

Abstract
Understanding the mechanisms governing the switch between hypoxia-induced adaptive and pathological transcription may reveal novel therapeutic targets for stroke. Using an in vitro hypoxia model that temporally separates these divergent responses, we found apoptotic signaling was preceded by a decline in c/EBP-β activity and was associated with markers of ER-stress including transient eIF2α phosphorylation, and the delayed induction of the bZIP proteins ATF4 and CHOP-10. Pretreatment with the eIF2α phosphatase inhibitor salubrinal blocked the activation of caspase-3, indicating that ER-related stress responses are integral to this transition. Delivery of either full-length, or a transcriptionally inactive form of c/EBP-β protected cultures from hypoxic challenge, in part by inducing levels of the anti-apoptotic protein Bcl-2. These data indicate that the pathologic response in cortical neurons induced by hypoxia involves both the loss of c/EBP-β-mediated survival signals and activation of pro-death pathways originating from the endoplasmic reticulum.
Keywords: c/EBP-β, bZIP, Hypoxia, Neuron, Delayed death, Apoptosis
Introduction
Neuroprotective trials for stroke have focused mainly on molecular targets activated early in the pathological signaling cascade. However, evidence suggest that cell-autonomous delayed apoptotic signaling plays a critical role in determining the ultimate burden of neuron loss after injury (Abe et al., 1995; Schwamm et al., 1998). Hypoxia is a potent stimulus for de novo gene expression, and sub-lethal hypoxic stress can enhance cell survival through the regulated expression of factors such as erythropoietin and vascular endothelial growth factor (Dirnagl et al., 2003; Jones and Bergeron, 2001). This process, also referred to as ischemic preconditioning, is supported in part by the activation of the hypoxia-inducible factor (HIF-1α) and an array of immediate early transcription factors with diverse biological functions including c-Jun and Egr-1/Krox-24 (Collaco-Moraes et al., 1994; Herdegen and Leah, 1998; Hsu et al., 1993). Stroke-induced gene expression also plays a critical role in promoting delayed neuron loss after ischemia (Honkaniemi et al., 1996). In this regard, pre-treatment with the macromolecular synthesis inhibitor cycloheximide confers neuroprotection (Du et al., 1996; Gwag et al., 1995). Data from in vitro studies and models of global ischemia indicate that this death mechanism is cell-autonomous. And from a therapeutic perspective, identification of the key regulatory nodes in hypoxia signaling networks that discriminate between these divergent transcriptional programs would be advantageous.
One potential sensor capable of triggering both adaptive and pathologic signaling after stroke is the endoplasmic reticulum (ER), shown previously to influence other diseases affecting the central nervous system (Kaufman, 2002; Rao et al., 2004). The physiologic changes associated with ischemia also activate stress-sensing proteins resident in the ER, which in turn stimulate adaptive transcription via the unfolded protein response (UPR) (Harding et al., 2002). For example, translational arrest induced by PERK-mediated phosphorylation of the translation initiation factor eIF2α at Ser51 is associated with cell survival and occurs in neurons within the ischemic penumbra (Kumar et al., 2001; Liu et al., 2006; Mengesdorf et al., 2002). Similarly, the bZIP transcription factor ATF6 and the inositol-requiring transmembrane kinase and endonuclease-1α (IRE-1α) regulate the expression of BiP/GRP78 and other factors that enhance the folding capacity of the ER. However, hyper-activation of ER-stress pathways can have negative consequences. Prolonged eIF2α inactivation induces the protein phosphatase regulatory subunit GADD34, which reverses eIF2α-mediated translational inhibition promoting programmed cell death (Brush et al., 2003). As such, phosphatase inhibitors like salubrinal that prolong translational arrest are protective against ER-stress (Boyce et al., 2005; Sokka et al., 2007). And while activation of CHOP-10 may enhance mitochondrial function through the direct regulation of heat shock proteins including mtDnaJ and ClpP, deletion of CHOP-10 is neuroprotective after stroke (Tajiri et al., 2004; Zhao et al., 2002). Lastly, activated caspase-3, caspase-12 and several BH3 proteins (i.e., Bcl-2, Bax, PUMA and others) associate with, and link the ER to the cellular apoptotic signaling machinery (Masud et al., 2007; Rao et al., 2004; Reimertz et al., 2003). A better understanding of the interplay between hypoxia, ER-stress signaling and the factors controlling downstream transcriptional responses to hypoxia could have significant implications for the treatment of stroke.
In the current study we characterized a translation-dependent in vitro model of hypoxia-induced neuronal apoptosis. By defining the temporal boundaries separating adaptation from the commitment to cell death, we sought to identify the factors required to activate neuronal death following prolonged hypoxic stress. In the current work, we report a novel cell survival function for the bZIP factor c/EBP-β, and show that the loss of c/EBP-β activity precedes the onset of cell death promoted in part by stress signals emanating from the endoplasmic reticulum. Furthermore, based on the observed delayed induction of the heterodimeric factors ATF4 and CHOP-10, we propose a model in which hypoxia-induced ER-stress responses shift the activity of the bZIP protein network from an initial adaptive response, towards a pro-apoptotic transcriptional program.
Results
Chronic hypoxia induces delayed neuronal death in cortical neurons
Classical oxygen-glucose deprivation (OGD) produces acute necrosis in mature cortical cultures, however sub-lethal challenge or OGD performed in the presence of glutamate antagonists can trigger a delayed form of neuron death that is dependent on gene expression (Gwag et al., 1995). To study the role of gene expression in the delayed loss of neurons after stroke, we developed an in vitro model of hypoxia-induced neuronal apoptosis using dissociated embryonic cortical cultures. By DIV7, most cells in culture express NeuN and have developed a dense network of β-III tubulin positive axonal projections (data not shown). Exposure to hypoxia (0.5% O2) induces procaspase-3 cleavage and nuclear pyknosis in up to 40% of neurons (Fig. 1A). Time course analyses indicate that both phenotypes depend on the duration of hypoxia (Fig. 1B) and not on media glucose concentrations provided cultures are maintained at or above 10 mM prior to the onset of hypoxia (Fig. S1). Nuclear pyknosis plateaued 24 h after the onset of hypoxia and was followed by the delayed uptake of trypan blue beginning 48 h post-exposure consistent with the phenomenon of secondary necrosis (Fig. S2) (Ankarcrona et al., 1995). And while pre-treatment with a combination of the glutamate receptor antagonists MK-801 (10 μM) and CNQX (100 μM) had no effect on neuron survival, addition 1 μg/ml of the translation inhibitor cycloheximide conferred complete protection against hypoxia-induced pyknosis (Fig. 1C). These data suggest that hypoxia alone is sufficient to induce a cell-autonomous death program in a subset of dissociated cortical neurons that requires de novo gene expression.
Fig. 1.
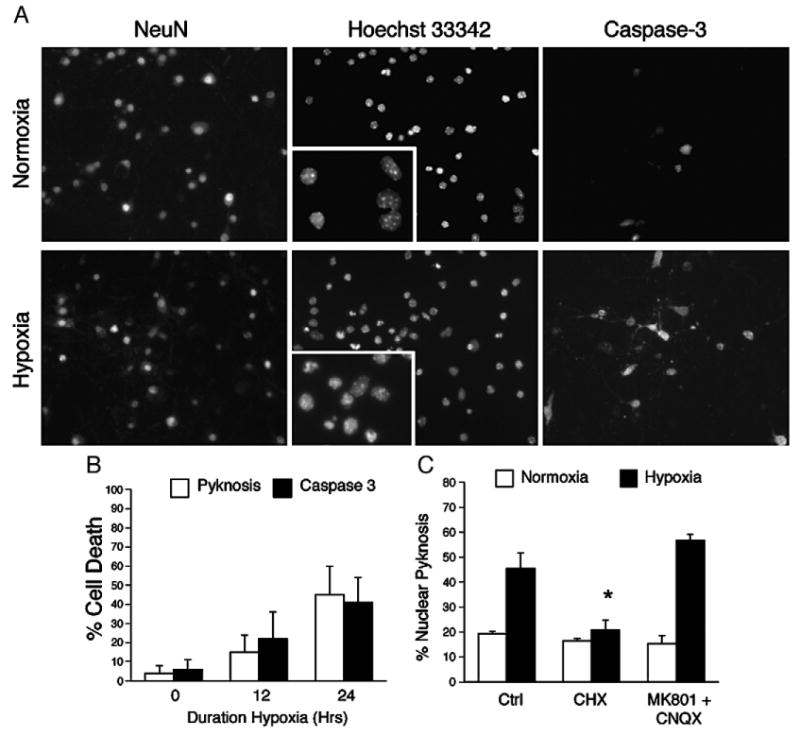
Chronic hypoxia activates apoptosis in dissociated neuronal cultures. (A) Continuous hypoxia induces nuclear pyknosis and caspase-3 cleavage in DIV7 cortical neurons. Neuronal cultures were exposed to hypoxia (0.5% O2) for 18 h and analyzed by fluorescence microscopy for expression of the neuronal marker NeuN, nuclear pyknosis using the DNA dye Hoechst 33342, and the cleavage of caspase-3. (B) Nuclear pyknosis approximates caspase-3 cleavage in vitro. (C) Hypoxia-induced neuron death requires de novo translation. Cultures were pretreated with cycloheximide (CHX 1 μg/ml, 3.5 μM) or a combination of the glutamate receptor antagonists MK-801 (100 μM) and CNQX (10 μM), and exposed hypoxia (0.5% O2, 18 h; filled bars) and analyzed for nuclear pyknosis versus control (21% O2; open bars) sister cultures. Data represent the average±S.D. of results from six non-overlapping fields across replicates (n=4; *=P<0.05).
