

Abstract
Lapatinib is active at the ATP-binding site of tyrosine kinases that are associated with the human epidermal growth factor receptor (EGFR, Her-1, or ErbB1) and Her-2. It is conceivable that lapatinib may inhibit the function of ATP-binding cassette (ABC) transporters by binding to their ATP-binding sites. The aim of this study was to investigate the ability of lapatinib to reverse tumor multidrug resistance (MDR) due to overexpression of ABCB1 and ABCG2 transporters. Our results showed that lapatinib significantly enhanced the sensitivity to ABCB1 or ABCG2 substrates in cells expressing these transporters although a small synergetic effect was observed in combining lapatinib and conventional chemotherapeutic agents in parental sensitive MCF-7 or S1 cells. Lapatinib alone, however, did not significantly alter the sensitivity of non-ABCB1 or non-ABCG2 substrates in sensitive and resistant cells. Additionally, lapatinib significantly increased the accumulation of doxorubicin or mitoxantrone in ABCB1 or ABCG2 overexpressing cells and inhibited the transport of methotrexate and E217βG by ABCG2. Furthermore, lapatinib stimulated the ATPase activity of both ABCB1 and ABCG2 and inhibited the photolabeling of ABCB1 or ABCG2 with [125I]Iodoarylazidoprazosin in a concentration-dependent manner. However, lapatinib did not affect the expression of these transporters at mRNA or protein levels. Importantly, lapatinib also strongly enhanced the effect of paclitaxel on the inhibition of growth of the ABCB1-overexpressing KBv200 cell xenografts in nude mice. Overall, we conclude that lapatinib reverses ABCB1- and ABCG2-mediated MDR by directly inhibiting their transport function. These findings may be useful for cancer combinational therapy with lapatinib in the clinic.
Keywords: multidrug resistance, ABCB1/P-gp, ABCG2/BCRP/MXR, EGFR tyrosine kinase inhibitor, lapatinib
Introduction
Multidrug resistance (MDR) is a major obstacle to successful chemotherapy treatment. MDR often results due to overexpression of ATP-binding cassette (ABC) transporters. In the human genome, 48 different ABC transporters have been identified and are divided into seven subfamilies (A–G) based on sequence similarities (1). So far, the major members of the ABC transporters leading to MDR in cancer cells include ABC subfamily B member 1 (ABCB1, also called P-glycoprotein, P-gp), ABC subfamily C members (ABCCs, MRPs) and ABC subfamily G member 2 (ABCG2, also called breast cancer resistance protein, BCRP; mitoxantrone resistance protein, MXR, and placenta-specific ABC transporter, ABCP). These membrane proteins actively pump out a wide range of structurally and functionally diverse amphipathic anticancer drugs from the inside of tumor cells thereby decreasing their intracellular drug accumulation and resulting in chemotherapeutic drug resistance (1-4). Furthermore, each transporter can translocate unique compounds in addition to some overlapping substrates (1). Drugs transported by ABCB1 include hydrophobic compounds, either uncharged or slightly positively charged, including most chemotherapeutic agents such as Vinca alkaloids, anthracyclines, epipodophyllotoxins and taxanes (5). A few of the ABCC subfamily members have been shown to confer MDR to organic anion compounds and phase II metabolic products, as well as some natural product chemotherapeutic agents, antifolates and nucleotide analogs (6). The spectrum of chemotherapeutic agents transported by ABCG2 includes anthracyclines, mitoxantrone, camptothecin-derived and indolocarbazole topoisomerase I inhibitors, methotrexate and flavopiridol (7).
The ErbB/HER family of receptor tyrosine kinases (RTK) mediate the transphosphorylation of tyrosine residues in the cytoplasmic domain of tyrosine kinase receptors via homodimerization or heterodimerization on ligand activation (8). In human tumors, the epidermal growth factor receptor (EGFR, Her-1, Erb B1) and the other three members of the EGF receptor family, human epidermal receptor type 2, 3, 4 (Her-2, -3 and -4), are often overexpressed, dis-regulated or mutated, and these abnormal alterations of EGFR activate a series of intracellular protein kinase signaling pathways such as the Ras/mitogen activated protein kinase (MAPK), phosphatidyl inositol 3-kinase (PI3K), signal transducer and activator of transcription (STAT), protein kinase C (PKC) and phospholipase D pathways, promoting tumor growth and progression including the promotion of proliferation, angiogenesis, invasion, metastasis and inhibition of apoptosis (8, 9). Therefore, blockade of EGFR or/and Her-2 activation may be able to suppress cancer cell growth and progression. Lapatinib (Tykerb, GW572016) is an orally active small molecule which is a novel member of the family of kinase inhibitors that inhibits the tyrosine kinases of Her-2 and EGFR. In preclinical studies, lapatinib was not cross-resistant to trastuzumab (10, 11). In clinical studies, lapatinib in combination with capecitabine has shown promising results compared with capecitabine alone in a phase III trial of HER-2–positive metastatic breast cancer patients whose cancers become unresponsive to trastuzumab or to other therapies (12). Lapatinib in combination with tamoxifen effectively inhibited cell proliferation and restored tamoxifen sensitivity in ER-positive, breast cancer models with demonstrated resistance to tamoxifen (13, 14). Several randomized phase I and II studies (EGF104383, EGF104535, EGF30001, EGF105767, EGF100161 and EGF1025801) are ongoing to compare paclitaxel/docetaxel plus lapatinib as first-line treatment for Her-2-overexpressing breast cancer patients. But the action and mechanisms underlying the lapatinib-induced chemosensitivity of conventional chemotherapeutic agents in cancer cells remain to be elucidated. Gefitinib, an inhibitor of the tyrosine kinase activity of Her-1, has been reported to interact with ABCG2 and ABCB1 and to reverse ABCB1- and/or ABCG2-mediated MDR by directly inhibiting their drug pump function in cancer cells (15). Furthermore, ABCG2-transduced cells were found to be resistant to gefitinib (16), and expression of ABCG2, but not its nonfunctional mutant, protects EGFR signaling-dependent tumor cells from death on exposure to gefitinib, and this protection was reversed by the ABCG2-specific inhibitor Ko143 (15). These reports strongly suggested that ABCG2 can actively pump gefitinib out of the cells. In our previous study, we also found that erlotinib was also able to antagonize ABCB1- and ABCG2-mediated MDR, suggesting that it might be a substrate of these two transporters (17). Lapatinib is also a potent and reversible inhibitor that acts at the ATP binding site of the tyrosine kinase domains of both EGFR and Her-2. It is conceivable that lapatinib may inhibit functions of ABC transporters by binding to their ATP-binding sites. These have spurred on efforts to investigate whether lapatinib can enhance the efficacy of conventional chemotherapeutic drugs via interaction with ABC transporters in MDR cancer cells and tumor xenograft model.
