crosslinking.20,22
Resistance artery remodeling induced by chronic vasoconstriction is driven by extracellular TG2 and blocked by suppressing TG catalytic activity.1 TG2 expression and release are induced in vitro by nitric oxide, the nitric oxide–derived oxidant peroxynitrite, retinoic acid signaling, certain inflammatory cytokines, and thrombin17,21,23,24 and in vivo in macrophages and SMCs in atherogenesis.25,26 TG2 limits both atherosclerotic lesion size and necrotic core expansion.25
Calcification decreases artery wall compliance, and arterial calcification is linked to excess mortality in hyperphosphatemic renal failure, diabetes mellitus, and atherosclerosis.5 Here, we identify that TG2 is critical for programming calcification by cultured SMCs in response to Pi donor treatment and BMP-2 and Pi donor–induced calcification in aortic ring explants in organ culture.
Materials and Methods
An expanded Materials and Methods section that contains details on mouse aortic SMC and explant isolation and culture, immunohistochemical experiments, RT-PCR, data collection, and statistics is available in the online data supplement at http://circres.ahajournals.org.
Animals
In vitro analyses used tissues of congenic TG2−/− mice and congenic TG2−/− mice, originally on a hybrid C57BL6/129SVJ background27 and crossed for more than 9 generations onto C57BL6 background.
Murine SMC Studies
Primary SMCs were isolated at 2 months of age from mouse aortas,8 and cells were carried on laminin for 2 passages before experimentation, unless otherwise indicated.
Results
TG2 Critically Mediates Induction of Both Chondro–Osseous Differentiation and Calcification by Cultured SMCs
SMC pericellular matrix alterations in diseased arteries promote changes in basal SMC contractile differentiation, mimicked by placing arterial SMCs in culture without a fibrillar collagen, laminin, or endothelial cell substratum.3 Here, we removed third-passage mouse aortic SMCs from laminin for 72 hours in culture and observed mRNA for TG2 and several TG isoenzymes to be expressed by normal cultured SMCs, with TG5 mRNA upregulated in TG2−/− SMCs (Figure 1A). TG2 expression was required for presence of the majority (≈ 80%) of TG catalytic activity in unstimulated mouse aortic SMCs, and PDGF and all-trans retinoic acid (ATRA) failed to induce TG activity in TG2−/− SMCs, unlike the case for TG2−/− SMCs (Figure 1B). Under these conditions, TG2 release into conditioned medium was stimulated by PDGF and ATRA in wild-type SMCs, but TG2 was undetectable in both conditioned medium (Figure 1C) and cell lysates (not shown) of TG2−/− SMCs. In freshly isolated primary mouse SMCs and aortas, we observed basal expression of several TG isoenzymes in congenic TG2−/− and TG2−/− mice, with upregulated TG5 mRNA in freshly isolated TG2−/− SMCs and aortas (Figure IA and IB in the online data supplement).
Figure 1.
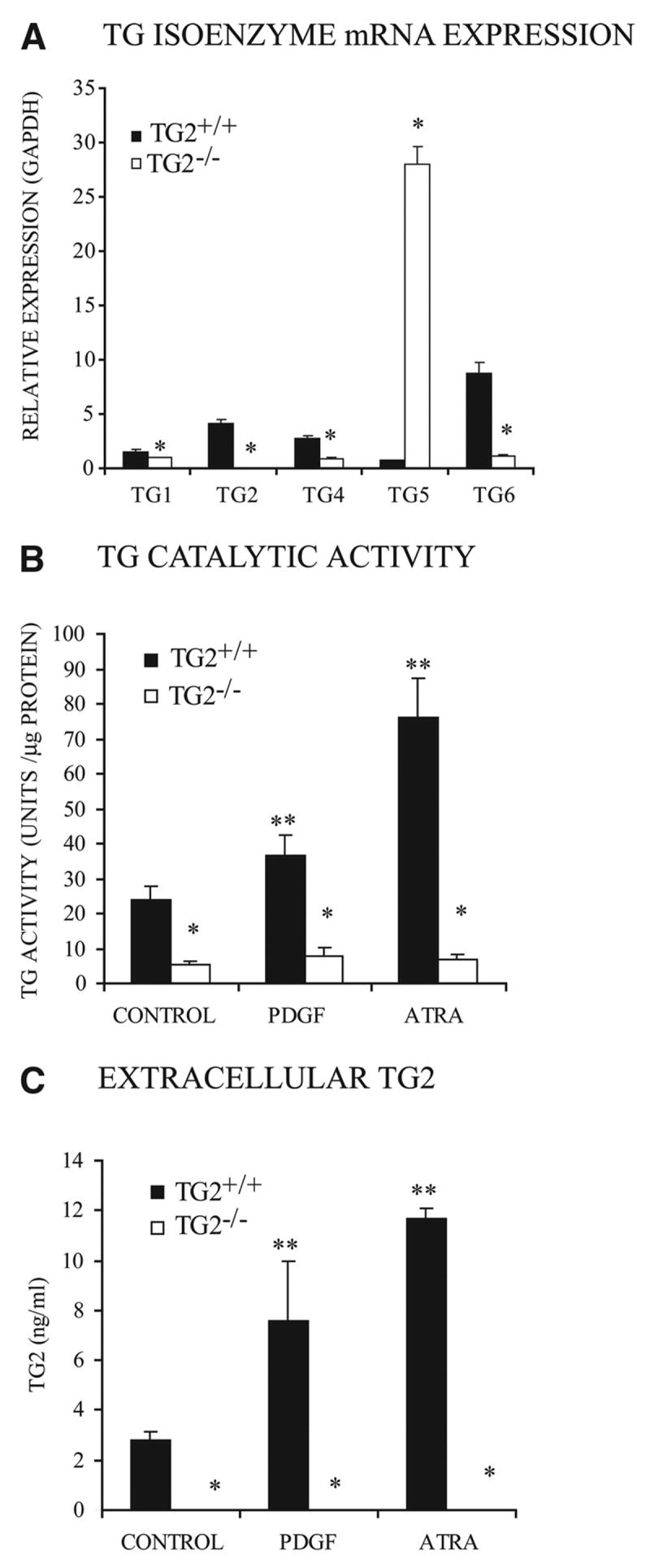
TG isoenzyme expression, TG catalytic activity, and TG2 release in cultured mouse SMCs. We cultured aliquots of 5×104 third-passage congenic TG2−/− and TG2−/− mouse aortic SMCs per well (in a 24-well plate) for 72 hours. A, Expression patterns of TG isoenzymes. Here, and in quantitative PCR experiments below, SYBR green–based mRNA copy quantification compared individual target mRNA to GAPDH mRNA copy levels for each experiment. Data were pooled from 4 experiments, replicates of 3. *P<0.001 for TG2−/− vs TG2−/− by independent samples t test with Bonferroni correction. B, TG catalytic-specific activity in cell lysates. C, TG2 released into SMC conditioned media in response to 10 ng/mL PDGF or 10 nmol/L ATRA, assayed by ELISA. For B and C, Data were pooled from 4 experiments, replicates of 3. *P<0.001 for TG2−/− vs TG2−/−, **P<0.001 for increases induced by PDGF and ATRA by ANOVA with post hoc Tukey test.