Defining adaptive and pathologic phases of gene expression in hypoxic cultures
Ischemic tolerance induced by sub-lethal challenge confers delayed neuroprotection through the expression of mRNA species with adaptive properties (Dirnagl et al., 2003). To test whether the onset of cell loss in our model was associated with the sequential expression of adaptive and pathologic genes, we performed a time course analysis of gene expression using quantitative RT-PCR against transcriptional targets previously validated using in vivo stroke models (Kim et al., 2004; Pirianov et al., 2007; Zhang et al., 2007). Selected genes included those associated with either ischemic preconditioning and survival (VEGF, Hexokinase II and the type-1 glucose transporter), or apoptosis (BNIP3, PUMA and NOXA). Results demonstrate that the adaptive expression began early (3–6 h) and remained elevated, while apoptotic transcription was delayed in onset (9–12 h) in keeping with the observed kinetics of cell death (12–18 h) (Fig. 2). If the observed transcriptional profiles are representative of the population response to hypoxia, these results indicate neurons are capable of mounting separable adaptive and pathological responses. Alternatively, if embryonic cortical cultures recapitulate patterns of selective vulnerability observed in vivo, it is possible that adaptive and pro-apoptotic gene expressions occur in distinct cellular populations. Either way, the existence of qualitatively and temporally ditinct transcriptional responses to hypoxic stress provide a framework to study the genetic networks regulating their activation.
Fig. 2.
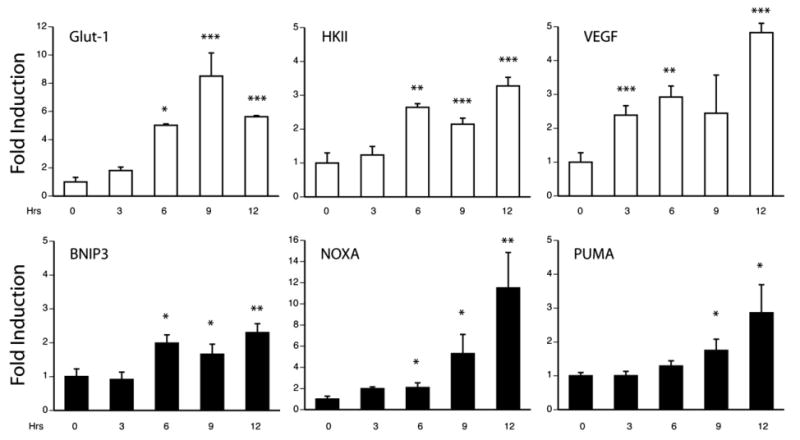
Transcriptional profiling by quantitative RT-PCR defines adaptive and pathologic phases of gene expression. Neuronal cultures were made hypoxic for 3, 6, 9 and 12 h, total RNA was harvested and analyzed by quantitative PCR for the adaptive targets vascular endothelial growth factor (VEGF), hexokinase II (HKII) and the glucose transporter (Glut-1) (open bars), or the pro-apoptotic factors BNIP3, NOXA and PUMA (filled bars). Data are expressed as fold-induction relative to normoxic samples and expressed as the average±S.D. of results from sister wells (n=3; *=P<0.05, **=P<0.01, ***=P<0.001).
Defining the role of ER-stress responses in hypoxia-induced delayed neuronal loss
The endoplasmic reticulum has emerged as an important sensor of cellular stress in diseases affecting the central nervous system including stroke (Rao et al., 2004). Shared, as well as ER-stress specific pathways have evolved to respond to diverse stimuli including hypoxia, amino acid starvation, hypoglycemia and perturbations in calcium homeostasis (Fig. 3A). The proximate sensors of ER-stress include PERK, ATF6 and IRE-1α, which exist in an inactive form bound to the ER chaperonin GRP78/BiP (Sommer and Jarosch, 2002). Exposure to physiologic stress enhances the release and homo-dimerization of these factors activating their latent signaling capacity. For example, PERK-mediated phosphorylation of eIF2α at Ser51 triggers the arrest of cap-dependent translation, an event that is both cytoprotective under conditions of ER-stress and is associated with adaptive responses in the ischemic penumbra (Harding et al., 2000; Mengesdorf et al., 2002). This and other strategies, including the regulated proteolysis and release of the transcriptionally active amino terminal fragment of ATF6, have evolved to regulate the expression of nuclear transcriptional targets with both adaptive and pathological effects on the cell.
Fig. 3.
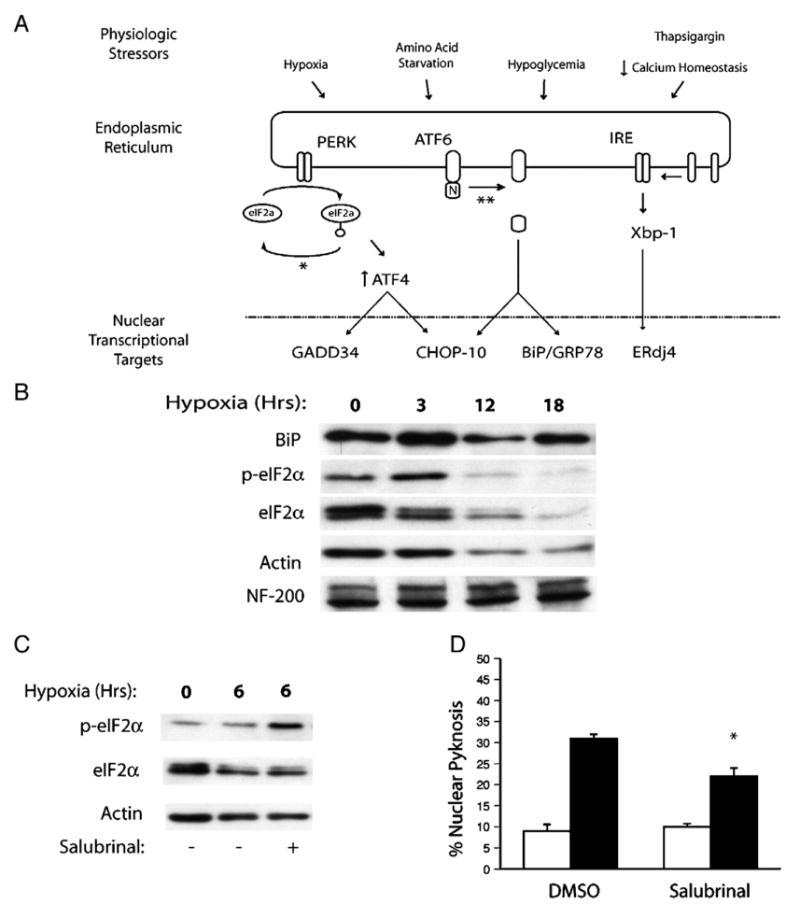
The unfolded protein response regulates neuron survival after chronic hypoxia. (A) Schematic representation of the physiologic stressors associated with ischemic stress, the proximate shared (eIF2α) and specific (ATF6, IRE/Xbp-1) ER sensors responsive to these cues and their downstream nuclear transcriptional targets. The sites of action of the pharmacological inhibitors salubrinal (*) and the serine protease inhibitor AEBSF (**) are indicated. (B) Western analysis indicates that hypoxia stimulates the transient induction of BiP/GRP78 and phosphorylation of eIF2α. Pretreatment with the phosphatase inhibitor salubrinal (10 μM) enhances eIF2α phosphorylation in hypoxic neurons (C), and (D) limits hypoxia-induced neuronal pyknosis in vitro (18 h, 0.5% O2; filled bars). Data for control (open bars) and hypoxic samples and expressed as the average±S.D. (n=3 wells; *=P<0.01).
The observation that expression of the pro-apoptotic Bcl-2 family members NOXA and PUMA were induced in our cultures suggested that ER-stress responses might be involved in propagating delayed apoptotic signals after hypoxic challenge (Li et al., 2006). To assess this, we analyzed hypoxic neuronal lysates by western blotting for markers related to signaling through the shared and ER-stress specific pathways. Hypoxia induced a transient rise in BiP/GRP78 protein, transient phosphorylation of the elongation factor eIF2α at Ser51, a decline in total eIF2α levels, and a relative increase in the amount of the low molecular weight hypo-phosphorylated eIF2α species (Fig. 3B). While levels of actin also declined over time consistent with global translational arrest, neurofilament levels remained stable. The kinetics of eIF2α phosphorylation supported a causal link between the loss of eIF2α inactivation and delayed cell death after hypoxia. To test this, we pretreated cultures with the eIF2α phosphatase inhibitor salubrinal, and compared eIF2α post-translational modification with post-hypoxic neuron survival (Figs. 3C and D). Results indicate that 10 μM salubrinal extended eIF2α inactivation by 3 h and conferred a modest survival benefit compared to DMSO treated control cultures (22±2% vs 31±1%, P<0.01). Interestingly, 10 μM salubrinal did not extend hypoxia-induced eIF2α phosphorylation beyond 6 h (Fig. 4A). Although not directly tested, this observation suggests that hypoxia induced the expression of GADD34, which promotes eIF2α dephosphorylation through direct interactions with protein phosphatase 1c (PP1c) (Brush et al., 2003; Novoa et al., 2001).
Fig. 4.