Materials and Methods
Materials
[125I]-Iodoarylazidoprazosin (IAAP) (2,200 Ci/mmol) and [3H]-E217βG (40.5 Ci/mmol) were obtained from PerkinElmer Life Sciences. [3H]-Mitoxantrone (4 Ci/mmol) and [3H]-methotrexate (23 Ci/mmol) were purchased from Moravek Biochemicals, Inc. Lapatinib and topotecan were products of Glaxo Smith Co. Erlotinib was purchased from Chemie Tek, Inc. Dulbecco’s modified Eagle’s medium (DMEM) and RPMI 1640 were products of Gibco BRL. Monoclonal antibody C-219 (against ABCB1) was supplied by the Signet Laboratories Inc. Monoclonal antibodies including ABCB1 (sc-8313), ABCG2 (sc-25256), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (sc-20357) were products of Santa Cruz Biotechnology Inc. Anti-MAP Kinase 1/2 (Erk1/2), p-ERK, p-AKT antibodies were purchased from Kangchen Co. (Shanghai, China). Akt antibody was a product of Cell Signaling Technology Inc. (Danvers, MA). R-Phycoerythrin (PE)-conjugated mouse monoclonal anti-human EGFR antibody and mouse IgG2b κ isotype control were obtained from BD Bioscience (San Jose, CA). Phycoerythrin (PE)-conjugated mouse monoclonal anti-human Her2 antibody and mouse IgG2B isotype control were from R&D Systems (Minneapolis, MN). Fumitremorgin C (FTC) was synthesized by Thomas McCloud, Developmental Therapeutics Program, Natural Products Extraction Laboratory, NIH (Bethesda, MD). Doxorubicin, paclitaxel, mitoxantrone, 6-mercaptopurine, 3-(4, 5-dimethylthiazol-yl)-2, 5- diphenyllapatinibrazolium bromide (MTT), rhodamine 123 and other chemicals were obtained from Sigma Chemical Co.
Cell lines and cell culture
The following cell lines were cultured in essential medium containing 10% fetal bovine serum at 37°C in the presence of 5% CO2: the human breast carcinoma cell line MCF-7 and its doxorubicin-selected derivative ABCB1 overexpression MCF-7/adr (18); the human epidermoid carcinoma cell line KB and its vincristine-selected derivative ABCB1 overexpressing KBv200 (19); the colon carcinoma cell line S1 and its mitoxantrone-selected derivative ABCG2 overexpression S1-M1-80 (20). HEK293/pcDNA3.1, ABCG2-482-R5, ABCG2-482-G2, and ABCG2-482-T7 cells were established by selection with G418 after transfecting HEK293 with either empty pcDNA3.1 vector or pcDNA3.1 vector containing full length ABCG2 coding either arginine (R), glycine (G), or threonine (T) at amino acid 482 position, respectively, and were cultured in medium with 2 mg/ml of G418 (21). All cells were grown in drug-free culture media for >2 weeks before assay.
Cytotoxicity assay
The MTT assay was used to access cytotoxicity (22, 23). In detail, cells were grown in 96-well microtiter plates. To determine the toxicity of lapatinib, various concentrations of lapatinib diluted with medium were added into the wells. To test the effect of lapatinib on the chemosensitivity of cancer cells, lapatinib was added to the medium with various concentrations of doxorubicin in MCF-7, MCF-7/adr, MX or topotecan in S1 or S1-M1-80 cells, respectively, mitoxantrone and cisplatin in HEK293/pcDNA3.1, ABCG2-482-R5, ABCG2-482-G2, and ABCG2-482-T7 cells. The concentrations required to inhibit growth by 50% (IC50) were calculated from survival curves using the Bliss method (24). The degree of resistance was calculated by dividing the IC50 for the MDR cells by that of the parental sensitive cells. The degree of the reversal of MDR was calculated by dividing the IC50 for cells with the anticancer drug in the absence of lapatinib by that obtained in the presence of lapatinib.
Experimental animals
Athymic nude mice, 5-6 weeks old and weighting 18-23 g (Center of Experimental Animals, Sun Yat-Sen University), were used for the KBv200 cell xenografts. All animals were provided with sterilized food and water.
MDR human carcinoma xenografts
The KBv200 cell xenograft model was established as described by Chen et al (25). Briefly, KBv200 cells grown in vitro were harvested and implanted subcutaneously (s.c.) under the shoulder in the nude mice. When the tumors reached a mean diameter of 0.5 cm, the mice were randomized into 4 groups and treated with one of the following regimens: 1) saline (q3d × 4); 2) paclitaxel (18 mg/kg i.p., q3d × 4); 3) lapatinib (100 mg/kg, p.o., q3d × 4), and 4) paclitaxel (18 mg/kg, i.p., q3d × 4) + lapatinib (100 mg/kg, p.o., q3d × 4 given 1 h before giving paclitaxel). The body weight of the animals was measured every 3 days in order to adjust the drug dosage. The two perpendicular diameters (A and B) were recorded every 3 days and tumor volume (V) was estimated according to the formula (25):
The curve of tumor growth was drawn according to tumor volume and time of implantation. The mice were anesthetized and killed when the mean of tumor weights was over 1 g in the control group. Tumor tissue was excised from the mice and weighted. The rate of inhibition (IR) was calculated according to the formula (25):
Doxorubicin and mitoxantrone accumulation
The intracellular doxorubicin accumulation in ABCB1overexpessing MCF-7/adr cells and their parental sensitive MCF-7 cells was examined by flow cytometry (26). The logarithmically growing cells were treated with 0.625, 1.25, or 2.5 μM lapatinib at 37°C for 3 h. Then 10 μM doxorubicin was added to the medium and the incubation continued for another 3 h. The cells were then collected, centrifuged and washed twice with cold PBS containing 10 μM verapamil. Cells were resuspended in 200 μl PBS and then analyzed by flow cytometry (Beckman-coulter, Elite), excitation 488 nm (argon laser) for the mean fluorescence intensity (MFI) of intracellular doxorubicin. The relative value of drug accumulation was identified by dividing the MFI for each measurement by that of the ABCB1 expressing cells. The accumulation of mitoxantrone in ABCG2 transfected cells was measured using [3H]-mitoxantrone. Confluent cells in 24-well plates were preincubated with or without lapatinib for 1 h at 37°C. To measure drug accumulation, the cells were then incubated with 0.2 μmol/L [3H]-mitoxantrone for 2 h in the presence or absence of lapatinib at 37°C. After washing three times with ice-cold PBS, the cells were typsinized and lysed in 10 mM lysis buffer (pH 7.4, containing 1% Triton X-100 and 0.2% SDS). Each sample was placed in scintillation fluid and radioactivity was measured in a Packard TRI-CARB® 1900CA liquid scintillation analyzer from Packard Instrument Company, Inc (Downers Grove, IL, USA).