Formation of multicellular nodules that calcified (assessed by von Kossa or Alizarin red S staining for deposited Pi and Ca2+, respectively) was blunted in TG2−/− SMCs, which here were removed from laminin and stimulated with 50 μg/mL ascorbate and the Pi donor/TNAP substrate β-glycerolphosphate (2.5 mmol/L) (Figure 2A through 2C). We also observed attenuated calcification by TG2−/− SMCs, which was partially rescued by exogenous, soluble recombinant wild-type TG2 (100 ng/mL), an effect shared by the K173L GTP binding site mutant of TG218 and the FXIIIA TG isoenzyme but not by TG catalytic site dead C277G TG2 mutant (Figure 2D).18 FXIIIA is expressed by adventitial macrophages,28 but we verified absence of FXIIIA expression by cultured aortic SMCs (supplemental Figure II).
Figure 2.

Decreased chondro–osseous nodule formation and matrix calcification by TG2−/− SMCs. Second-passage mouse aortic SMCs (5×104 per well in 24-well plate) (A through C) or 1×105 per well in a 12-well plate (D) were cultured in media for up to 14 days with added 2.5 mmol/L β-glycerolphosphate and 50 μg/mL ascorbate to stimulate calcification. A and B, SMCs were fixed with 4% paraformaldehyde for 10 min and then washed with H2O (A) or PBS (B). A, To detect Pi deposited in the matrix, fixed cultures were stained with von Kossa and counterstained with nuclear fast red (day 14 results shown). B, To visualize matrix Ca2+ deposition, Alizarin red S staining was performed (day 7 results shown). A and B are representative of 4 experiments, cells pooled from 12 animals each genotype, each experiment. C, Each 24-well dish (assessed as in A) was examined microscopically for the total number of von Kossa–positive nodules found per 9-mm2 well. Data were pooled from 4 experiments, replicates of 3. *P<0.001 for TG2−/− vs TG2−/− by independent samples t test with Bonferroni correction. D, To assess TG2 structure/function in calcification, SMCs were cultured with 100 ng/mL recombinant soluble wild-type (WT) TG2 (sTG2), TG catalytic site (C277G), GTP binding site mutant (K173L) TG2, or WT FXIIIA TG isoenzyme for 1 to 10 days. Ca2+ deposition was determined after decalcification in 0.6 N HCl using phenolsulphonphthalein to bind free Ca.2 Data were pooled from 4 experiments, replicates of 3. *P<0.01 for TG2−/− vs TG2−/− with recombinant isoenzymes, **P<0.01 for TG2−/− vs TG2−/− by ANOVA with post hoc Tukey test.
In Pi-stimulated calcification, 29 TG2+/+ but not TG2−/− SMCs developed increased mRNA for the transcription factor Msx2, which promotes maintenance of multipotentiality and osteoblastogenesis but suppresses chondrogenesis,10 of TNAP, of Pit-1, of the osteoblast master transcription factor runx2, and of the chondrocyte-specific extracellular matrix constituent aggrecan (Figure 3, top). In contrast, TG2−/− SMCs developed increased mRNA expression of the artery calcification inhibitors OPN, MGP, and OPG (Figure 3, bottom). Extracellular PPi and PPi-generating nucleotide pyrophosphatase phosphodiesterase-specific activity were not significantly altered in TG2−/− SMCs, but TNAP-specific enzyme activity more than doubled in TG2+/+ while remaining unchanged in TG2−/− SMCs over 10 days in culture (not shown). OPN (at days 1 to 7) and OPG (at days 1 to 17) were markedly increased in the conditioned media of TG2−/− relative to TG2+/+ SMCs stimulated to calcify (Figure 4A and 4B). In the absence of either stimulation by a Pi donor or of further culture, freshly isolated TG2−/− mouse aortic SMCs and aortas demonstrated minimal differences relative to TG2+/+ samples for mRNA of the same promoters and inhibitors of calcification, except for more OPG in TG2−/− aortas (supplemental Figures III and IV).
Figure 3.

Changes in calcification regulatory gene expression in TG2−/− SMCs in response to Pi donor treatment. For quantitative PCR, total RNA was collected from 5×104 second-passage SMCs in media with 2.5 mmol/L β-glycerolphosphate and 50 μg/mL ascorbate added at day 0, with results expressed relative to GAPDH. A, Decreased expression of chondro–osseous and calcification-promoting genes (TNAP, Msx2, runx2, aggrecan, Pit-1) in TG2−/− SMCs. Data were pooled from 3 experiments, replicates of 2. *P<0.005 for TG2+/+ vs TG2−/− by independent samples t test with Bonferroni correction. B, Increased expression of physiological arterial calcification inhibitors (MGP, OPN, OPG) in TG2−/− SMCs. Data were pooled from 3 experiments, replicates of 2. *P<0.001for TG2+/+ vs TG2−/− by independent samples t test with Bonferroni correction.
Figure 4.
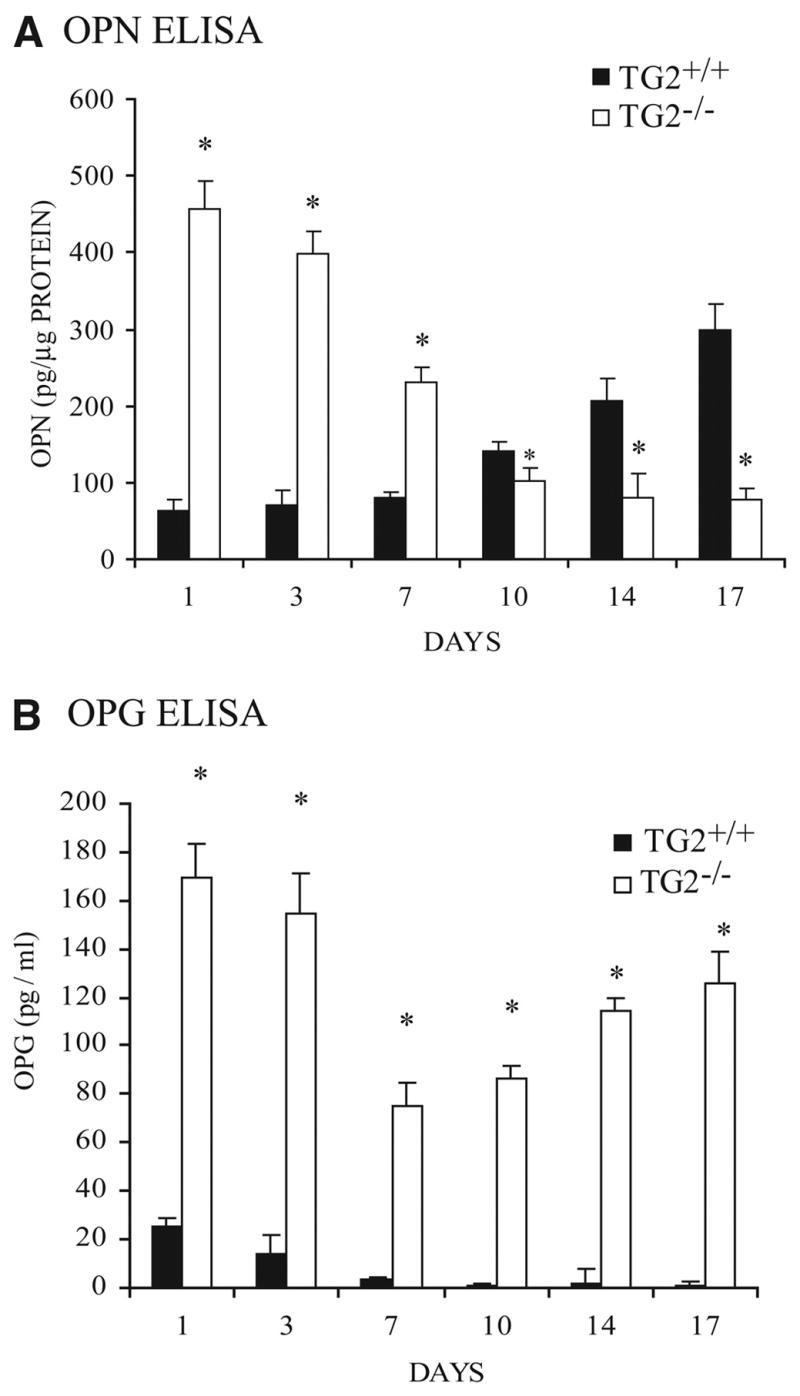
TG2 modulates extracellular OPN and OPG levels. A and B, Aliquots of 5×104 second-passage SMCs per well in 24-well plates were cultured up to 17 days with added 2.5 mmol/L β-glycerolphosphate and 50 μg/mL ascorbate, and conditioned media were collected for OPN (A) and OPG (B) ELISA. Data were pooled from 3 experiments, replicates of 3. *P<0.001 for TG2+/+ vs TG2−/− by independent samples t tests with Bonferroni correction.