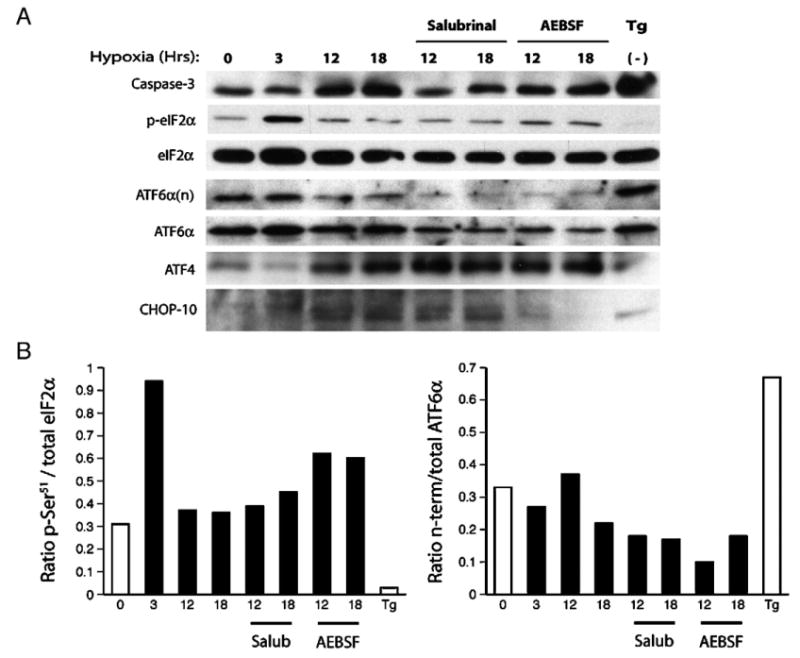
Hypoxia-induced ER-stress responses regulate neuron survival and the delayed expression of the bZIP factors ATF4 and CHOP-10. (A) Protein lysates were harvested from normoxic (0 Hr) or hypoxic cultures (3–18 Hrs; 0.5% O2) and analyzed by western blotting for caspase-3 cleavage, eIF2α phosphorylation, proteolytic cleavage of ATF6, release of the transcriptionally active ATF6 amino-terminus (ATF6α(n)), and induction of the bZIP proteins ATF4 and CHOP-10. Sister cultures were also treated with salubrinal (10 μM), the serine protease inhibitor AEBSF (100 μM) or the Ca2+-ATPase inhibitor thapsigargin (Tg; 1 μg/ml) and exposed to hypoxia (excluding Tg treated samples) as indicated. (B) Western blots were quantified using densitometry and the data are expressed histogram form as the ratios of p-eIF2α/total eIF2α and ATF6α(n)/total ATF6α.
To better understand the relationship between hypoxia and ER-dependent signaling, we next analyzed hypoxia's effects on the shared (eIF2α) and ER-specific signaling involving ATF6, and the downstream factors ATF4 and CHOP-10. To demonstrate these pathways could be activated in vitro, normoxic cultures were treated with the Ca2+-ATPase pump inhibitor thapsigargin (Tg). Tg (1 μg/ml) induced eIF2α dephosphorylation, ATF6 cleavage (detected by release of the amino terminal domain ATF6α(n)), expression of the pro-apoptotic bZIP factor CHOP-10, and the robust activation of caspase-3 (Fig. 4A). While hypoxia alone induced transient eIF2α phosphorylation, triggered the delayed induction of CHOP and ATF4, and stimulated cleavage of caspase-3, it had only a minor effect on ATF6 proteolysis and release of the transcriptionally active amino terminal fragment (Fig. 4B, right). Consistent with our prior results, salubrinal blocked caspase-3 cleavage, enhanced ATF4 expression, and reduced the basal expression and cleavage of ATF6 without influencing levels of CHOP-10. In contrast, the serine protease inhibitor AEBSF disrupted ATF6 processing and CHOP-10 expression, it had only moderate effects on caspase-3 cleavage (Ma et al., 2002; Okada et al., 2003). These data argue that the shared unfolded protein response involving eIF2α is a key intermediate in the activation of delayed apoptotic signaling in this model, while ATF6-mediated responses appear less important in this regard. Moreover, the data demonstrating persistent CHOP-10 expression despite salubrinal treatment suggest that CHOP-10 expression alone is not sufficient to stimulate apoptotic signaling after hypoxic stress.
To identify additional transcription factors that could interface with the ER-signaling apparatus, we queried functional protein association networks using the STRING (Search Tool for Recurring Instances of Neighboring Genes) algorithm. This application draws upon empirically derived public data sets and constructs a map of interacting factors with greatest statistical relevance based on an input set of query proteins (von Mering et al., 2007). In addition to ATF4, CHOP-10 and eIF2α, we included GADD34 in our search given its suspected activity in our system. Results confirmed several expected relationships including ATF4-mediated GADD34 expression, the association of p38MAPK with CHOP-10, and protein–protein interactions between the immediate early genes c-Jun and Fos (Fig. 5A). What was most intriguing was the set of interactions uncovered among the bZIP transcription factors ATF4, CHOP-10, c-Jun and c/EBP-β. And while c-Jun, ATF4 and CHOP have been linked with cellular responses to hypoxia, c/EBP-β's role in hypoxic signaling has not been established (Blais et al., 2004; Ron and Habener, 1992). To investigate this further, we analyzed c-Jun and c/EBP-β expression by western blotting and found that hypoxia stimulated the transient induction of Jun and its downstream transcriptional target c/EBP-β before any appreciable induction of ATF4 was seen (Lin et al., 2002). We also observed that hypoxia induced the robust and transient phosphorylation of c/EBP-β at Thr188 (Fig. 5B), which is required for efficient function of the transcription activation domain (Piwien-Pilipuk et al., 2002). The decline in c/EBP-β protein levels was insensitive to both salubrinal and AEBSF treatment, suggesting that the mechanisms regulating c/EBP-β expression and/or stability are likely PERK and ATF6 independent. This complex pattern of c-Jun, c/EBP-β, CHOP-10 and ATF4 regulation suggested that perhaps shifts in bZIP stoichiometry could drive the formation of pathogenic heterodimers triggering delayed apoptosis in hypoxic neurons (Ohoka et al., 2005; Vinson et al., 2002). Reports describing CHOP and ATF4 driven expression of the Akt inhibitor Trib3 and its role in apoptosis support this idea (Ohoka et al., 2005).
Fig. 5.
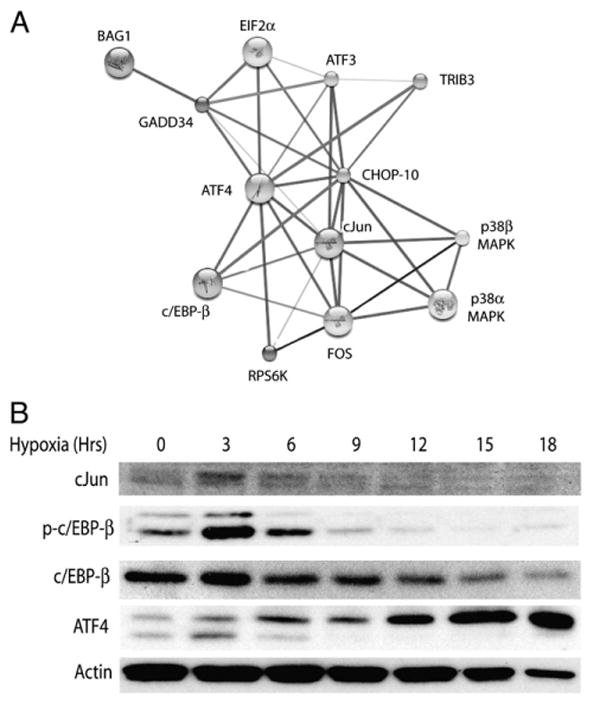
Dynamic regulation of the bZIP family of transcription factors in hypoxic neurons. (A) The STRING protein–protein interaction algorithm (http://string.embl.de/) implicates the bZIP factors c/EBP-β and c-Jun in the regulated activity of ATF4 and CHOP-10. (B) Western analysis of hypoxic neuronal cultures for c-Jun, total and phosphorylated c/EBP-β (Thr188).
To further explore the relationship between hypoxia and the regulation of c/EBP-β activity, we analyzed c/EBP-β expression patterns in cortical neurons by immunocytochemistry using an antibody recognizing the transcriptionally active species phosphorylated at Thr188 (p-c/EBP-β). When compared to the marker NeuN, we found that p-c/EBP-β was ubiquitously expressed and localized to the nucleus of neurons under basal conditions (Fig. 6A). We also determined whether hypoxia-induced nuclear export was involved in the mechanism of c/EBP-β downregulation as described previously in primary mouse hepatocytes exposed to TNFα (Buck et al., 2001). Our analysis indicated that while hypoxia increased immunoreactivity in the neuritic processes, the majority of p-c/EBP-β remained confined to the nucleus. We also observed differences in the relative levels of p-c/EBP-β across individual neurons exposed to hypoxia and hypothesized this could be mechanistically related to the phenomenon of selective vulnerability. To address this possibility, hypoxic cultures were analyzed for the accumulation of pyknotic nuclei relative to either the high or low levels of p-c/EBP-β expression observed. While indirect, the data indicate that the maintenance of c/EBP-β phosphorylation at Thr188 was associated with greater post-hypoxic survival (72±12% vs 15±17%, P<0.05; Fig. 6B), suggesting the early induction and persistence of activated c/EBP-β in culture is an adaptive response to hypoxic challenge.
Fig. 6.