Preparation of membrane vesicles and total cell lysates
Membrane vesicles were prepared by the nitrogen cavitation method as previously described (17, 27). Vesicles were stored at -80°C until ready for use. To prepare the total cell lysates, cells were harvested and rinsed twice with PBS. Cell extracts were prepared with RIPA buffer (1×PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 100 μg/ml phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin) for 30 min with occasional rocking, and clarified by centrifugation at 12,000 × g at 4°C for 15 min. The supernatant containing total cell lysates was stored at -80°C until it was ready for use. The protein concentration was determined by Bradford method. High Five insect cells (Invitrogen, CA, USA) were infected with the recombinant baculovirus carrying the human ABCB1 or ABCG2 cDNAs with a His6 tag at the C-terminal end [BV-MDR1(His6)] or [BV-(His6)-ABCG2] as described previously and the membrane vesicles of High Five insect cells were prepared as previously described (28) and stored at -70°C.
In vitro transport assays
Transport assays were performed essentially using the rapid filtration method as previously described (17, 29). Membrane vesicles were incubated with various concentrations of lapatinib for 1 h on ice, and then transport reactions were carried out at 37°C for 10 min in a total volume of 50 μl medium (membrane vesicles 10 μg, 0.25 M sucrose, 10 mM Tris-HCl, pH 7.4, 10 mM MgCl2, 4 mM ATP or 4 mM AMP, 10 mM phosphocreatine, 100 μg/ml creatine phosphokinase, and 0.5 μM [3H]-methotrexate or 0.25 μM [3H]-E217βG). Reactions were stopped by the addition of 3 ml of ice-cold stop solution (0.25 M sucrose, 100 mM NaCl, and 10 mM Tris-HCl, pH 7.4). During the rapid filtration step, samples were passed through 0.22 μm GVWP filters (Millipore Corporation, Billerica, MA) presoaked in the stop solution. The filters were washed three times with 3 ml of ice-cold stop solution. Radioactivity was measured by the use of a liquid scintillation counter.
ATPase assay of ABCB1 and ABCG2
The Vi-sensitive ATPase activity of ABCB1 and ABCG2 in the membrane vesicles of High Five insect cells was measured as previously described (30). The membrane vesicles (10 μg of protein) were incubated in ATPase assay buffer (50 mM MES, pH 6.8, 50 mM KCl, 5 mM sodium azide, 2 mM EGTA, 2 mM dithiothreitol, 1 mM ouabain, and 10 mM MgCl2) with or without 0.3 mM vanadate at 37°C for 5 min, then incubated with different concentrations of lapatinib at 37°C for 3 min. The ATPase reaction was induced by the addition of 5 mM Mg-ATP, and the total volume was 0.1 ml. After incubation at 37°C for 20 min, the reactions were stopped by loading 0.1 ml of 5% SDS solution. The liberated Pi was measured as described previously (17, 30).
Photoaffinity labeling of ABCB1 and ABCG2 with [125I]-IAAP
The photoaffinity labeling of ABCB1 and ABCG2 with [125I]-IAAP was performed as previously described (17, 31). We have used the crude membranes from MCF7/Flv1000 cells expressing R482 ABCG2 and membrane vesicles of High Five insect cells expressing ABCB1 for photolabeling experiments. The membranes (50 μg of protein) were incubated at room temperature with different concentrations of lapatinib in the ATPase assay buffer with [125I]-IAAP (7 nM) for 5 min under subdued light. The samples were photo-cross-linked with 365 nm UV light for 10 minutes at room temperature. ABCG2 was immunoprecipitated using BXP21 antibody (32) while ABCB1 was immunoprecipitated as described previously except that C219 antibody was used (30). The samples were subjected to SDS-PAGE using a 7% Tris-acetate NuPAGE gel, the gels were dried and exposed to Bio-Max MR film (Eastman Kodak Co.) at -70°C for 8-12 h. The radioactivity incorporated into the ABCB1 or ABCG2 band was quantified using the STORM 860 PhosphorImager system and ImageQuaNT (Molecular Dynamics, CA).
Reverse transcription-PCR
The cells were treated with lapatinib for 48 h. Total cellular RNA was isolated by Trizol Reagent (Gibco BRL, USA) RNA extraction kit following manufacturer instruction. cDNA libraries were prepared from drug-sensitive and MDR cells. Reverse transcription was done with reverse transcriptase (Promega Corp., Madison, WI). Oligonucleotide primers for ABCB1, ABCG2 and GAPDH were synthesized commercially (Invitrogen Co., China). The primers used were ABCB1, sense primer, 5’-CCCATCATTGCAATAGCAGG-3’, antisense primer, 5’-GTTCAAACTTCTGCTCCTGA-3’; ABCG2, sense primer, 5’-TGGCTGTCATGGCTTCAGTA-3’, antisense primer, 5’-GCCACGTGATTCTTCCACAA-3’; GAPDH, sense primer, 5’- GAAGGTGAAGGTCGGAGTC-3’, antisense primer, 5’-GAAGATGGTGATGGGATTTC-3’. Using the GeneAmp PCR system 9700 (PE Applied Biosystem, Foster City, CA), reactions were carried out for ABCB1, ABCG2 and GAPDH at 94°C for 2 min for initial denaturation, and then at 94°C for 30 s, 56°C for 30 s, and 72°C for 1 min. After 35 cycles of amplification, additional extensions were done at 72°C for 10 min. Products were resolved and examined by 2% agarose gel electrophoresis (24).
Western blot analysis
To determine whether lapatinib affects the expression of ABCB1 or ABCG2, the cells were incubated with different concentrations of lapatinib for 48 h. To test whether lapatinib is able to block Akt or Erk1/2 phosphorylation, we incubated cells with different concentrations of lapatinib (0.625~2.5 μM) for different periods of time (12~48 h). Then the cells were harvested and rinsed twice with PBS. Cell extracts were prepared by incubating with buffer (1× PBS, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 100 μg/ml phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin) for 30 min with occasional rocking, and clarified by centrifugation at 12,000 × g for 15 min at 4°C. Identical amounts (100 μg of protein) of cell lysates were incubated at 37C for 20 min and resolved by SDS-PAGE and electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes. After being incubated in blocking solution containing 5% non-fat milk in TBST buffer (10 mM Tris-HCL (pH 8.0), 150 mM NaCl, and 0.1% Tween 20) for 1 h at 4°C, membranes were incubated with the appropriately diluted primary antibody. The expression of GAPDH was used as a loading control. The membranes were then incubated for 1 h with HRP-conjugated secondary antibody at 1:1000 dilution. Proteins were detected by the enhanced chemiluminescence detection system (Amersham, Aylesbury, UK). Protein expression was quantified by Scion Image software (Scion Co, USA) (33).