To rule out compensatory effects attributable to germline TG2 depletion that limited calcification by SMCs, we used short hairpin RNA transfection in human aortic SMCs to knock down TG2 expression by >80%, associated with ≈55% to 65% loss of total TG catalytic activity in the 72 hours after transfection (Figure 5A and 5B). The acquired TG2 depletion blunted calcification in response to both the Pi donor and BMP-2 (10 ng/mL) in human SMCs (Figure 5C).
Figure 5.
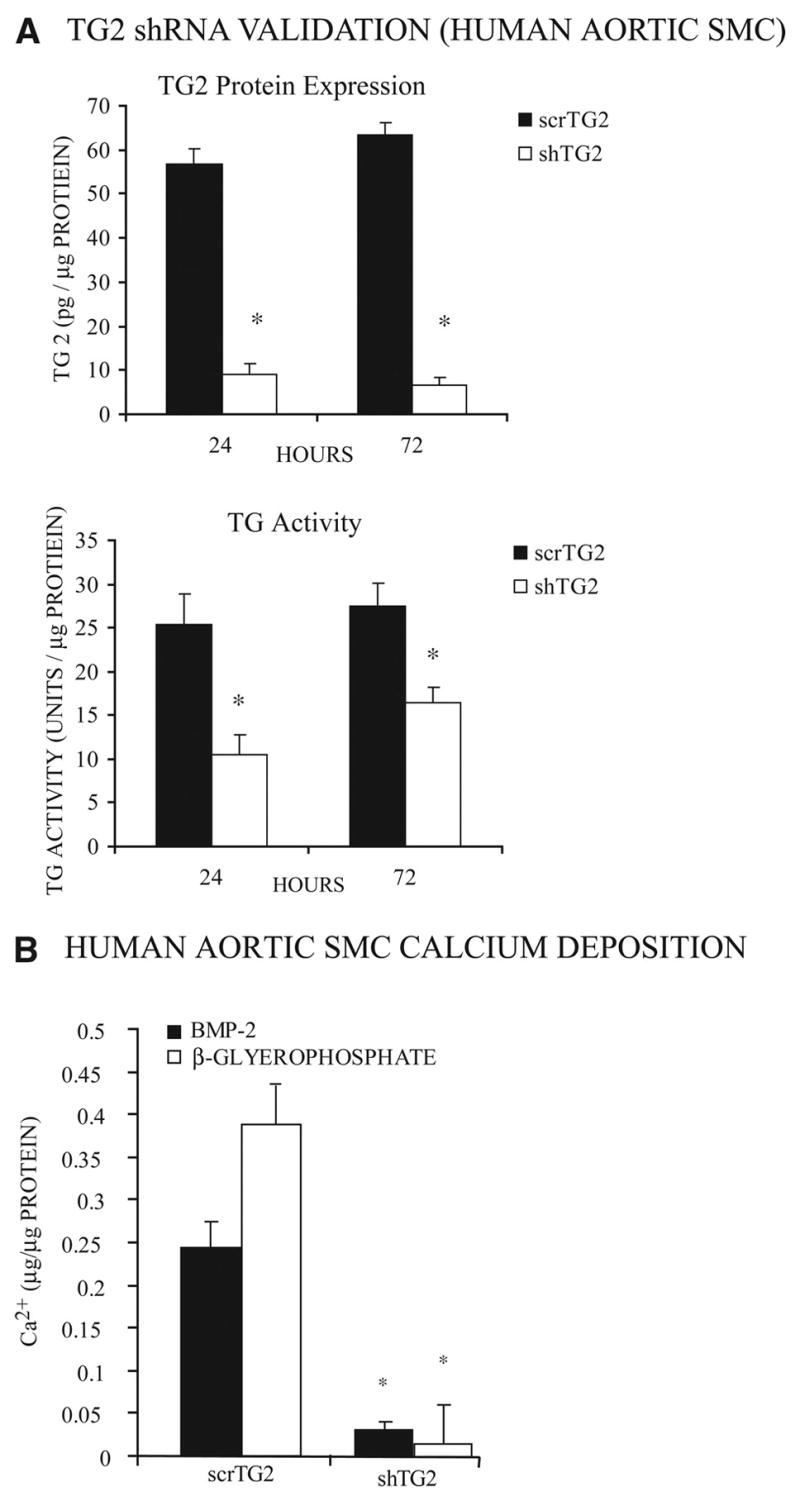
Both BMP-2– and Pi donor–induced calcification are inhibited in human aortic SMCs. A, To verify the effectiveness of TG2 depletion by short hairpin RNA (shTG2) and scrambled control (scrTG2) cDNA transfections, human aortic SMC lysates from aliquots of 3×105 cells/well in a 6-well plate were collected 24 and 72 hours after transfection. Total cellular TG2 protein determined by sandwich ELISA was normalized to total protein. Additionally, TG catalytic activity was determined. Data were pooled from 3 experiments, replicates of 3. *P<0.001 by independent samples t tests with Bonferroni correction. B, Human aortic SMCs were transfected with shTG2 and scrTG2, and after 12 hours, 10 ng/mL BMP-2, or 2.5 mmol/L β-glycerolphosphate and 50 μg/mL ascorbate, was added for 72 hours, followed by determination of Ca2+ deposition as above. Data were pooled from 3 experiments, replicates of 3. *P<0.001 by independent samples t test with Bonferroni correction.
TG2 “gain of function” in human SMCs via treatment with nanomolar (100 ng/mL) recombinant soluble TG218 suppressed OPN production by >50% (Figure 6A). Hence, we qualitatively assessed OPN expression by immunocytochemistry for TG2−/− mouse SMCs plated on 1 μg/cm2 murine laminin to promote maintenance of contractile differentiation state, with or without additional precoating of the plate with TG2 (100 ng/mL for 10 min), followed by 4 washes with PBS. Under these conditions, where no TG2 was provided by SMCs, there was substantial TG2 retention in the laminin matrix and the TG2 pretreatment of the laminin substratum suppressed OPN expression (Figure 6B).