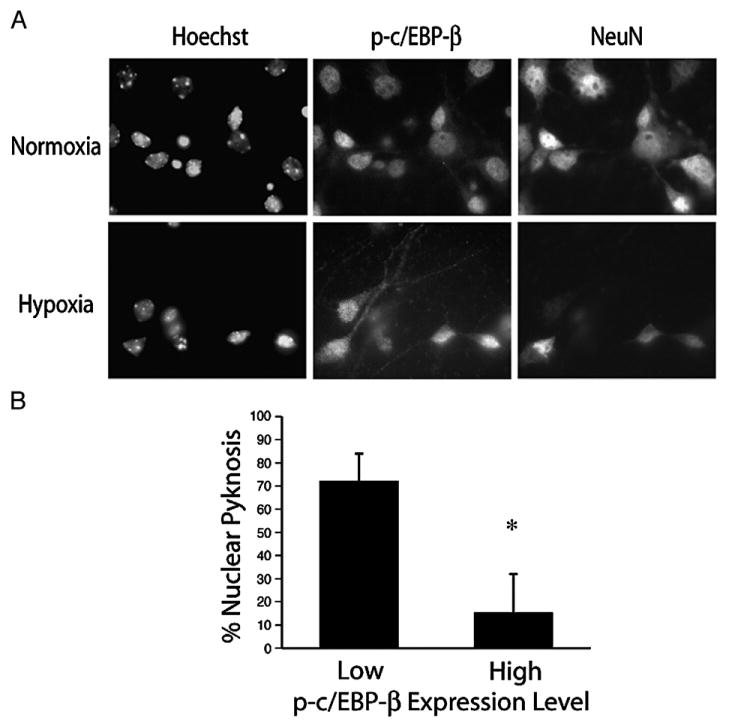
Persistent expression of phosphorylated c/EBP-β Thr188 is a marker for neuron survival following hypoxic stress. (A) Dissociated cortical cultures were exposed to hypoxia (18 h, 0.5% O2) and analyzed by ICC for c/EBP-β Thr188 and the neuronal marker NeuN. (B) Nuclear morphology was determined for neurons expressing either high or low levels of p-c/EBP-β as indicated. Data represent the average±S.D. of results from five non-overlapping fields (*=P<0.05).
Expression of c/EBP-β protects cortical neurons from delayed apoptosis
If the stoichiometry of bZIP heterodimerization governs neuron survival after hypoxic challenge, we hypothesized that the apoptotic switch could be blocked by upregulating c/EBP-β expression. To this end, we generated a bi-cistronic herpes amplicon vectors expressing eGFP, and one of three different cDNAs including full-length c/EBP-β, a dominant-negative form of c/EBP-β (ΔTAD) lacking the transcriptional transactivation domain, or a control vector expressing luciferase (LUC). After confirming equivalent transduction across samples (Fig. 7A), cultures were screened for their relative susceptibility to hypoxia-induced nuclear pyknosis. Results show that transduction with wild-type c/EBP-β virus reduced nuclear pyknosis roughly 4-fold relative to cultures receiving control virus (6.7±1.6% vs 29.7±1.3%, P<0.01; Fig. 7B). Transduction with the ΔTAD construct was also protective albeit to a lesser degree (20.2±4.1% vs 29.7±1.3%, P<0.05). Western blot analysis confirmed the expression of full-length (WT) and the 15-kDa truncated form (ΔTAD) of c/EBP-β in transduced lysates (Fig. 7C). An additional species representing the internally translated, transcriptionally inactive c/EBP-β inhibitor of (LIP) was also identified consistent with previous reports (Fig. 7C) (Calkhoven et al., 2000). In keeping with the pyknosis counts, western analysis demonstrated that in comparison to control cultures, both c/EBP-β constructs attenuated caspase-3 cleavage. We also observed that while eGFP levels declined in control and ΔTAD-transduced cultures, c/EBP-β delivery maintained eGFP expression, which we consider to be a surrogate marker of neuron survival (Fig. 7C, middle). These data indicate that the loss of c/EBP-β activity promotes the switch towards apoptotic signaling in hypoxic cortical neurons.
Fig. 7.
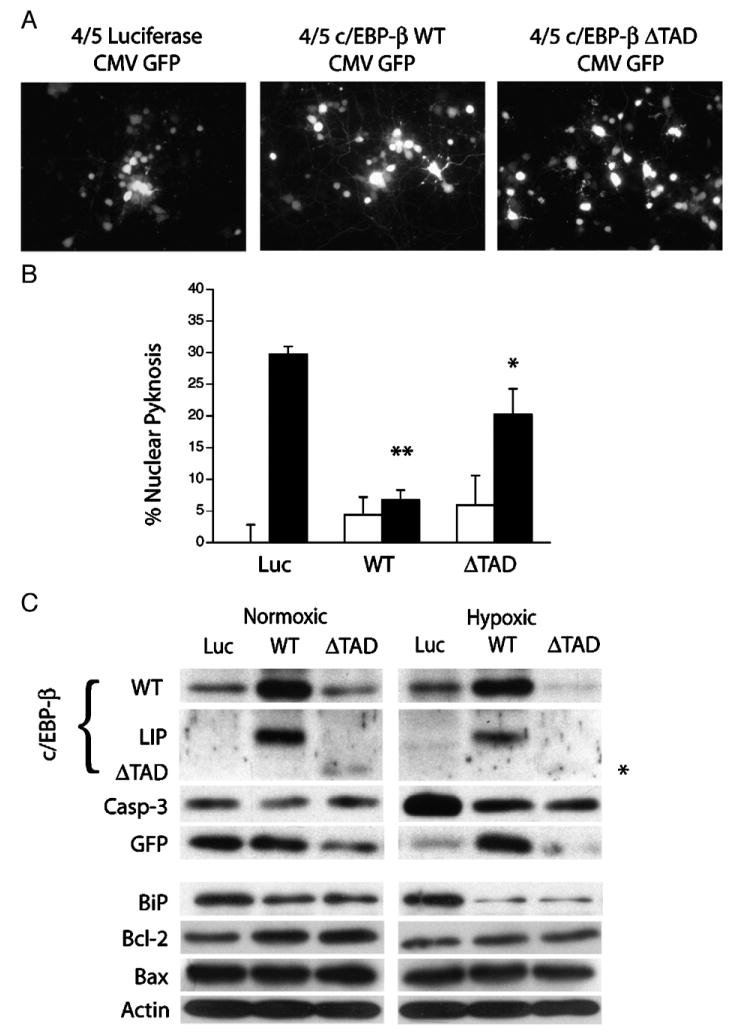
Viral-mediated expression of c/EBP-β supports neuron survival after hypoxic challenge. (A) The bi-cistronic herpes amplicon vector HSVprPucCMVeGFP identifies neurons expressing luciferase (LUC), full-length c/EBP-β (WT), or a truncated form of c/EBP-β (ΔTAD). (B) c/EBP-β expression promotes neuron survival after hypoxic challenge. DIV7 cultures were transduced with the expression constructs as indicated and analyzed for levels of nuclear pyknosis after 18 h of normoxic (open bars) or hypoxic (closed bars) conditions. The data represent the average (±S.D.) pyknosis amongst transduced neurons from five non-overlapping fields (n=3; *=P<0.05, **=P<0.01). (C) Expression of full-length or truncated c/EBP-β (ΔTAD) limit hypoxia-induced caspase-3 cleavage and enhance Bcl-2 levels. Cortical neurons were transduced with wild-type, dominant-negative (ΔTAD) and control virus 12 h prior to hypoxic challenge and subjected to 18 h of hypoxia (0.5% O2). Lysates were harvested and analyzed by western blotting for the expression of the full-length, internally translated inhibitory fragment (LIP), the ΔTAD fragment (asterisk), activated caspase-3, GFP, BiP/GRP78, Bcl-2, and Bax.
Since c/EBP-β induction has been linked with activation of the ER-stress response (Chen et al., 2004a), and c/EBP-β transactivates several mitochondrial heat shock proteins (Zhao et al., 2002), we hypothesized that c/EBP-β might also regulate the expression of the ER chaperonin BiP/GRP78. Unexpectedly, western analysis demonstrated that delivery of either c/EBP-β or ΔTAD reduced the basal expression of BiP, an effect exaggerated by exposure to hypoxia (Fig. 7). This result suggested that either c/EBP-β-LIP or the ΔTAD construct indirectly reduced BiP expression by repressing the CHOP-10 promoter and reducing the amount of CHOP-10 available to transactivate BiP (McCullough et al., 2001). Interestingly, c/EBP-β can also inhibit the transcriptional activity of the tumor-suppressor p53 by blocking its ability to bind DNA (Schneider-Merck et al., 2006). Since CHOP-10 and p53 also inhibit the expression of the pro-survival protein Bcl-2, we hypothesized that c/EBP-β promoted neuron survival by blocking trans-repression of Bcl-2 (McCullough et al., 2001; Miyashita et al., 1994). Western analysis on cultures transduced with either control, full-length or ΔTAD expressing virus revealed that while levels of actin and the pro-apoptotic factor Bax were unaffected, delivery of either full-length c/EBP-β or ΔTAD, but not the control vector induced levels of Bcl-2 protein under normoxic conditions.