Flow cytometry
Single cell suspensions were prepared by the addition of 0.5 mM EDTA followed by three washes with an isotonic PBS buffer (supplemented with 0.5% BSA). For EGFR flow cytometric analysis, approximately 1×106 MCF-7, MCF-7/adr, S1 and S1-M1-80 cells (100 μl) were incubated at 4°C for 45 min with 20 μl of R-PE-conjugated anti-human EGFR reagent. Following this incubation, cells were washed twice with PBS buffer (supplemented with 0.5% BSA) and the supernatant was discarded. Finally, the cells were resuspended in 400 μl PBS buffer for flow cytometric analysis. Isotype control samples were treated in an identical manner with PE-labeled mouse IgG2b κ antibody. For Her2 expression analysis, 10 μl of PE-conjugated anti-human ErbB2 regent was mixed with 25 μl of cells (4×106 cells/ml). After incubating for 45 min at 4°C, the cells were washed twice with PBS buffer (supplemented with 0.5% BSA) and resuspended in 400 μl PBS buffer for flow cytometric analysis. Isotype control samples were treated in an identical manner with PE-labeled mouse IgG2B antibody (34).
Statistical analysis
All experiments were repeated at least three times and statistical significance determined using the Student’s t-test. Significance was determined at P<0.05.
Results
Effect of lapatinib and chemotherapeutic agents in various MDR cells and their parental cells
We examined the cytotoxic effects of lapatinib alone in different cell lines using the MTT assay. More than 90% of cells were viable at concentrations of lapatinib up to 2.5 μM in MCF-7, MCF-7/adr, S1, and S1-M1-80 cells (Figure S1, supplementary data). In contrast, lapatinib at 10 μM had virtually no cytotoxic effects on HEK293 cells (Figure S2). The cytotoxic effect of chemotherapeutic agents in MCF-7, MCF-7/adr, S1, and S1-M1-80 cells in the presence of 0.625, 1.25, or 2.50 μM lapatinib was tested. The mean IC50 values of chemotherapeutic agents in various pairs of sensitive and resistant cells in different concentrations of lapatinib are shown in Table 1. In ABCB1-overexpressing MCF-7/adr cells, lapatinib produced a significant dose-dependent increase in the cytotoxicity of doxorubicin in MCF-7/adr cells. In contrast, lapatinib only produced a ~2-fold sensitization to doxorubicin in the parental MCF-7 cells. Importantly, lapatinib, at the lowest concentration tested (0.625 μM) was still able to reverse resistance to doxorubicin at 6.5-fold in MCF-7/adr cells. When MCF-7 and MCF-7/adr cells were incubated with the specific ABCG2 inhibitor FTC at 2.5 μM, we found that FTC did not significantly affect the toxicity of doxorubicin in either MCF-7 or MCF-7/adr cell lines (Table 1). This result indicated that lapatinib reverses the resistance of MCF-7/adr cells by interacting with ABCB1. Lapatinib also significantly decreased resistance to mitoxantrone and topotecan in ABCG2-overexpressing S1-M1-80 cells. In addition, a small synergetic effect was also observed for the combination of lapatinib with either topotecan or mitoxantrone in the parental S1 cells but FTC did not significantly enhance the toxic effects of mitoxantrone in parental S1 cells (Table 1). These results suggest that lapatinib strongly enhances the sensitivity of ABCB1 and ABCG2 overexpressing MDR cells to conventional chemotherapeutic agents, but has only a slight effect in the parental cells.
Table 1. Effect of lapatinib on reversing ABCB1- and ABCG2-mediated drug resistance.
Cell survival was determined by MTT assay as described in “Materials and Methods”. Data are the means ± SD of at least three independent experiments performed in triplicate. The fold-reversal of MDR was calculated by dividing the IC50 for cells with the anticancer drug in the absence of lapatinib or FTC by that obtained in the presence of lapatinib or FTC. * and ** represent P<0.05 and P<0.01 respectively, for values versus that obtained in the absence of lapatinib.
| IC50 ± SD (μM) (fold-reversal) | ||||
|---|---|---|---|---|
| MCF-7 | MCF-7/adr (ABCB1) | |||
| Doxorubicin | 0.334±0.026** | (1.0) | 11.80±2.524** | (1.0) |
| +0.625 μM Lapatinib | 0.148±0.016* | (0.443) | 1.805±0.201** | (6.5) |
| +1.25 μM Lapatinib | 0.062±0.029* | (1.86) | 1.336±0.114** | (8.8) |
| +2.5 μM Lapatinib | 0.155±0.013* | (0.464) | 0.875±0.148** | (13.5) |
| +2.5 μM FTC | 0.365±0.072 | (1.1) | 14.50±2.57 | (1.23) |
| S1 | S1-M1-80 (ABCG2) | |||
| Mitoxantrone | 0.204±0.051** | (1.0) | 39.67±9.14** | (1.0) |
| +0.625 μM Lapatinib | 0.055±0.021** | (3.7) | 0.736±0.250** | (53.9) |
| +1.25 μM Lapatinib | 0.054±0.019** | (3.8) | 0.493±0.185** | (90.4) |
| +2.5 μM Lapatinib | 0.035±0.012** | (5.9) | 0.268±0.174** | (148) |
| +2.5 μM FTC | 0.167±0.012 | (1.2) | 0.244±0.091** | (163) |
| Topotecan | 0.084±0.045** | (1.0) | 40.48±17.24** | (1.0) |
| +0.625 μM Lapatinib | 0.015±0.007** | (5.6) | 0.658±0.206** | (61.5) |
| +1.25 μM Lapatinib | 0.014±0.005** | (6.0) | 0.437±0.094** | (92.6) |
| +2.5 μM Lapatinib | 0.012±0.006** | (7.0) | 0.428±0.091** | (94.6) |
Recent studies have shown that mutations at amino acid 482 in ABCG2 affect the substrate and antagonist specificity of ABCG2 (21, 35). Therefore, we investigated whether lapatinib would reverse ABCG2-mediated resistance to mitoxantrone in cells transfected with either the wild-type (R482) or mutant (R482G and R482T) forms of ABCG2. As shown in Table 2, the IC50 values for mitoxantrone in three ABCG2 transfected cell lines ABCG2-482-R5, ABCG2-482-G2 and ABCG2-482-T7 cells were significantly greater than those in their parental cell line HEK293/pcDNA3.1 cells. Lapatinib, at 2.5 and 10 μM, significantly reduced the IC50 value for mitoxantrone and reversed resistance to mitoxantrone in cells expressing either wild-type or mutant ABCG2. In addition, the reversal effect produced by lapatinib at 10 μM was similar to that of the specific ABCG2 inhibitor FTC at 2.5 μM and better than that of another EGFR TK inhibitor erlotinib at 10 μM (Table 2). There was no significant difference in the IC50 values for mitoxantrone in the presence or absence of lapatinib in HEK293/pcDNA3 cells (Table 2). In addition, lapatinib did not significantly alter the IC50 values of cisplatin which is not a substrate of ABCG2 in any of the cell lines. These results suggest that lapatinib specifically enhances the sensitivity of ABCG2 substrates in cells expressing either wild-type or mutant R482G/T ABCG2.