Figure 6.
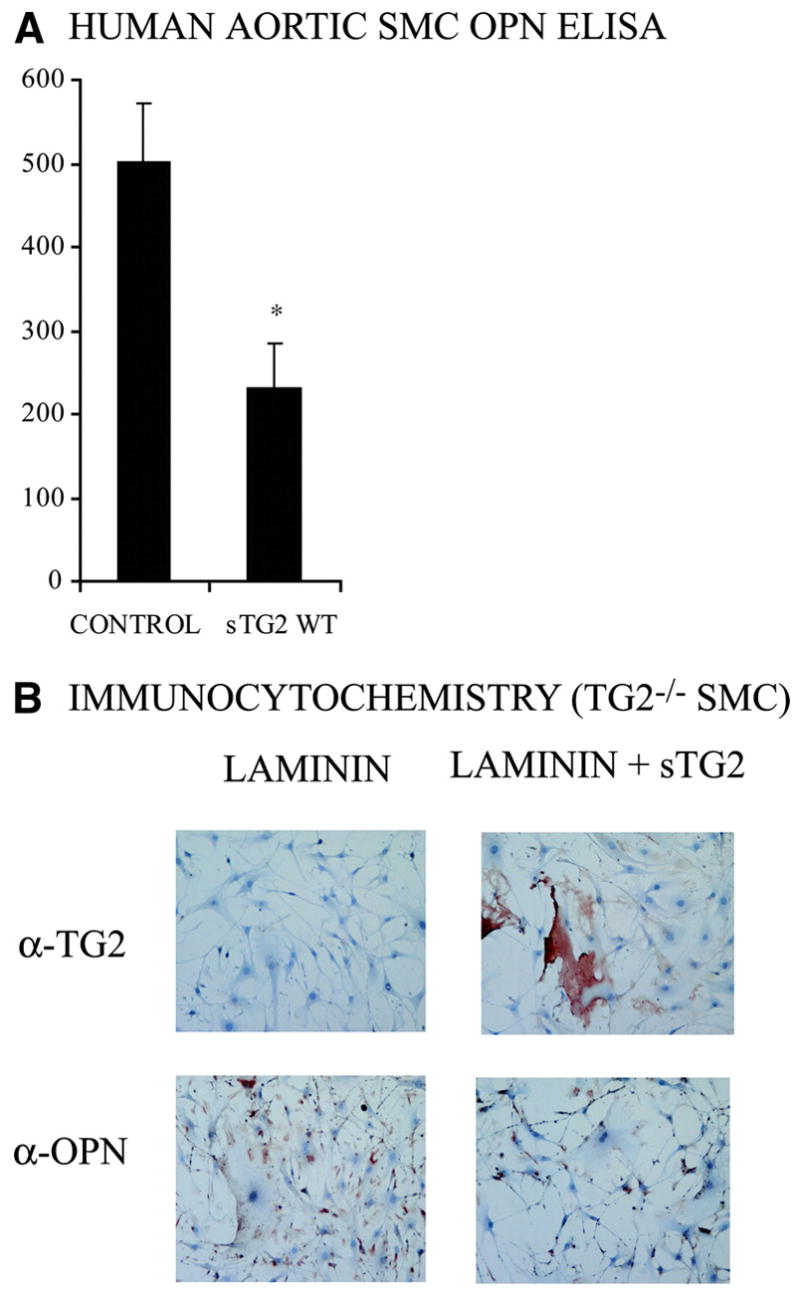
Exogenous TG2 modulates OPN expression by SMCs. A, Aliquots of 5×104 human aortic SMCs were cultured for 72 hours with and without 100 ng/mL recombinant soluble WT TG2 (sTG2), and conditioned media were assayed for OPN by ELISA. Data were pooled from 3 experiments. *P<0.001 by independent samples t test. B, Where indicated, recombinant WT sTG2 (100 ng/mL) was added to laminin-coated coverslips for 10 min at 37°C, followed by 4 washes in PBS to remove TG2 unincorporated into the laminin substratum, followed by plating and culturing of aliquots of 1×105 TG2−/− SMCs for 24 hours before fixation with 4% paraformaldehyde. Immunocytochemistry assessed TG2 and OPN. Data are representative of 3 experiments.
Failure of Stimulated Induction of Chondro–Osseous Differentiation and Calcification by TG2-Deficient SMCs Is Not Attributable to Enhanced Retention of Contractile Differentiation
We next tested whether failure of stimulated induction of chondro–osseous differentiation in TG2−/− SMCs was attributable to enhanced retention of contractile differentiation or predisposition to synthetic differentiation, given that OPN and MGP expression are associated with SMC synthetic differentiation.3,30,31 We confirmed TG2+/+ SMCs to robustly express the prerequisite for spreading type I collagen and to spread when on fibronectin but to retain contractile differentiation on laminin (Figure 7).32 In contrast, TG2−/− SMCs robustly expressed type I collagen and spread both on fibronectin and laminin (Figure 7). Primary TG2−/− SMCs cultured on laminin for 5 days also developed decreased expression of the contractile differentiation associated mR-NAs Notch-3, myocardin, and smooth muscle α-actin and myosin heavy chain, whereas expression increased for collagen I and OPN expression and the stereotypic synthetic differentiation marker myosin light chain kinase (MLCK) 210-kDa isoform (supplemental Figure V).3 There were only minimal TG2 deficiency–related differences among these same markers of contractile and synthetic differentiation in freshly isolated aortic SMCs without further culture and in whole aortas (supplemental Figure VIA and VIB).
Figure 7.

TG2−/− SMCs fail to retain contractile differentiation on laminin. Aliquots of 1×105 second passage mouse aortic SMCs were cultured for 72 hours on 1 μg/cm2 laminin (to maintain contractile differentiation) or fibronectin (to promote synthetic differentiation). After 24 hours, SMCs were fixed and immuno-histochemically stained for type I collagen. Data are representative of 5 experiments.
TG2 Promotes Pi-Stimulated Calcification in Aortas in Organ Culture
To validate the physiological and translational significance of deficient Pi-induced calcification in TG2−/− SMCs, we adapted a rat aortic ring organ culture model to mouse samples.33 We cultured aortic 2- to 3-mm ring explants for 7 days in medium supplemented with 7 U/mL alkaline phosphatase and 2.5 mmol/L sodium Pi and first stained for chondrocyte-specific type IX/XI collagen expression,8 which we observed to be induced in TG2+/+ but not in TG2−/− explants (Figure 8A). Induced 45Ca incorporation and free Ca2+ both were suppressed in TG2−/− aortas (Figure 8B and 8C), and the TG2 catalytic site-specific irreversible inhibitor Boc-DON-Gln-Ile-Val-OMe34 (at 10 μmol/L) inhibited Pi-induced calcification by TG2+/+ aortic explants by ≈50% under conditions in which TG2−/− aortic explants demonstrated ≈75% less calcification (supplemental Figure VII).
Figure 8.