Discussion
The role of c/EBP-β in the neuronal response to hypoxic stress
c/EBP-β belongs to a family of related bZIP transcription factors and promotes the differentiation of multiple cell types including neural progenitors. During development, c/EBP-β signaling biases precursor cells to commit to a neural rather than glial lineage (Menard et al., 2002). c/EBP-β activity also induces axonal sprouting after axotomy, and, not surprising, regulates the expression of the neuronal markers NeuN, GAP43 and βIII tubulin (Nadeau et al., 2005). Of note, we have observed that hypoxia-induced loss of c/EBP-β is associated with a decline in both NeuN and β-III tubulin in vitro. c/EBP-β also appears to be a transcriptional target of the immediate early gene c-Jun (Lin et al., 2002). This is particularly intriguing given the observed early and transient induction of c-Jun in our model, and given reports linking Jun expression with neurite outgrowth and cell survival in neuronal cell lines (Dragunow et al., 2000; Leppa et al., 2001). Although we do not provide direct evidence for the activation IRE-1α signaling, c/EBP-β harbors an Xbp-1 response element in the genomic region corresponding to its 3′UTR, which induces c/EBP-β expression following amino acid starvation (Chen et al., 2004a). c/EBP-β also activates the asparagine synthetase (ASNS) promoter linking c/EBP-β with ER pathways controlling amino acid metabolism (Chen et al., 2004b). These reports suggest several potential mechanisms responsible for the observed induction of c/EBP-β in cortical neurons after hypoxic stress. Data from knockout studies also indicate that c/EBP-β plays an essential role in regulating carbohydrate metabolism (Croniger et al., 2001; Liu et al., 1999). It is interesting to speculate that in addition to activating mitochondrial heat shock responses though CHOP-10, and responding to amino acid limitation in cooperation with ATF4, c/EBP-β may function as an effector loop linking ER responses to hypoglycemia with the mobilization of energy stores (Scheuner et al., 2001).
How then does c/EBP-β support neuron survival? Although not directly confirmed, the relative benefit of the full-length over the ΔTAD construct suggests that c/EBP-β may directly induce the expression of pro-survival genes. Microarray analyses indicate that c/EBP-β regulates a class of genes involved in polyamine synthesis including ornithine decarboxylase (ODC) and spermidine acetyl-transferase (Cortes-Canteli et al., 2004). Both enzymes are important for developing and mature neurons, and the observation that knock-down of ODC exacerbates stroke size following middle cerebral artery occlusion suggest these encode some survival function as well (Raghavendra Rao et al., 2001). And as our data suggest, high levels of c/EBP-β could also inhibit the pro-apoptotic activity of p53 through trans-repression domains located in the carboxy-terminus of both proteins as previously reported (Schneider-Merck et al., 2006). Alternatively, c/EBP-β may indirectly promote survival by disrupting the formation of pathogenic bZIP heterodimeric complexes (Fig. 8). Under basal conditions, c/EBP-β interacts with bZIP partners involved in cellular differentiation and inflammation. Upon stimulation with moderate levels of hypoxia, c/EBP-β and CHOP-10 transactivate the mitochondrial heat shock proteins Cpn10 and Cpn60 and other targets involved in adaptive stress responses (Zhao et al., 2002). However, with prolonged hypoxic stress, falling c/EBP-β levels would favor the formation of pathological bZIP complexes (i.e., ATF4:CHOP) capable of activating pro-apoptotic targets like the Akt inhibitor Tribbles 3 (TRB3) (Ohoka et al., 2005). And, since c/EBP-β represses activity of the CHOP-10 promoter (Fawcett et al., 1996), loss of c/EBP-β expression could favor pro-apoptotic signaling through the accumulation of CHOP-10. We do not yet know which mechanisms govern the loss of c/EBP-β activity after hypoxic stress. While the transient induction of c-Jun offers one potential explanation, the accelerated decay rate of c/EBP-β protein compared with either neurofilament or actin suggest this factor may be subject to targeted degradation. In this regard, we speculate that hypoxia regulated activity of the TRB3 homolog Tribbles 2 (TRB2), which binds to and induces c/EBP-β degradation, may play a role (Naiki et al., 2007). If this were the case, one would predict that disruption of the TRB2-c/EBP-β axis should promote neuron survival under conditions of prolonged hypoxic stress.
Fig. 8.
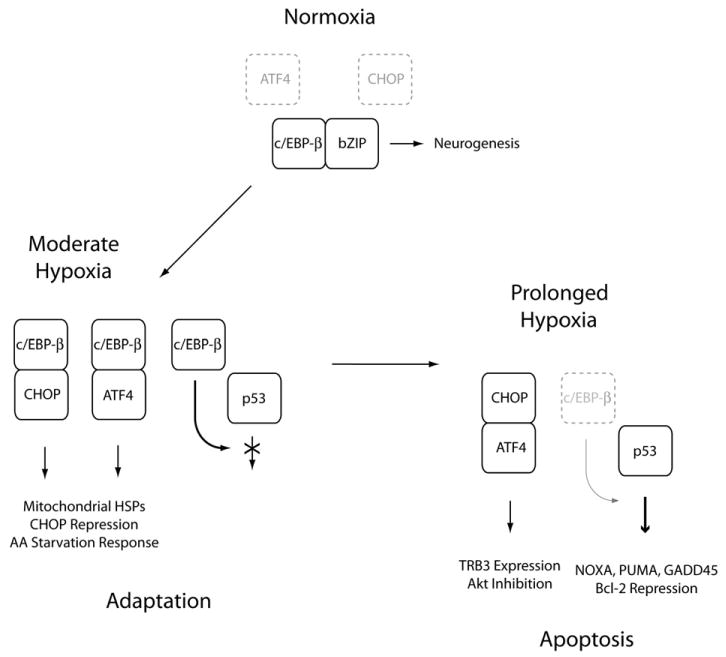
Hypoxia-mediated loss of neuronal c/EBP-β activity promotes the adaptive to apoptotic transition. This schematic illustrates our conceptual model of how attenuated c/EBP-β activity promotes the adaptive to apoptotic transition in hypoxic neurons. Under normoxic conditions, c/EBP-β associates with other neuronal bZIP heterodimeric partners to regulate neurogenesis. Under conditions of sub-lethal hypoxic stress, ER-stress induces the expression of ATF4 and CHOP-10, which associate with c/EBP-β to drive the expression of mitochondrial heat shock proteins and genes involved in the amino acid (AA) starvation response. c/EBP-β also functions as an inhibitory factor repressing the expression of CHOP-10 as well as directly trans-repressing the expression of pro-apoptotic p53 targets. Under conditions of severe hypoxic stress, c/EBP-β levels decline allowing for the formation of pathological bZIP heterodimers (i.e., ATF4:CHOP-10) and the activation of p53-dependent latent pro-apoptotic transcriptional activity.
The role of ER-stress signaling in hypoxia-induced apoptotic signaling
The mammalian unfolded protein response is comprised of both ER-stress specific pathways (including Ire1/XBP-1 and ATF-6), and those activated by other cellular stresses (i.e., PERK-independent eIF2α phosphorylation). The transcriptional targets downstream of these signaling pathways can also be deemed intrinsic (BiP and Xbp-1), extrinsic (i.e., ATF4 and GADD34), or mixed (CHOP-10, HERPUD) with respect to their dependence on ER-stress responses for transactivation (Ma and Hendershot, 2004). Our initial interest in pursuing the ER as the proximate hypoxic sensor responsible for the apoptotic switch in our model was stimulated by reports describing the ability of intraperitoneal cycloheximide to inhibit delayed neuron death after transient focal ischemia (Du et al., 1996). Aside from the possible role in limiting pro-death gene expression, it seemed plausible that cycloheximide could also confer protection by reducing client protein overload and attenuating the activation of pro-death signaling following prolonged ER-stress. In our model, hypoxia appeared to regulate both systems, although the shared pathway involving eIF2α had a greater effect on neuron survival. While salubrinal lowered caspase-3 activation to control levels, it was less effective in blocking nuclear pyknosis, Reports describing caspase-3-independent induction of EndoG activity by BNIP3 offer one potential explanation for this observation and reinforce the notion that combination therapies for stroke will likely be required (Zhang et al., 2007). It is possible that other non-ER kinases like PKR regulate eIF2α phosphorylation and neuron survival after hypoxic stress (Blais et al., 2004). Likewise, the physiologic stressors that activate PERK also activate the AMPK/TSC2/mTOR pathway (Liu et al., 2006). Since pharmacologic inhibition of AMPK signaling ameliorates neuron loss in stroke, determining whether AMPK activity also regulates the apoptotic switch in our model merits further consideration (McCullough et al., 2005).
A dual role for CHOP-10 in the neuronal stress response to hypoxia?
CHOP-10 (GADD153) is a 29-kDa protein initially characterized as a negative inhibitor of c/EBP-β involving adipocyte differentiation (Ron and Habener, 1992). And although CHOP has been associated with neuronal apoptosis after stroke, we observed that in the presence of salubrinal, induction of CHOP-10 protein was not sufficient to cause apoptosis. Aside from its ability to transactivate a group of adaptive genes (Aldridge et al., 2007), CHOP is a known substrate of both p38 and casein II kinase (CKII) (Maytin et al., 2001; Ubeda and Habener, 2003; Wang and Ron, 1996). But, while post-translational modification (PTM) of the CHOP transcriptional activation domain modulates its transcription activation potential, there is no evidence as of yet linking PTMs with target gene selectivity. Presumably, promiscuous heterodimerization between CHOP-10, c-Jun, ATF4 and other family members plays some role in activating CHOP's latent apoptotic activity (Ohoka et al., 2005; Ubeda et al., 1999; Vinson et al., 2002). Studies geared towards identifying the relevant phospho-acceptor sites and kinase pathways that converge on CHOP-10 should provide insight to this problem.