Table 2. Effect of Lapatinib on reversing ABCG2-mediated resistance to mitoxantrone and cisplatin.
Cell survival was determined by MTT assay as described in “Materials and Methods”. Data are the means ± SD of at least three independent experiments performed in triplicate. The fold-reversal of MDR was calculated by dividing the IC50 for cells with the anticancer drug in the absence of inhibitor by that obtained in the presence of inhibitor. ** represents P<0.01, for values versus that obtained in the absence of inhibitor.
| Compounds | IC50 ± SD (μM) (fold-reversal) |
|||
|---|---|---|---|---|
| HEK293/pcDNA3.1 | ABCG2-482-R5 | ABCG2-482-G2 | ABCG2-482-T7 | |
| Mitoxantrone | 0.043±0.004(1.0) | 0.694±0.021(1.0) | 1.683±0.004(1.0) | 1.060±0.022(1.0) |
| + Lapatinib 2.5 μM | 0.044±0.003(1.1) | 0.177±0.023(3.9)** | 0.219±0.003(7.7)** | 0.192±0.024(5.5)** |
| + Lapatinib 10 μM | 0.047±0.003(1.1) | 0.065±0.022(10.7)** | 0.070±0.006(24.0)** | 0.070±0.002(15.1)** |
| + FTC 2.5 μM | 0.042±0.006(1.0) | 0.071±0.011(9.8)** | 0.102±0.023(16.5)** | 0.087±0.001(12.2)** |
| + Erlotinib 10 μM | 0.042±0.003(1.0) | 0.085±0.002(8.2)** | 0.082±0.001(20.5)** | 0.075±0.004(14.1)** |
| Cisplatin | 1.85±0.175(1.0) | 1.89±0.240(1.0) | 2.05±0.265(1.0) | 1.83±0.031(1.0) |
| + Lapatinib 10 μM | 2.16±0.233(1.2) | 1.59±0.252(1.2) | 2.16±0.173(1.0) | 1.51±0.119(1.2) |
Lapatinib enhances the accumulation of chemotherapeutic agents in MDR cells overexpressing ABCB1 and ABCG2
The results above indicated that lapatinib could enhance the sensitivity of MDR cells to certain chemotherapeutic agents. The mechanism by which this occurs is unknown. Therefore, we examined its effects on doxorubicin accumulation in ABCB1 expressing MCF-7/adr cells and parental MCF-7 cells. Fig. 1A illustrates the effect of lapatinib on the accumulation of doxorubicin in the MCF-7/adr and MCF-7 cells. Doxorubicin accumulation was significantly higher (4.2-fold) in the sensitive MCF-7 cells than in the MDR MCF-7/adr cells. In contrast, the level of doxorubicin accumulation in the drug-sensitive MCF-7 cells was unaffected by lapatinib concentrations of 0.625, 1.25 or 2.5 μM of lapatinib. In the absence of lapatinib, the level of doxorubicin accumulation was low in MCF-7/adr cells and lapatinib restored the level of doxorubicin accumulation to that of the parental cells in a dose-dependent manner. The intracellular accumulation of doxorubicin was 1.5-, 2.9-, 3.6-fold higher in MCF-7/adr cells in the presence of 0.625, 1.25 or 2.5 μM of lapatinib, respectively. As depicted in Fig. 1B, in all cells overexpressing ABCG2, lapatinib at 1 μM and 2.5 μM produced a concentration-dependent increase in the intracellular accumulation of [3H]-mitoxantrone, and the effects of lapatinib at 2.5 μM were similar to that of FTC at 2.5 μM. However, lapatinib did not significantly alter the intracellular accumulation of [3H]-mitoxantrone in HEK293/pcDNA3.1 cells. These results demonstrate that lapatinib was able to increase intracellular accumulation of chemotherapeutic agents in cells expressing ABCB1 or ABCG2.
Figure 1. Effect of lapatinib on the accumulation of doxorubicin or [3H]-mitoxantrone and the transport of [3H]-methotrexate.
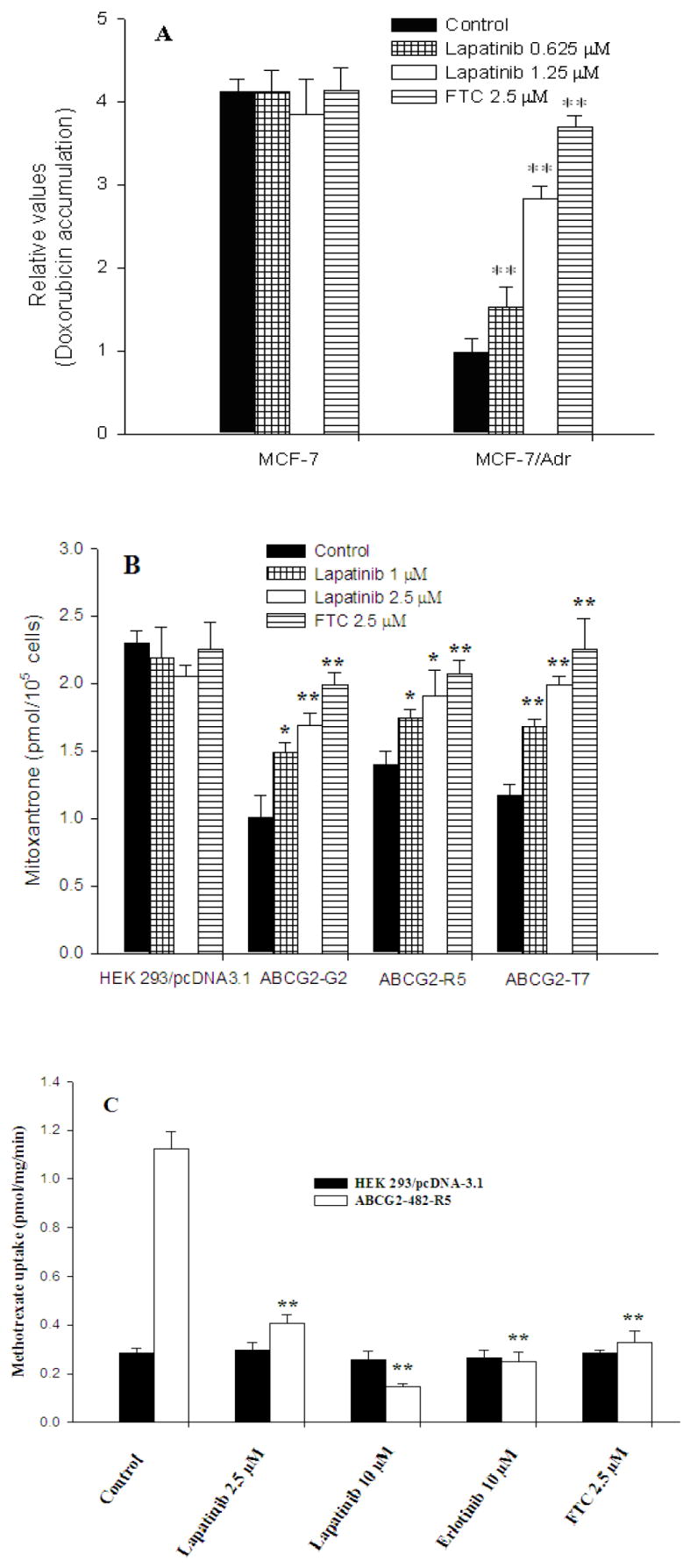
Accumulation of doxorubicin (A) in ABCB1 overexpressing MCF-7/adr cells and their parental MCF-7 cells. Accumulation of [3H]-mitoxantrone (B) in ABCG2 overexpressing transfected cells and empty-vector transfected cells. The transport of [3H]-methotrexate (C) in membrane vesicles from HEK293/pcDNA3.1 and ABCG2-482-R5 cells. All were measured as the described in the “Materials and Methods”. Data points represent the means ± SD of triplicate determinations. * and ** represent P < 0.05 and P < 0.01 respectively, for values versus those in the control group. Experiments were repeated at least three times, and a representative experiment is shown.