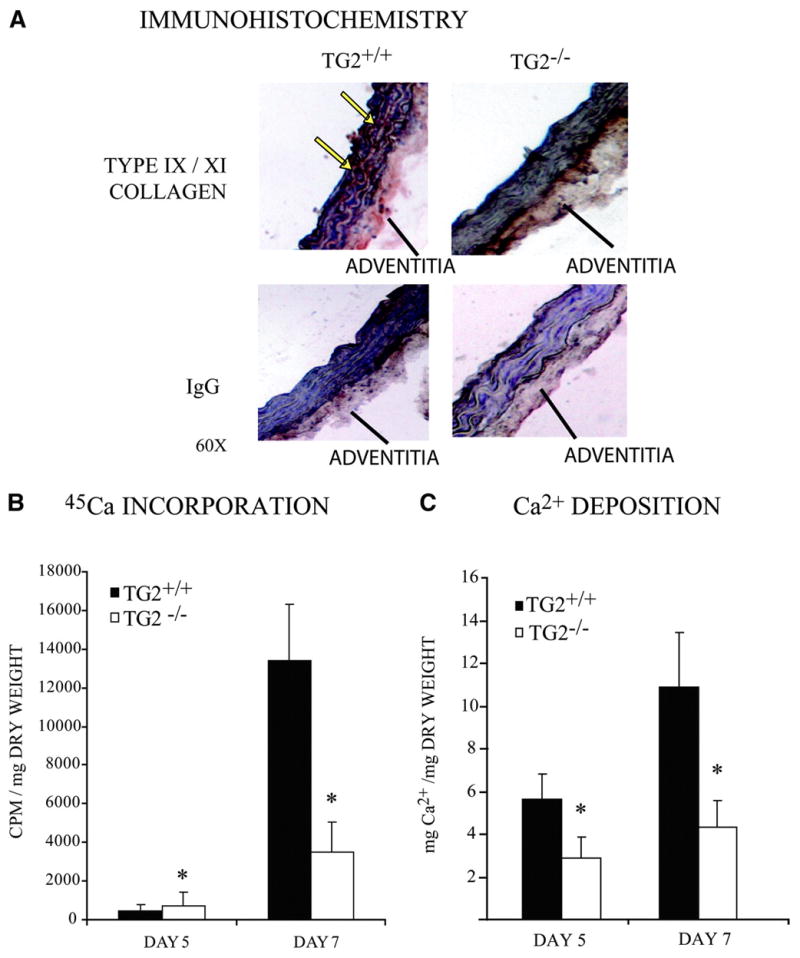
Decreased calcification in TG2−/− mouse aortic ring explant cultures. Isolated whole aortas were cut into 2- to 3-mm slices and cultured in growth media supplemented with 2.5 mmol/L NaPi and 7 U/mL alkaline phosphatase. A, Frozen sections were immunohistochemically stained for chondrocyte-specific type IX/XI collagen. B, Aortic cultures were incubated as above for 4 and 6 days before addition of 0.3 μCi/mL 45Ca for 24 hours. Aortas were collected on days 5 and 7, washed 3 times with PBS, dried, and weighed, and incorporated 45Ca cpm was quantified. Data were pooled from 10 animals, replicates of 3. *P<0.005 by independent samples t test with Bonferroni correction. C, Free Ca2+ deposition per milligram of dry weight in aortic ring explants was determined by phenolsulphonphthalein binding as above. Data were pooled from 12 animals, 2 aortic cultures per time point. *P<0.001 by independent samples t test with Bonferroni correction. Data were pooled from 5 animals, replicates of 3.
Discussion
In this study, we demonstrated that TG2 is essential for Pi-induced programming of SMC transition to chondro–osseous differentiation and that TG2 played a central role in calcification by cultured SMCs and aortic ring explants in organ culture. Remarkably, with TG2 deficiency, there was failure to upregulate several genes associated with chondro–osseous differentiation, coupled with sharply increased expression of the physiological artery calcification inhibitors MGP, OPN, and OPG in response to Pi donor treatment. Pi functions as a raw material for deposition of crystalline basic calcium phosphate within SMC-derived matrix vesicles and in the “hole regions” of fibrillar type I collagen.35 Additionally, Pi that is taken up by sodium-dependent cotransport via Pit-136 (and in some conditions Pit-2 in mineralization-competent cells including SMCs37) functions as a signaling molecule and growth regulator in osteogenic development.38
Importantly, the calcification response of TG2-deficient SMCs was attenuated in response to not only a Pi donor but also to BMP-2, whose expression is elevated in atherosclerotic lesions.39 BMP-2 promotes calcification by SMCs and myofibroblasts in part through Msx2 and Wnt signaling pathways.10 However, BMP-2 also induces Pit-1 expression in osteoblastic cells, a mechanism essential to driving matrix calcification.40 We observed Pit-1 mRNA and Pi-generating TNAP mRNA expression and enzyme activity to be suppressed in TG2−/− SMCs.
Central to the effects of Pi in chondro–osseous cell differentiation are regulation of mitogen-activated protein kinase signaling and modulation of expression of multiple genes that regulate calcification, exemplified by rapid induction of OPN.38 We observed the OPN induction response of TG2−/− SMCs to Pi donor treatment to be increased. However, exaggerated OPN expression also was observed in TG2−/− SMCs without donor treatment. We did not Pi measure Pi uptake by TG2-deficient SMCs in this study. It remains possible that basal sodium-dependent Pi uptake mediated by Pit-1 (or sodium-independent Pi uptake37) could be altered in TG2-deficient SMCs, for example, by loss of TG2 effects on cytosolic and plasma membrane protein–protein interactions in putative macromolecular complexes that move Pi. Even so, the capacity of exogenous catalytically active TG2 and FXIIIA to partially restore calcification responses of TG2−/− SMCs to Pi donor treatment and the observed partial suppression of calcification in aortic explants by a peptide-based TG2-specific catalytic site inhibitor are noteworthy. Our observations, combined with the grossly normal skeletal development of TG2−/− mice,17,27 argue that paracrine and autocrine effects of secreted, catalytically active TG2 on SMCs promote transition to calcifying chondro–osseous nodule formation in SMCs.
TG-induced transamidation of both extracellular matrix and plasma membrane proteins likely contributes substantially to SMC chondro–osseous differentiation and calcification, because treatment of type I collagen with TG2 promotes collagen compaction,1 and osteoblasts grown on type I collagen crosslinked by TG2 differentiate more quickly than on native untreated collagen.41 Furthermore, TG2 transamidation of OPN promotes OPN dimerization and crosslinking to collagen42 and alters the capacity of OPN to regulate calcium-containing crystal deposition in the extracellular matrix.43 The physiological significance of the relationship between TG2 and OPN in calcified arteries was recently highlighted by the discovery that most of the OPN extracted from MGP−/− mouse aortas is polymerized in association with TG2 expression and isopeptide bond formation.44
Adhesion to the basement membrane protein laminin (or to collagen I fibrils) normally promotes retention of contractile SMC differentiation.3 However, cultured TG2−/− SMCs developed synthetic differentiation on laminin. Significantly, TG2 transamidation of RhoA places RhoA in a constitutively active state, and RhoA maintains SMC contractile differentiation partly by suppressing the activity of Akt,45 a serine/threonine kinase downstream of phosphatidylinositol 3-kinase that is stimulated by multiple receptor tyrosine kinases, functions as a cell survival and growth promoter, and also inhibits calcification by SMCs.46 However, fibronectin-binding integrin coreceptor activity, GTPase signaling activity, and phospholipase Cδ1 binding by TG218,20,47 each could modulate differentiation of SMCs. For example, guanine nucleotide-bound TG2 binds the cytoplasmic tail peptide GFFKR motif in α integrin subunits, including α1, -5, -V, and -IIb, and thereby inhibits fibroblast migration.47
In this study, we complemented SMC culture studies with aortic organ culture experiments that yielded very similar results in response to stimulation with a Pi donor. Removal of SMCs from their physiological extracellular matrix for cell culture experiments herein was informative for SMC responses to matrix alterations and stress, but, significantly, we observed few robust differences between normal and freshly isolated TG2-deficient aortas and aortic SMCs for the chondro–osseous, and contractile and synthetic differentiation, markers and regulators studied. Thus, primary functions of TG2 in the aorta and other vessels1,28 likely become evident when arteries remodel in response to injury.