Defining molecular markers of selective vulnerability
Hypoxia-induced neuronal loss in vivo exhibits spatial and temporal heterogeneity, and the magnitude and mechanism of cell injury is directly proportional to the severity of the ischemic insult (Banasiak and Haddad, 1998). Transient global ischemia is associated with the selective loss of discrete neuronal populations including CA1 pyramidal neurons in the hippocampus, layer V cortical neurons and Purkinje cells within the cerebellum. As we observed, hypoxia also induced the death of a subset of vulnerable neurons in vitro. While the mechanisms regulating selective neuronal vulnerability are incompletely understood, intrinsic differences in both the constitutive and regulated expression of calcium binding proteins, cell surface receptors, profiles of receptor desensitization and intrinsic differences in antioxidant defenses have been proposed (Brorson et al., 1995; Jiang et al., 2004; Tombal et al., 2002). Although not directly evaluated in the present study, we found that the restricted expression of the transcriptionally active p-c/EBP-β Thr188 species was a marker of neuronal survival in hypoxic cultures. These data suggest that a comprehensive analysis of other bZIP proteins regulated by hypoxia in cortical neurons could provide important clues regarding the pathways controlling selective vulnerability after stroke. However, while such reductionist systems facilitate modeling cell-autonomous apoptotic responses to hypoxia, additional work using in vivo models will be required to establish that these pathways influence neuron survival following either focal or global ischemia.
In summary, we have characterized an in vitro model of continuous hypoxia and demonstrate its utility in dissecting the molecular events involved in delayed neuronal death. Moreover, we provide evidence that the regulated activity of the bZIP factor c/EBP-β is a critical determinant of neuron loss following hypoxia. Future studies related to the mechanisms controlling c/EBP-β's stability, the pathways involved in c/EBP-β-mediated neuroprotection, and the second messenger systems controlling the latent apoptotic potential of related bZIP proteins like CHOP-10 could lead to the development of novel therapies for stroke.
Experimental methods
Reagents
Actinomycin D, cycloheximide, Hoechst 33342, polyethelenimine (PEI), sodium borate, the protease inhibitor cocktail (P1754), DMSO, protease inhibitor cocktail, sodium dithionite and l-glutamine were purchased from Sigma-Aldrich (St. Louis, MO). CNQX, MK801 and glutamic acid were purchased from RBI, Inc. (Natick, MA). Cell culture grade 0.25% trypsin–EDTA, trypan blue, Neurobasal® media and B27 supplement (AO-plus) were purchased from GIBCO/Invitrogen (Carlsbad, California). The eIF2α phosphatase inhibitor salubrinal, the serine protease inhibitor AEBSF and Thapsigargin were purchased from Calbiochem (San Diego, CA) and stock solutions were prepared with DMSO. Paraformaldehyde was purchased from Mallinckrodt-Baker (Phillipsburg, NJ).
Primary neuronal cultures
All protocols were approved by the University of Rochester committee on animal resources (UCAR), and complied with relevant federal guidelines. Cortices from C57BL/6 mice (E15.5) were dissected free of meninges and transferred to ice-cold Ca2+/Mg2+-free D-PBS, followed by incubation in 0.25% trypsin (1 ml/hemisphere) for 15 min at room temperature. Trypsin was removed by rinsing 3× with MEM. The tissue was triturated and plated in Neurobasal media containing B27 supplement, glutamic acid, and glutamate according to manufacturer's instructions. Neurons were seeded at 1×105 cells on 12 mm cover slips (Fischer Scientific, Pittsburgh, PA) or to 60 mm tissue culture plates (Corning Costar, Corning, NY) at a density of 2.5×106 cells/well (Brewer, 1995). Surfaces were coated with PEI diluted 1:500 in sodium borate buffer (150 mM; pH 8.0) and washed 3× with sterile water before use.
Hypoxic exposures and cell death measurements
Exposures were carried out using a humidified, triple gas incubator (Model 3310, Thermo-Forma, Marietta, OH) calibrated against a dissolved oxygen probe (Microelectrodes, Bedford, NH). Cultures were fixed with 4% paraformaldehyde in PBS (pH 7.4) for 30 min at room temperature, stained with 5 μM Hoechst 33342, and mounted in mowiol for visualization of pyknosis under UV fluorescence. Trypan blue exclusion assays were performed according to the manufacturer's instructions. Immunocytochemical detection for cleaved caspase was performed as described below. Significance testing was performed by Student's T-tests. P-values <0.05 were considered significant.
Expression profiling and genetic pathway analysis
Total RNA was harvested using the Micro to Midi Total RNA Purification System (Invitrogen, Carlsbad, CA) and transcribed using the superscript III cDNA synthesis kit (Stratagene, La Jolla, CA). PrimerExpress® was used to design primer-probes which were synthesized (IDT, Coralville, IA) for the following targets: VEGF (Fwd 5′-GACTCGGAATCTCTTGGTGAGTG-3′, Rev 5′-AGGAAGGTGAAGCCCGGA-3′, Probe 5′-TGGGCAGAGCGCCACCAGC-3′), Glut-1 (Fwd 5′-TCCTGTTGCCCTTCTGCC-3′, Rev 5′-GGTTCTCCTCGTTACGATTGATG-3′, Probe 5′-CGAGAGCCCCCGCTTCCTGC-3′), Hexokinase II (Fwd 5′-GACTCGGAATCTCTTGGTGAGTG-3′, Rev 5′-AGGAAGGTGAAGCCCGGA-3′, Probe 5′-TGGGCAGAGCGCCACCAGC-3′), NIX (Fwd 5′- ACTTGTTGTGTTGCTGCTCGA -3′, Rev 5′- GAGACTGGAAGCGGCACG -3′, Probe 5′- AGCCGG ATACTGTCGTCCTGCGG -3′), NOXA (Fwd 5′- GGCCTCAAACTCCACCTGC -3′, Rev 5′- CCCAG TGATGCTGGCACTT-3′, Probe 5′- TTGCCTCCCGGGTGCTGGG -3′), PUMA (Fwd 5′- GAGCGGCGGAGACAAGAA -3′, Rev 5′- GAGATTGTACATGACCCTCCAGG -3′, Probe 5′- AGCAGCATCGA CACCGACCCTCA -3′), and BNIP3 (Fwd 5′-GCAGGGCTCCTGGGTAGAA -3′, Rev 5′- GACGGAGG CTGGAACGCT -3′, Probe 5′- TGCACTTCAGCAATGGCAATGGGAG -3′). Each 25 μl reaction contained the following: 1× TaqMan® master mix, 10 μM of forward and reverse primers, 5 μM of probe and 5 μl of pre-diluted (1:20) control or test cDNA and run on an ABI-7700 thermocycler (Applied Biosystems Inc, Foster City, CA). RT (−) reactions were analyzed in parallel to monitor for genomic DNA contamination. Data represent the average absolute fold-induction±standard deviation (n=3) using cDNA standards spanning 5-log order dilution.
To analyze putative protein–protein networks activated in our hypoxic model we used the STRING algorithm (http://string.embl.de/). Association data are based on a combination of experimental data, as well as text mining and database resources (including the COG database, Ensembl, and RefSeq) (von Mering et al., 2003, 2007). Using the multiple name option, targets for the human proteins c/EBP-β, ATF4, DDIT3 and PPP1R15A were selected. Within the confidence view second tier associations were incorporated using the ‘more’ tab. Nodes in the network represent the individual protein species while the thickness of the connecting lines indicate the confidence of the interactions depicted.
Western blotting
Whole cell lysates were harvested from 2.5×106 neurons as follows: cells were washed 1× with ice-cold PBS and incubated in RIPA buffer (Tris–HCl: 50 mM, pH 7.4; 1% NP-40; 0.25% Na-deoxycholate: 150 mM NaCl; 1 mM EDTA, 1× protease inhibitor cocktail) on ice for 10 min. Samples were sonnicated and centrifuged at 12,000 rpm/30 s/4 °C in a bench top centrifuge and supernatants were stored at −70C. 15 μL aliquots were resolved by SDS PAGE (8–15%), transferred to PVDF membranes (Millipore, Bedford, MA) and blocked for 1 h at room temperature in wash buffer (50 mM Tris, 0.9% NaCl, 0.05% tween-20) containing 5% non-fat dry milk. Primary antibodies were added with constant agitation at 4 °C (1–12 h), blots were rinsed thee times in wash buffer and incubated with HRP conjugated secondary antibody (1 h diluted 1:2000; Santa Cruz Biotechnology), washed again and detected by ECL detection (Renaissance; Amersham Pharmacia Biotech, Inc., Piscataway, NJ). The following antibodies were used: βIII-tubulin and NeuN (Chemicon, Temecula, CA); activated caspase-3, c-Jun, eIF2α, p-eIF2α (ser51) and p-c/EBP-β(Thr188) were obtained from Cell Signaling Technologies (Beverly, MA); ATF4 (AVIVA, San Diego, CA); c/EBP-β (C-19), Bcl-2 (C-2), Bax (N-20), and Chop-10 (B-3) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Immunocytochemical analysis and image acquisition
Cells were gently rinsed with chilled PBS and fixed for 30 min at 4 °C with 4% PFA in PBS. Cells were then rinsed 2× with PBS containing 0.05% TritonX-100 and permeabilized for 30 min/4 °C in blocking solution (0.15 M NaCl, 20 mM Tris–Hcl, pH 7.5, 4.5% NFDM, 0.1% Triton X-100). Cover slips were inverted on 70 μl droplets of primary antibody diluted 1:100 in triton/blotto on Parafilm for 1 h at room temperature. Cells were rinsed 3× with blotto (0.05% triton X-100) and incubated with conjugated secondary antibodies at RT for 1 h (1:2000; Alexa dyes, Molecular Probes). Samples were rinsed 3× in PBS (0.05% triton X-100) and mounted in mowiol for fluorescence analysis. Images were acquired using the Coolsnap Fx camera (Roper Bioscience), mounted to a Zeiss Axioscope equipped with Plan-Neofluor objectives. Image archiving, normalization and pseudocoloring and densitometry measurements used to analyze results from western blotting experiments were performed using ImagJv1.36 (http://rsb.info.nih.gov/ij/).