Lapatinib inhibits the transport of [3H]-methotrexate and [3H]-E217βG by wild-type ABCG2
To further confirm the effect of lapatinib on the transport activity of ABCG2, we used membrane vesicles prepared from HEK293/pcDNA3 and ABCG2-482-R5 cells to perform inhibition experiments. We chose these two cell lines as the rate of ATP-dependent uptake of [3H]-methotrexate, an antifolate anticancer drug and a substrate of ABCG2 in membranes isolated from HEK293/pcDNA3.1 cells was significantly different from membrane vesicles of ABCG2-482-R5 cells, but not from membrane vesicles of ABCG2-482-G2 and ABCG2-482-T7 cell lines (17). The effect of lapatinib on the transport of methotrexate by ABCG2 was shown in Fig. 1C. The rates of [3H]-methotrexate uptake were significantly inhibited by lapatinib in a concentration-dependent manner. Furthermore, the inhibitory effect of lapatinib on methotrexate transport by ABCG2 membrane vesicles is comparable to that of erlotinib and FTC (Fig. 1C). In addition, lapatinib produced a concentration-dependent inhibition of [3H]-E217βG, another substrate of ABCG2 (data not shown). These transport results suggest that lapatinib inhibits the transport of [3H]-methotrexate and [3H]-E217βG in wild-type ABCG2-482-R5 expressing cells.
Lapatinib activates the ATPase activity of ABCB1 and ABCG2
The drug-efflux function of ABCB1 and ABCG2 is linked to ATP hydrolysis which is stimulated in the presence of ABCB1 and ABCG2 substrates. To assess the effect of lapatinib on the ATPase activity of ABCB1 and ABCG2, we measured ABCB1- and ABCG2-mediated ATP hydrolysis using various concentrations of lapatinib under conditions that suppressed the activity of other major membrane ATPases. As shown in Fig. 2, lapatinib affected the ATPase activity of ABCB1 (Fig. 2A) and ABCG2 (Fig. 2B) in a concentration-dependent manner. Furthermore, the maximum ATPase activities of ABCB1 and ABCG2 in the presence of lapatinib were up to 42.9 ± 1.9 and 64.9 ± 1.7 nmoles Pi/mg protein/min, respectively. Interestingly, lapatinib significantly stimulates the ATPase activities of ABCG2 at extremely low concentrations. This is not easy observed in Fig. 2B; Consequently, only the low concentrations of lapatinib affecting the ATPase of ABCG2 are presented in the Inset of Fig. 2B. These data indicated that lapatinib may be a substrate of ABCB1 and ABCG2.
Figure 2. Effect of lapatinib on the ATPase activity of ABCB1 and ABCG2 and the [125I]-IAAP photoaffinity labeling of ABCB1 and ABCG2.
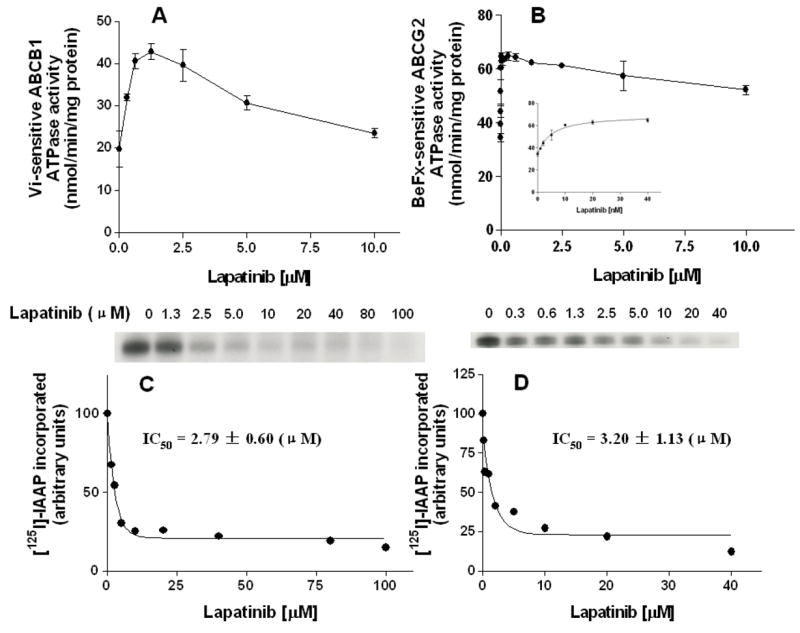
The Vi-sensitive ATPase activity of ABCB1 (A) and beryllium-fluoride-sensitive ATPase activity of ABCG2 (B), and the effect of lower concentrations of lapatinib on ABCG2-ATPase is shown in Inset of Fig. 2B. The ATP hydrolysis in membrane vesicles was determined with different concentrations of lapatinib, as described in “Materials and Methods” section. The basal ATPase activity (in the absence of lapatinib) was subtracted from the activities obtained at the indicated lapatinib concentrations and the graph was fitted by non-linear least-squares regression analysis using GraphPad Prism version 2.0. The values represent the means ± S.E.M. from at least three independent experiments. The photoaffinity labeling of ABCB1 (C) and ABCG2 (D) with [125I]-IAAP was performed using indicated concentrations of lapatinib as described in the “Materials and Methods” section. The radioactivity incorporated into ABCB1 and ABCG2 was determined by exposing the gel to an X-ray film at -70°C. Panels C and D show the autoradiograms and quantification of incorporation of IAAP into the ABCB1 and ABCG2 band respectively, from at least three independent experiments.