Limitations of this study included lack of study of signal transduction mechanisms. We did not determine whether transamidation and deamidation of extracellular matrix proteins20 by TG2 modulates matrix–cell communication, SMC condensation into nodules, and hydroxyapatite growth to promote calcification. Significantly, crosslinking of fibronectin by TG2 on the cell surface promotes activation of RhoA,48 and increased RhoA activity stimulates chondrogenesis.49 We did not test whether SMC intracellular TG2 accounted for incomplete reconstitution by exogenous TG2 of calcification by TG2−/− SMCs, let alone the only partial inhibition of aortic explant calcification by micromolar Boc-DON-Gln-Ile-Val-OMe. SMC transdifferentiation to osteoblasts and chondrocytes has been described previously4 but is not universally accepted. It remains possible that expansion of small numbers of pericytes, vascular stem cells, and adventitial myofibroblasts within SMC preparations and their transition to osteoblastic differentiation5,9,11,12 contributed to calcification events described. We did not explore why differences appear to exist between TG isoenzyme expression patterns in small mesenteric arteries28 and, described here, in the aorta and whether this relates to vascular calcification. We also have not assessed direct impact on differentiation and function of TG2-deficient SMCs of TG5, a TG isoenzyme first discovered in epidermis that regulates keratinocyte differentiation but also is expressed outside of the skin.50 Last, we have not yet extended these studies to in vivo analyses of arterial calcification.
Our results provide further evidence for TG2 modulating the nature of the SMC differentiation response and phenotypic features of arterial responses to injury,1,28 such as patterns of intima and media repair and remodeling. Our findings reveal that indirect effects complement direct effects of TG2 on SMCs in artery repair. These include regulation of expression of OPG, an inhibitor of both atherosclerotic lesion progression and calcification,16 and suppression of the expression of OPN, which promotes matrix metalloproteinase-9 activation, induces oxidative stress and matrix metalloproteinase-2 expression in SMCs, and is proatherogenic.51 Effects of exogenous TG2 (and FXIIIA) on SMCs here are significant because both SMCs and cells that interact with SMCs, including endothelial cells and macrophages, could release TG2 in normal and diseased arteries, and activated macrophages can release FXIIIA.25,52,53 Significantly, atorvastatin promotes endothelial TG2 expression.54 It would be of interest to assess the role of TG2 in stabilization of atherosclerotic plaques by statins.
SMCs are heterogeneous within atherosclerotic lesions.55,56 Furthermore, TG2 catalytic activity can become deficient via decreased TG2 expression, increased TG2 proteolysis, and increased conversion of TG2 to the GTP-bound form.20 Therefore, our results may point to a new paradigm in which differential effects of clones of TG2-sufficient versus TG2-deficient SMCs modulate the phenotype of arterial repair in atherosclerosis and other forms of arterial injury. In essence, although TG2 mediates arterial remodeling to vasoconstriction1 and limits the size and possibly necrotic core expansion of atherosclerotic lesions,25 robust TG2 release in the course of artery wall repair has the potential to promote calcification. The normal developmental phenotype of TG2−/− mice and the capacity of pharmacological TG inhibition specific for TG2 to inhibit Pi-induced aortic explant calcification buttress the translational potential of our findings for arterial calcification, particularly that associated with hyperphosphatemia in chronic kidney disease.
Supplementary Material
Acknowledgments
We gratefully acknowledge Paul Clopton (San Diego Veterans Affairs Medical Center) for biostatistical analyses.
Sources of Funding
Supported by grants from the Department of Veterans Affairs and the NIH (National Heart, Lung, and Blood Institute; National Institute of Arthritis and Musculoskeletal and Skin Diseases; HL077360, AR54135, and AR049366) (to R.A.T.).
Footnotes
Disclosures
R.A.T. serves as consultant and receives research support from TAP Pharmaceuticals.
References
- 1.Bakker EN, Buus CL, Spaan JA, Perree J, Ganga A, Rolf TM, Sorop O, Bramsen LH, Mulvany MJ, Vanbavel E. Small artery remodeling depends on tissue-type transglutaminase. Circ Res. 2005;96:119–126. doi: 10.1161/01.RES.0000151333.56089.66. [DOI] [PubMed] [Google Scholar]
- 2.Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004;15:237–254. doi: 10.1016/j.cytogfr.2004.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Raines EW, Koyama H, Carragher NO. The extracellular matrix dynamically regulates smooth muscle cell responsiveness to PDGF. Ann N Y Acad Sci. 2000;902:39–51. doi: 10.1111/j.1749-6632.2000.tb06299.x. [DOI] [PubMed] [Google Scholar]
- 4.Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, Schinke T, Karsenty G, Giachelli CM. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and down-regulation of smooth muscle lineage markers. Circ Res. 2001;89:1147–1154. doi: 10.1161/hh2401.101070. [DOI] [PubMed] [Google Scholar]
- 5.Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 2004;24:1161–1170. doi: 10.1161/01.ATV.0000133194.94939.42. [DOI] [PubMed] [Google Scholar]
- 6.Speer MY, Chien YC, Quan M, Yang HY, Vali H, McKee MD, Giachelli CM. Smooth muscle cells deficient in osteopontin have enhanced susceptibility to calcification in vitro. Cardiovasc Res. 2005;66:324–333. doi: 10.1016/j.cardiores.2005.01.023. [DOI] [PubMed] [Google Scholar]
- 7.Yang H, Curinga G, Giachelli CM. Elevated extracellular calcium levels induce smooth muscle cell matrix mineralization in vitro. Kidney Int. 2004;66:2293–2299. doi: 10.1111/j.1523-1755.2004.66015.x. [DOI] [PubMed] [Google Scholar]
- 8.Johnson K, Polewski M, van Etten D, Terkeltaub R. Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1−/− mice. Arterioscler Thromb Vasc Biol. 2005;25:686–691. doi: 10.1161/01.ATV.0000154774.71187.f0. [DOI] [PubMed] [Google Scholar]