Amplicon constructs, virus production and protection experiments
The herpes amplicon plasmid CMVeGFP–HSVprPuc co-expressing the enhanced GFP protein was constructed as follows. The amplicon vector HSVprPuc was linearized by digestion with AccI and ligated with a CMVeGFP fragment generated by PCR from the vector pEGFP-C2 (Clontech, Inc). The luciferase cDNA derived from the pGL3 series of reporter plasmids (Promega, Madison, WI) and the wild-type c/EBP-β and the amino terminal transactivation domain deletion mutant derived from the plasmids CMV-c/EBP-β and c/EBP-βΔTAD (kindly provided by R.M. Pope, Northwestern University Medical School) were sub-cloned into the bi-cistronic amplicon vector CMVeGFP–HSVprPuc. Herpes simplex virus (HSV-1) amplicon vector stocks were generated and titered as described previously (Geschwind et al., 1994). GFP titers were determined on NIH3T3 cells and ranged between 1–3×10ˆ8 gfu/ml with amplicon to helper ratios of 1:1. Neuronal cultures were at an MOI of 0.8 12 h prior to hypoxic exposure to allow for transgene expression. Neuronal transduction was confirmed by paraformaldehyde fixed cultures under FITC fluorescence.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.mcn.2008.01.014.
Acknowledgments
This work was supported by grants to MWH (NIH-MH12305 and NINDS-T32-NS007338), DR (NIH-NS046633) and to HJF (NIH-NS364201).
Footnotes
The authors report no commercial affiliations or conflicts of interest pertaining to this manuscript.
Publisher's Disclaimer: This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues.
Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited.
In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier's archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/copyright
References
- Abe K, Aoki M, Kawagoe J, Yoshida T, Hattori A, Kogure K, Itoyama Y. Ischemic delayed neuronal death. A mitochondrial hypothesis. Stroke. 1995;26:1478–1489. doi: 10.1161/01.str.26.8.1478. [DOI] [PubMed] [Google Scholar]
- Aldridge JE, Horibe T, Hoogenraad NJ. Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS ONE. 2007;2:e874. doi: 10.1371/journal.pone.0000874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Banasiak KJ, Haddad GG. Hypoxia-induced apoptosis: effect of hypoxic severity and role of p53 in neuronal cell death. Brain Res. 1998;797:295–304. doi: 10.1016/s0006-8993(98)00286-8. [DOI] [PubMed] [Google Scholar]
- Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, Wouters BG, Bell JC. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol. 2004;24:7469–7482. doi: 10.1128/MCB.24.17.7469-7482.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, Kaufman RJ, Ma D, Coen DM, Ron D, Yuan J. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Brewer GJ. Serum-free B27/neurobasal medium supports differentiated growth of neurons from the striatum, substantia nigra, septum, cerebral cortex, cerebellum, and dentate gyrus. J Neurosci Res. 1995;42:674–683. doi: 10.1002/jnr.490420510. [DOI] [PubMed] [Google Scholar]
- Brorson JR, Manzolillo PA, Gibbons SJ, Miller RJ. AMPA receptor desensitization predicts the selective vulnerability of cerebellar Purkinje cells to excitotoxicity. J Neurosci. 1995;15:4515–4524. doi: 10.1523/JNEUROSCI.15-06-04515.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brush MH, Weiser DC, Shenolikar S. Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1 alpha to the endoplasmic reticulum and promotes dephosphorylation of the alpha subunit of eukaryotic translation initiation factor 2. Mol Cell Biol. 2003;23:1292–1303. doi: 10.1128/MCB.23.4.1292-1303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck M, Zhang L, Halasz NA, Hunter T, Chojkier M. Nuclear export of phosphorylated C/EBPbeta mediates the inhibition of albumin expression by TNF-alpha. EMBO J. 2001;20:6712–6723. doi: 10.1093/emboj/20.23.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calkhoven CF, Muller C, Leutz A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 2000;14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- Chen C, Dudenhausen EE, Pan YX, Zhong C, Kilberg MS. Human CCAAT/enhancer-binding protein beta gene expression is activated by endoplasmic reticulum stress through an unfolded protein response element downstream of the protein coding sequence. J Biol Chem. 2004a;279:27948–27956. doi: 10.1074/jbc.M313920200. [DOI] [PubMed] [Google Scholar]
- Chen H, Pan YX, Dudenhausen EE, Kilberg MS. Amino acid deprivation induces the transcription rate of the human asparagine synthetase gene through a timed program of expression and promoter binding of nutrient-responsive basic region/leucine zipper transcription factors as well as localized histone acetylation. J Biol Chem. 2004b;279:50829–50839. doi: 10.1074/jbc.M409173200. [DOI] [PubMed] [Google Scholar]
- Collaco-Moraes Y, Aspey BS, de Belleroche JS, Harrison MJ. Focal ischemia causes an extensive induction of immediate early genes that are sensitive to MK-801. Stroke. 1994;25:1855–1860. doi: 10.1161/01.str.25.9.1855. discussion 1861. [DOI] [PubMed] [Google Scholar]
- Cortes-Canteli M, Wagner M, Ansorge W, Perez-Castillo A. Microarray analysis supports a role for ccaat/enhancer-binding protein-beta in brain injury. J Biol Chem. 2004;279:14409–14417. doi: 10.1074/jbc.M313253200. [DOI] [PubMed] [Google Scholar]
- Croniger CM, Millward C, Yang J, Kawai Y, Arinze IJ, Liu S, Harada-Shiba M, Chakravarty K, Friedman JE, Poli V, Hanson RW. Mice with a deletion in the gene for CCAAT/enhancer-binding protein beta have an attenuated response to cAMP and impaired carbohydrate metabolism. J Biol Chem. 2001;276:629–638. doi: 10.1074/jbc.M007576200. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Dragunow M, Xu R, Walton M, Woodgate A, Lawlor P, MacGibbon GA, Young D, Gibbons H, Lipski J, Muravlev A, Pearson A, During M. c-Jun promotes neurite outgrowth and survival in PC12 cells. Brain Res Mol Brain Res. 2000;83:20–33. doi: 10.1016/s0169-328x(00)00191-1. [DOI] [PubMed] [Google Scholar]
- Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: a role for apoptosis? J Cereb Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- Fawcett TW, Eastman HB, Martindale JL, Holbrook NJ. Physical and functional association between GADD153 and CCAAT/enhancer-binding protein beta during cellular stress. J Biol Chem. 1996;271:14285–14289. doi: 10.1074/jbc.271.24.14285. [DOI] [PubMed] [Google Scholar]
- Geschwind MD, Lu B, Federoff HJ. Expression of neurotrophic genes from herpes simplex virus type 1 vectors: modifying neuronal phenotype. Methods Neurosci. 1994;21:462–482. [Google Scholar]
- Gwag BJ, Lobner D, Koh JY, Wie MB, Choi DW. Blockade of glutamate receptors unmasks neuronal apoptosis after oxygen-glucose deprivation in vitro. Neuroscience. 1995;68:615–619. doi: 10.1016/0306-4522(95)00232-8. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, Zeng H, Ron D. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]
- Herdegen T, Leah JD. Inducible and constitutive transcription factors in the mammalian nervous system: control of gene expression by Jun, Fos and Krox, and CREB/ATF proteins. Brain Res Brain Res Rev. 1998;28:370–490. doi: 10.1016/s0165-0173(98)00018-6. [DOI] [PubMed] [Google Scholar]
- Honkaniemi J, Massa SM, Breckinridge M, Sharp FR. Global ischemia induces apoptosis-associated genes in hippocampus. Brain Res Mol Brain Res. 1996;42:79–88. doi: 10.1016/s0169-328x(96)00121-0. [DOI] [PubMed] [Google Scholar]
- Hsu CY, An G, Liu JS, Xue JJ, He YY, Lin TN. Expression of immediate early gene and growth factor mRNAs in a focal cerebral ischemia model in the rat. Stroke. 1993;24:I78–I58. [PubMed] [Google Scholar]
- Jiang X, Mu D, Manabat C, Koshy AA, Christen S, Tauber MG, Vexler ZS, Ferriero DM. Differential vulnerability of immature murine neurons to oxygen–glucose deprivation. Exp Neurol. 2004;190:224–232. doi: 10.1016/j.expneurol.2004.07.010. [DOI] [PubMed] [Google Scholar]
- Jones NM, Bergeron M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J Cereb Blood Flow Metab. 2001;21:1105–1114. doi: 10.1097/00004647-200109000-00008. [DOI] [PubMed] [Google Scholar]
- Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110:1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Ahn HJ, Ryu JH, Suk K, Park JH. BH3-only protein Noxa is a mediator of hypoxic cell death induced by hypoxia-inducible factor 1alpha. J Exp Med. 2004;199:113–124. doi: 10.1084/jem.20030613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Azam S, Sullivan JM, Owen C, Cavener DR, Zhang P, Ron D, Harding HP, Chen JJ, Han A, White BC, Krause GS, DeGracia DJ. Brain ischemia and reperfusion activates the eukaryotic initiation factor 2alpha kinase, PERK. J Neurochem. 2001;77:1418–1421. doi: 10.1046/j.1471-4159.2001.00387.x. [DOI] [PubMed] [Google Scholar]