Lapatinib affects the photo-labeling of ABCB1 and ABCG2 with [125I]-IAAP
ABCB1 and ABCG2 can be photo-labeled by a photoaffinity analog of prazosin, [125I]-IAAP, and their substrates as well as inhibitors can compete for [125I]-IAAP labeling of ABCB1 and ABCG2 (32). We therefore examined the photo-labeling of ABCB1 and ABCG2 with [125I]-IAAP by incubating membrane vesicles in the presence of various concentrations of lapatinib in order to primarily understand the physical interaction of lapatinib with the substrate interaction sites of ABCB1 and ABCG2. As indicated in Fig. 2, lapatinib strongly inhibited the photoaffinity labeling of ABCB1 (Fig. 2C) and ABCG2 (Fig. 2D) with [125I]-IAAP in a concentration-dependent manner. The concentration of lapatinib required for 50% inhibition of photo-labeling of ABCB1 and ABCG2 with [125I]-IAAP was 2.8 ± 0.6 μM and 3.2 ± 1.1 μM, respectively. The results suggest that lapatinib binds to both the ABCB1 and ABCG2 substrate-binding site(s) with high affinity.
EGFR and Her-2 status and effect of lapatinib on the blockade of Akt and Erk1/2 phosphorylation
Using the MTT assay as an index of cytotoxicity, we observed that lapatinib alone does not produce significant cytotoxic effects in MCF-7 and S1 cell lines. However, non-toxic concentrations of lapatinib significantly enhance the cytotoxic effects of doxorubicin in MCF-7 cells, while FTC does not significantly enhance the cytotoxic effects of doxorubicin in MCF-7 cells (Table 1). To determine whether the relative lack of cytotoxicity produced by lapatinib is related to the expression of EGFR and/or Her2, we used flow cytometry to detect EGFR and Her-2 in MCF-7 and S1 cell lines (Fig. 3B). Calu-3, a positive control cell line expressed relative high levels of both EGFR (66.6 ± 2.8 %) and Her-2 (97.5 ± 1.2 %). The expression level of EGFR in S1 cells (65.0 ± 4.9 %) is significantly higher than that in S1-M1-80 cells (37.6 ± 2.5 %) while the expression level of Her-2 in S1 cells (29.3 ± 3.1%) is significantly lower than that in S1-M1-80 cells (49.3 ± 6.6 %). MCF-7 cell expressed low levels of EGFR (10.8 ± 2.7 %), whereas the MCF-7/adr cell line showed high expression (65.0 ± 4.5 %). However, the MCF-7 (28.8 ± 2.2 %) and MCF-7/adr (8.2 ± 0.5 %) cell lines expressed low levels of Her-2. These results indicated that lapatinib potentates the cytotoxic effects of anticancer drugs independent of the level of EGFR and Her-2 expression. Furthermore, we tested whether the concentrations of lapatinib that we used in our experiments can inhibit the phosphorylation of Akt or Erk1/2. As shown in Fig. 3A, lapatinib did not significantly block the phosphorylation of Akt and Erk1/2 in any of the four cell sublines. This result suggested that lapatinib-induced enhancement of the cytotoxicity of chemotherapeutic agents in MCF-7, MCF-7/adr, S1 and S1-M1-80 cells is not due to its antagonism of EGFR and Her-2 receptors.
Figure 3. The effect of lapatinib on blockade of Akt and Erk1/2 phosphorylation (A), the expression level of EGFR and Her-2 (B), the expression of ABCB1 and ABCG2 genes (C) and on the expression level of ABCB1 and ABCG2 protein (D).
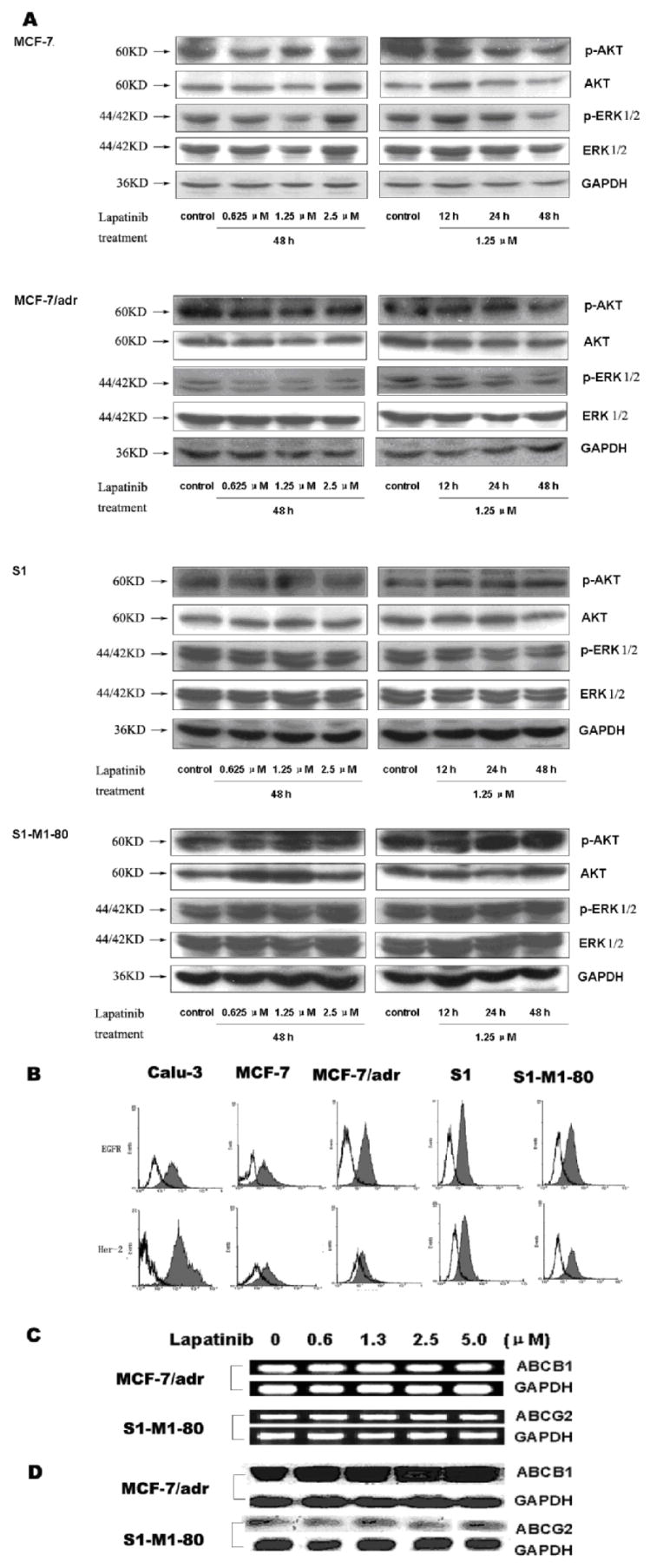
In figure 3A, equal amount protein from various cells was loaded for Western blot as described in the “Materials and Methods”. In 3B, Caul-3 cells were used as a positive control of EGFR and Her-2 expressing cells. Flow cytometric analysis was performed as described in the “Materials and Methods”. In 3C, MCF-7/adr and S1-M1-80 cells were treated by lapatinib of various concentrations for 48 h. The mRNA levels of ABCB1 and ABCG2 were determined by RT-PCR described as in the “Materials and Methods”. In 3D, MCF-7/Adr and S1-M1-80 cells were treated by lapatinib of various concentrations for 48 h. Equal amounts of total cell lysates were used for loading and were detected by Western blotting as described in the “Materials and Methods”. All these experiments were repeated at least three times, and a representative experiment is shown in each panel.