- 9.Tintut Y, Alfonso Z, Saini T, Radcliff K, Watson K, Bostrom K, Demer LL. Multilineage potential of cells from the artery wall. Circulation. 2003;108:2505–2510. doi: 10.1161/01.CIR.0000096485.64373.C5. [DOI] [PubMed] [Google Scholar]
- 10.Shao JS, Cheng SL, Pingsterhaus JM, Charlton-Kachigian N, Loewy AP, Towler DA. Msx2 promotes cardiovascular calcification by activating paracrine Wnt signals. J Clin Invest. 2005;115:1210–1220. doi: 10.1172/JCI24140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Towler DA, Bidder M, Latifi T, Coleman T, Semenkovich CF. Diet-induced diabetes activates an osteogenic gene regulatory program in the aortas of low density lipoprotein receptor-deficient mice. J Biol Chem. 1998;273:30427–30434. doi: 10.1074/jbc.273.46.30427. [DOI] [PubMed] [Google Scholar]
- 12.Alexander MY, Wilkinson FL, Kirton JP, Rock CF, Collett GD, Jeziorska M, Smyth JV, Heagerty AM, Canfield AE. Identification and characterization of vascular calcification-associated factor, a novel gene upregulated during vascular calcification in vitro and in vivo. Arterioscler Thromb Vasc Biol. 2005;25:1851–1857. doi: 10.1161/01.ATV.0000175750.94742.46. [DOI] [PubMed] [Google Scholar]
- 13.Rutsch F, Ruf N, Vaingankar S, Toliat MR, Suk A, Hohne W, Schauer G, Lehmann M, Roscioli T, Schnabel D, Epplen JT, Knisely A, Superti-Furga A, McGill J, Filippone M, Sinaiko AR, Vallance H, Hinrichs B, Smith W, Ferre M, Terkeltaub R, Nurnberg P. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat Genet. 2003;34:379–381. doi: 10.1038/ng1221. [DOI] [PubMed] [Google Scholar]
- 14.Speer MY, McKee MD, Guldberg RE, Liaw L, Yang HY, Tung E, Karsenty G, Giachelli CM. Inactivation of the osteopontin gene enhances vascular calcification of matrix Gla protein-deficient mice: evidence for osteopontin as an inducible inhibitor of vascular calcification in vivo. J Exp Med. 2002;196:1047–1055. doi: 10.1084/jem.20020911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, Boyle WJ, Simonet WS. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, Schwartz SM, Giachelli CM, Rosenfeld ME. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE−/− mice. Arterioscler Thromb Vasc Biol. 2006;26:2117–2124. doi: 10.1161/01.ATV.0000236428.91125.e6. [DOI] [PubMed] [Google Scholar]
- 17.Johnson KA, van Etten D, Nanda N, Graham RM, Terkeltaub RA. Distinct transglutaminase 2-independent and transglutaminase 2-dependent pathways mediate articular chondrocyte hypertrophy. J Biol Chem. 2003;278:18824–18832. doi: 10.1074/jbc.M301055200. [DOI] [PubMed] [Google Scholar]
- 18.Johnson KA, Terkeltaub RA. External GTP-bound transglutaminase 2 is a molecular switch for chondrocyte hypertrophic differentiation and calcification. J Biol Chem. 2005;280:15004–15012. doi: 10.1074/jbc.M500962200. [DOI] [PubMed] [Google Scholar]
- 19.Nurminskaya M, Kaartinen MT. Transglutaminases in mineralized tissues. Front Biosci. 2006;11:1591–1606. doi: 10.2741/1907. [DOI] [PubMed] [Google Scholar]
- 20.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 21.Merz D, Liu R, Johnson K, Terkeltaub R. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J Immunol. 2003;171:4406–4415. doi: 10.4049/jimmunol.171.8.4406. [DOI] [PubMed] [Google Scholar]
- 22.Zemskov EA, Janiak A, Hang J, Waghray A, Belkin AM. The role of tissue transglutaminase in cell-matrix interactions. Front Biosci. 2006;11:1057–1076. doi: 10.2741/1863. [DOI] [PubMed] [Google Scholar]
- 23.Johnson K, Hashimoto S, Lotz M, Pritzker K, Terkeltaub R. Interleukin-1 induces pro-mineralizing activity of cartilage tissue transglutaminase and factor XIIIa. Am J Pathol. 2001;159:149–163. doi: 10.1016/S0002-9440(10)61682-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Auld GC, Ritchie H, Robbie LA, Booth NA. Thrombin upregulates tissue transglutaminase in endothelial cells: a potential role for tissue transglutaminase in stability of atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 2001;21:1689–1694. doi: 10.1161/hq1001.097063. [DOI] [PubMed] [Google Scholar]
- 25.Boisvert WA, Rose DM, Boullier A, Quehenberger O, Sydlaske A, Johnson KA, Curtiss LK, Terkeltaub R. Leukocyte Transglutaminase 2 Expression Limits Atherosclerotic Lesion Size. Arterioscler Thromb Vasc Biol. 2006;26:563–569. doi: 10.1161/01.ATV.0000203503.82693.c1. [DOI] [PubMed] [Google Scholar]
- 26.Haroon ZA, Wannenburg T, Gupta M, Greenberg CS, Wallin R, Sane DC. Localization of tissue transglutaminase in human carotid and coronary artery atherosclerosis: implications for plaque stability and progression. Lab Invest. 2001;81:83–93. doi: 10.1038/labinvest.3780214. [DOI] [PubMed] [Google Scholar]
- 27.Nanda N, Iismaa SE, Owens WA, Husain A, Mackay F, Graham RM. Targeted inactivation of Gh/tissue transglutaminase II. J Biol Chem. 2001;276:20673–20678. doi: 10.1074/jbc.M010846200. [DOI] [PubMed] [Google Scholar]
- 28.Bakker EN, Pistea A, Spaan JA, Rolf T, de Vries CJ, van Rooijen N, Candi E, VanBavel E. Flow-dependent remodeling of small arteries in mice deficient for tissue-type transglutaminase: possible compensation by macrophage-derived factor XIII. Circ Res. 2006;99:86–92. doi: 10.1161/01.RES.0000229657.83816.a7. [DOI] [PubMed] [Google Scholar]
- 29.Jono S, McKee MD, Murry CE, Shioi A, Nishizawa Y, Mori K, Morii H, Giachelli CM. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:e10–e17. doi: 10.1161/01.res.87.7.e10. [DOI] [PubMed] [Google Scholar]
- 30.Thyberg J, Hultgardh-Nilsson A, Kallin B. Inhibitors of ADP-ribosylation suppress phenotypic modulation and proliferation of smooth muscle cells cultured from rat aorta. Differentiation. 1995;59:243–252. doi: 10.1046/j.1432-0436.1995.5940243.x. [DOI] [PubMed] [Google Scholar]
- 31.Bonin LR, Madden K, Shera K, Ihle J, Matthews C, Aziz S, Perez-Reyes N, McDougall JK, Conroy SC. Generation and characterization of human smooth muscle cell lines derived from atherosclerotic plaque. Arterioscler Thromb Vasc Biol. 1999;19:575–587. doi: 10.1161/01.atv.19.3.575. [DOI] [PubMed] [Google Scholar]
- 32.Thyberg J. Differentiated properties and proliferation of arterial smooth muscle cells in culture. Int Rev Cytol. 1996;169:183–265. doi: 10.1016/s0074-7696(08)61987-7. [DOI] [PubMed] [Google Scholar]