- Leppa S, Eriksson M, Saffrich R, Ansorge W, Bohmann D. Complex functions of AP-1 transcription factors in differentiation and survival of PC12 cells. Mol Cell Biol. 2001;21:4369–4378. doi: 10.1128/MCB.21.13.4369-4378.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lee B, Lee AS. Endoplasmic reticulum stress-induced apoptosis: multiple pathways and activation of p53-up-regulated modulator of apoptosis (PUMA) and NOXA by p53. J Biol Chem. 2006;281:7260–7270. doi: 10.1074/jbc.M509868200. [DOI] [PubMed] [Google Scholar]
- Lin WC, Shen BJ, Tsay YG, Yen HC, Lee SC, Chang CJ. Transcriptional activation of C/EBPbeta gene by c-Jun and ATF2. DNA Cell Biol. 2002;21:551–560. doi: 10.1089/104454902320308924. [DOI] [PubMed] [Google Scholar]
- Liu S, Croniger C, Arizmendi C, Harada-Shiba M, Ren J, Poli V, Hanson RW, Friedman JE. Hypoglycemia and impaired hepatic glucose production in mice with a deletion of the C/EBPbeta gene. J Clin Invest. 1999;103:207–213. doi: 10.1172/JCI4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Hendershot LM. Herp is dually regulated by both the endoplasmic reticulum stress-specific branch of the unfolded protein response and a branch that is shared with other cellular stress pathways. J Biol Chem. 2004;279:13792–13799. doi: 10.1074/jbc.M313724200. [DOI] [PubMed] [Google Scholar]
- Ma Y, Brewer JW, Diehl JA, Hendershot LM. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. J Mol Biol. 2002;318:1351–1365. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Masud A, Mohapatra A, Lakhani SA, Ferrandino A, Hakem R, Flavell RA. Endoplasmic reticulum stress-induced death of mouse embryonic fibroblasts requires the intrinsic pathway of apoptosis. J Biol Chem. 2007;282:14132–14139. doi: 10.1074/jbc.M700077200. [DOI] [PubMed] [Google Scholar]
- Maytin EV, Ubeda M, Lin JC, Habener JF. Stress-inducible transcription factor CHOP/gadd153 induces apoptosis in mammalian cells via p38 kinase-dependent and -independent mechanisms. Exp Cell Res. 2001;267:193–204. doi: 10.1006/excr.2001.5248. [DOI] [PubMed] [Google Scholar]
- McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol. 2001;21:1249–1259. doi: 10.1128/MCB.21.4.1249-1259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough LD, Zeng Z, Li H, Landree LE, McFadden J, Ronnett GV. Pharmacological inhibition of AMP-activated protein kinase provides neuroprotection in stroke. J Biol Chem. 2005;280:20493–20502. doi: 10.1074/jbc.M409985200. [DOI] [PubMed] [Google Scholar]
- Menard C, Hein P, Paquin A, Savelson A, Yang XM, Lederfein D, Barnabe-Heider F, Mir AA, Sterneck E, Peterson AC, Johnson PF, Vinson C, Miller FD. An essential role for a MEK-C/EBP pathway during growth factor-regulated cortical neurogenesis. Neuron. 2002;36:597–610. doi: 10.1016/s0896-6273(02)01026-7. [DOI] [PubMed] [Google Scholar]
- Mengesdorf T, Proud CG, Mies G, Paschen W. Mechanisms underlying suppression of protein synthesis induced by transient focal cerebral ischemia in mouse brain. Exp Neurol. 2002;177:538–546. doi: 10.1006/exnr.2002.8002. [DOI] [PubMed] [Google Scholar]
- Miyashita T, Harigai M, Hanada M, Reed JC. Identification of a p53-dependent negative response element in the bcl-2 gene. Cancer Res. 1994;54:3131–3135. [PubMed] [Google Scholar]
- Nadeau S, Hein P, Fernandes KJ, Peterson AC, Miller FD. A transcriptional role for C/EBP beta in the neuronal response to axonal injury. Mol Cell Neurosci. 2005;29:525–535. doi: 10.1016/j.mcn.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Naiki T, Saijou E, Miyaoka Y, Sekine K, Miyajima A. TRB2, a mouse Tribbles ortholog, suppresses adipocyte differentiation by inhibiting AKT and C/EBPbeta. J Biol Chem. 2007;282:24075–24082. doi: 10.1074/jbc.M701409200. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, Sato R, Negishi M, Mori K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. J Biol Chem. 2003;278:31024–31032. doi: 10.1074/jbc.M300923200. [DOI] [PubMed] [Google Scholar]
- Pirianov G, Brywe KG, Mallard C, Edwards AD, Flavell RA, Hagberg H, Mehmet H. Deletion of the c-Jun N-terminal kinase 3 gene protects neonatal mice against cerebral hypoxic-ischaemic injury. J Cereb Blood Flow Metab. 2007;27:1022–1032. doi: 10.1038/sj.jcbfm.9600413. [DOI] [PubMed] [Google Scholar]
- Piwien-Pilipuk G, MacDougald O, Schwartz J. Dual regulation of phosphorylation and dephosphorylation of C/EBPbeta modulate its transcriptional activation and DNA binding in response to growth hormone. J Biol Chem. 2002;277:44557–44565. doi: 10.1074/jbc.M206886200. [DOI] [PubMed] [Google Scholar]
- Raghavendra Rao VL, Dogan A, Bowen KK, Dempsey RJ. Ornithine decarboxylase knockdown exacerbates transient focal cerebral ischemia-induced neuronal damage in rat brain. J Cereb Blood Flow Metab. 2001;21:945–954. doi: 10.1097/00004647-200108000-00007. [DOI] [PubMed] [Google Scholar]
- Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–380. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- Reimertz C, Kogel D, Rami A, Chittenden T, Prehn JH. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J Cell Biol. 2003;162:587–597. doi: 10.1083/jcb.200305149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant-negative inhibitor of gene transcription. Genes Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Schneider-Merck T, Pohnke Y, Kempf R, Christian M, Brosens JJ, Gellersen B. Physical interaction and mutual transrepression between CCAAT/enhancer-binding protein beta and the p53 tumor suppressor. J Biol Chem. 2006;281:269–278. doi: 10.1074/jbc.M503459200. [DOI] [PubMed] [Google Scholar]
- Schwamm LH, Koroshetz WJ, Sorensen AG, Wang B, Copen WA, Budzik R, Rordorf G, Buonanno FS, Schaefer PW, Gonzalez RG. Time course of lesion development in patients with acute stroke: serial diffusion- and hemodynamic-weighted magnetic resonance imaging. Stroke. 1998;29:2268–2276. doi: 10.1161/01.str.29.11.2268. [DOI] [PubMed] [Google Scholar]
- Sokka AL, Putkonen N, Mudo G, Pryazhnikov E, Reijonen S, Khiroug L, Belluardo N, Lindholm D, Korhonen L. Endoplasmic reticulum stress inhibition protects against excitotoxic neuronal injury in the rat brain. J Neurosci. 2007;27:901–908. doi: 10.1523/JNEUROSCI.4289-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer T, Jarosch E. BiP binding keeps ATF6 at bay. Dev Cell. 2002;3:1–2. doi: 10.1016/s1534-5807(02)00210-1. [DOI] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, Morioka M, Gotoh T, Hamada JI, Ushio Y, Mori M. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–415. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Tombal B, Denmeade SR, Gillis JM, Isaacs JT. A supramicromolar elevation of intracellular free calcium ([Ca(2+)](i)) is consistently required to induce the execution phase of apoptosis. Cell Death Differ. 2002;9:561–573. doi: 10.1038/sj.cdd.4400999. [DOI] [PubMed] [Google Scholar]
- Ubeda M, Habener JF. CHOP transcription factor phosphorylation by casein kinase 2 inhibits transcriptional activation. J Biol Chem. 2003;278:40514–40520. doi: 10.1074/jbc.M306404200. [DOI] [PubMed] [Google Scholar]
- Ubeda M, Vallejo M, Habener JF. CHOP enhancement of gene transcription by interactions with Jun/Fos AP-1 complex proteins. Mol Cell Biol. 1999;19:7589–7599. doi: 10.1128/mcb.19.11.7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinson C, Myakishev M, Acharya A, Mir AA, Moll JR, Bonovich M. Classification of human B-ZIP proteins based on dimerization properties. Mol Cell Biol. 2002;22:6321–6335. doi: 10.1128/MCB.22.18.6321-6335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional associations between proteins. Nucleic Acids Res. 2003;31:258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Mering C, Jensen LJ, Kuhn M, Chaffron S, Doerks T, Kruger B, Snel B, Bork P. STRING 7—recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 2007;35:D358–D362. doi: 10.1093/nar/gkl825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Ron D. Stress-induced phosphorylation and activation of the transcription factor CHOP (GADD153) by p38 MAP Kinase. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yang X, Zhang S, Ma X, Kong J. BNIP3 upregulation and EndoG translocation in delayed neuronal death in stroke and in hypoxia. Stroke. 2007;38:1606–1613. doi: 10.1161/STROKEAHA.106.475129. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Wang J, Levichkin IV, Stasinopoulos S, Ryan MT, Hoogenraad NJ. A mitochondrial specific stress response in mammalian cells. EMBO J. 2002;21:4411–4419. doi: 10.1093/emboj/cdf445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.mcn.2008.01.014.