- 33.Lomashvili KA, Cobbs S, Hennigar RA, Hardcastle KI, O’Neill WC. Phosphate-induced vascular calcification: role of pyrophosphate and osteopontin. J Am Soc Nephrol. 2004;15:1392–1401. doi: 10.1097/01.asn.0000128955.83129.9c. [DOI] [PubMed] [Google Scholar]
- 34.Madi A, Karpati L, Kovacs A, Muszbek L, Fesus L. High-throughput scintillation proximity assay for transglutaminase activity measurement. Anal Biochem. 2005;343:256–262. doi: 10.1016/j.ab.2005.05.034. [DOI] [PubMed] [Google Scholar]
- 35.Murshed M, Harmey D, Millan JL, McKee MD, Karsenty G. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005;19:1093–1104. doi: 10.1101/gad.1276205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res. 2006;98:905–912. doi: 10.1161/01.RES.0000216409.20863.e7. [DOI] [PubMed] [Google Scholar]
- 37.Villa-Bellosta R, Bogaert YE, Levi M, Sorribas V. Characterization of phosphate transport in rat vascular smooth muscle cells. Implications for vascular calcification. Arterioscler Thromb Vasc Biol. 2007;27:1030–1036. doi: 10.1161/ATVBAHA.106.132266. [DOI] [PubMed] [Google Scholar]
- 38.Beck GR., Jr Inorganic phosphate as a signaling molecule in osteoblast differentiation. J Cell Biochem. 2003;90:234–243. doi: 10.1002/jcb.10622. [DOI] [PubMed] [Google Scholar]
- 39.Bostrom K, Watson KE, Horn S, Wortham C, Herman IM, Demer LL. Bone morphogenetic protein expression in human atherosclerotic lesions. J Clin Invest. 1993;91:1800–1809. doi: 10.1172/JCI116391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Suzuki A, Ghayor C, Guicheux J, Magne D, Quillard S, Kakita A, Ono Y, Miura Y, Oiso Y, Itoh M, Caverzasio J. Enhanced expression of the inorganic phosphate transporter Pit-1 is involved in BMP-2-induced matrix mineralization in osteoblast-like cells. J Bone Miner Res. 2006;21:674–683. doi: 10.1359/jbmr.020603. [DOI] [PubMed] [Google Scholar]
- 41.Chau DY, Collighan RJ, Verderio EA, Addy VL, Griffin M. The cellular response to transglutaminase-cross-linked collagen. Biomaterials. 2005;26:6518–6529. doi: 10.1016/j.biomaterials.2005.04.017. [DOI] [PubMed] [Google Scholar]
- 42.Kaartinen MT, Pirhonen A, Linnala-Kankkunen A, Maenpaa PH. Cross-linking of osteopontin by tissue transglutaminase increases its collagen binding properties. J Biol Chem. 1999;274:1729–1735. doi: 10.1074/jbc.274.3.1729. [DOI] [PubMed] [Google Scholar]
- 43.Rosenthal AK, Gohr CM, Uzuki M, Masuda I. Osteopontin promotes pathologic mineralization in articular cartilage. Matrix Biol. 2007;26:96–105. doi: 10.1016/j.matbio.2006.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaartinen MT, Murshed M, Karsenty G, McKee MD. Osteopontin upregulation and polymerization by transglutaminase 2 in calcified arteries of matrix Gla protein-deficient mice. J Histochem Cytochem. 2007;55:375–386. doi: 10.1369/jhc.6A7087.2006. [DOI] [PubMed] [Google Scholar]
- 45.Guilluy C, Rolli-Derkinderen M, Tharaux PL, Melino G, Pacaud P, Loirand G. Transglutaminase-dependent RhoA activation and depletion by serotonin in vascular smooth muscle cells. J Biol Chem. 2007;282:2918–2928. doi: 10.1074/jbc.M604195200. [DOI] [PubMed] [Google Scholar]
- 46.Collett GD, Sage AP, Kirton JP, Alexander MY, Gilmore AP, Canfield AE. Axl/phosphatidylinositol 3-kinase signaling inhibits mineral deposition by vascular smooth muscle cells. Circ Res. 2007;100:502–509. doi: 10.1161/01.RES.0000258854.03388.02. [DOI] [PubMed] [Google Scholar]
- 47.Kang SK, Yi KS, Kwon NS, Park KH, Kim UH, Baek KJ, Im MJ. Alpha1B-adrenoceptor signaling and cell motility: GTPase function of Gh/transglutaminase 2 inhibits cell migration through interaction with cytoplasmic tail of integrin alpha subunits. J Biol Chem. 2004;279:36593–36600. doi: 10.1074/jbc.M402084200. [DOI] [PubMed] [Google Scholar]
- 48.Janiak A, Zemskov EA, Belkin AM. Cell surface transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src-p190RhoGAP signaling pathway. Mol Biol Cell. 2006;17:1606–1619. doi: 10.1091/mbc.E05-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woods A, Beier F. RhoA/ROCK signaling regulates chondrogenesis in a context-dependent manner. J Biol Chem. 2006;281:13134–13140. doi: 10.1074/jbc.M509433200. [DOI] [PubMed] [Google Scholar]
- 50.Aeschlimann D, Koeller MK, Allen-Hoffmann BL, Mosher DF. Isolation of a cDNA encoding a novel member of the transglutaminase gene family from human keratinocytes. Detection and identification of transglutaminase gene products based on reverse transcription-polymerase chain reaction with degenerate primers. J Biol Chem. 1998;273:3452–3460. doi: 10.1074/jbc.273.6.3452. [DOI] [PubMed] [Google Scholar]
- 51.Lai CF, Seshadri V, Huang K, Shao JS, Cai J, Vattikuti R, Schumacher A, Loewy AP, Denhardt DT, Rittling SR, Towler DA. An osteopontin-NADPH oxidase signaling cascade promotes pro-matrix metalloproteinase 9 activation in aortic mesenchymal cells. Circ Res. 2006;98:1479–1489. doi: 10.1161/01.RES.0000227550.00426.60. [DOI] [PubMed] [Google Scholar]
- 52.Akimov SS, Belkin AM. Cell surface tissue transglutaminase is involved in adhesion and migration of monocytic cells on fibronectin. Blood. 2001;98:1567–1576. doi: 10.1182/blood.v98.5.1567. [DOI] [PubMed] [Google Scholar]
- 53.Dardik R, Inbal A. Complex formation between tissue transglutaminase II (tTG) and vascular endothelial growth factor receptor 2 (VEGFR-2): proposed mechanism for modulation of endothelial cell response to VEGF. Exp Cell Res. 2006;312:2973–2982. doi: 10.1016/j.yexcr.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 54.Soehnlein O, Eskafi S, Schmeisser A, Kloos H, Daniel WG, Garlichs CD. Atorvastatin induces tissue transglutaminase in human endothelial cells. Biochem Biophys Res Commun. 2004;322:105–109. doi: 10.1016/j.bbrc.2004.07.087. [DOI] [PubMed] [Google Scholar]
- 55.Adams LD, Geary RL, Li J, Rossini A, Schwartz SM. Expression profiling identifies smooth muscle cell diversity within human intima and plaque fibrous cap: loss of RGS5 distinguishes the cap. Arterioscler Thromb Vasc Biol. 2006;26:319–325. doi: 10.1161/01.ATV.0000196647.45718.d6. [DOI] [PubMed] [Google Scholar]
- 56.Mahoney WM, Schwartz SM. Defining smooth muscle cells and smooth muscle injury. J Clin Invest. 2005;115:221–224. doi: 10.1172/JCI24272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.