Effect of lapatinib on the expression of mRNA and protein levels of ABCB1 and ABCG2
The reversal of ABC transporter-mediated MDR can be achieved either by decreasing transporter expression or by inhibiting function. Therefore, we determined the effect of lapatinib on the expression level of mRNA and protein levels using RT-PCR and Western blot, respectively. Our results showed that no marked difference in ABCB1 or ABCG2 expression at the mRNA (Fig. 3C) or protein level (Fig. 3D) was observed in MCF-7/adr cells (upper panel) or S1-M1-80 cells (lower panel) treated with lapatinib for 48 h compared to untreated cells. These results provide evidence that lapatinib does not affect the expression of ABCB1 and ABCG2. Thus, it mediates the reversal of MDR by inhibiting the function of ABCB1 and ABCG2.
Lapatinib reverses ABCB1-mediated MDR in vivo
We examined the efficacy of lapatinib in vivo to reverse the resistance to paclitaxel using an established KBv200 cell xenografts in nude mice. There was no significant difference in tumor size between animals treated with saline, lapatinib or paclitaxel alone. However, the combination of lapatinib and paclitaxel produced a significant greater inhibitory effect on tumor growth compared to animals treated with only saline, paclitaxel or lapatinib (p < 0.05) (Fig. 4) and the inhibition rate was 50.1%. In addition, at the doses tested, no mortality or significant decrease in body weight was associated with the combination treatments, suggesting that the combination regimen did not result in increased toxicity.
Figure 4. Potentiation of antitumor effects of paclitaxel by lapatinib in a xenograft model of KBv200 cells in athymic nude mice.
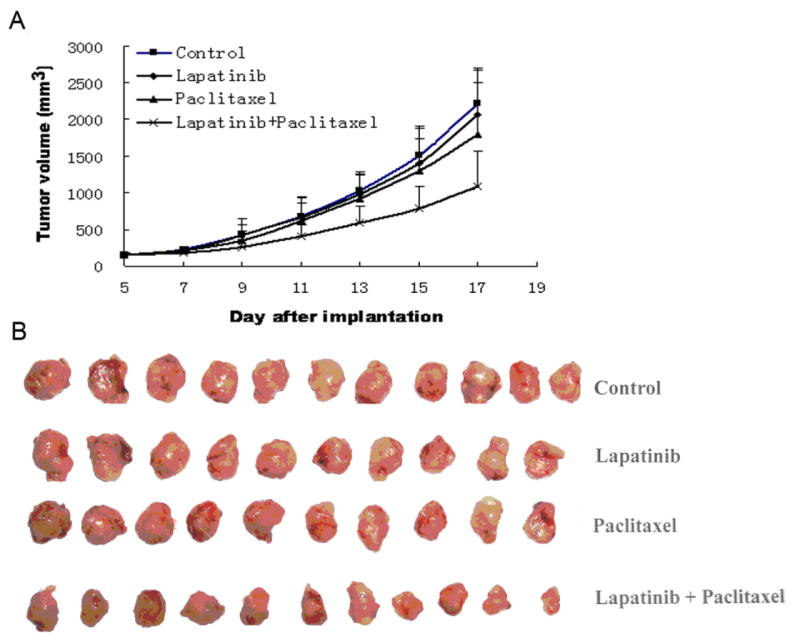
The experiment was carried out using athymic mice implanted subcutaneously (s.c.) with KBv200 cells. A, changes in tumor volume with time. Data represent the mean tumor volume for each group ± standard deviation in 10-11 mice after implantation. B, tumor sizes. The picture was taken on 17th day after implantation. Lapatinib dose was 100 mg/Kg, P.O., q3d×4, and paclitaxel (18 mg/kb, i.p., q3d×4) + (100 mg/Kg, P.O., q3d×4, given 1 h before paclitaxel administration) for combination.
Discussion
Lapatinib is an inhibitor of the intracellular tyrosine kinase domains of both the EGFR and Her-2 receptors. Mutations or dysregulation in these receptors have been shown to play a role in the development of certain cancers. Lapatinib was approved for use in combination with capecitabine for the treatment of patients with advanced or metastatic breast cancer whose tumors overexpressed HER-2 and who had received prior therapy with an anthracycline, a taxane, and trastuzumab. As the new tyrosine kinase inhibitors are being introduced into the clinic, a significant effort will be directed toward increasing the anticancer activity of conventional chemotherapeutic agents or restoring chemosensitivity of resistant cancer cells to conventional chemotherapeutic agents. Our results showed for the first time that lapatinib had potent reversing activity in both ABCB1- and ABCG2-expressing MDR cells in vitro. Lapatinib, however, had no significant reversal effect in ABCC4-overexpressing NIH3T3/ABCC4-2 cells and lung cancer resistant protein (LRP) overexpressing SW1573/2R1220 cells (unpublished data). Although lapatinib slightly enhanced the cytotoxicity of doxorubicin, mitoxantrone and topotecan in drug-sensitive MCF-7 and S1 cells, respectively, lapatinib significantly potentiated the cytotoxicity of conventional chemotherapeutic agents in ABCB1- and ABCG2- overexpressing MDR cells to a much greater extent. Furthermore, lapatinib did not significantly alter cellular sensitivity to non-ABCB1 or non-ABCG2 substrates. Although the concentrations of lapatinib used in the current study (up to 2.5 μM for MCF-7 and S1 cells) have been reported to be sufficient to block the EGFR signaling pathway, we did not observe any significant effect on the growth and survival of cells (Fig. S1 and S2). In addition, we found that 2.5 μM lapatinib does not block the phosphorylation of Akt and Erk1/2 (Fig. 3A) in MCF-7 and S1 cell lines. Thus, the potentiation of the cytotoxic effects of doxorubicin by lapatinib in MCF-7 cells may not be related to the antagonism of EGFR or Her-2 receptors. It is possible that this effect may be generated by a non-specific cytotoxic mechanism or other unknown action of the drug. In order to determine if the in vitro effects of lapatinib can be extended to an in vivo paradigm, we have examined the effect of lapatinib on the antitumor activity of paclitaxel in xenograft model in mouse. Indeed, our results indicated that the combination of lapatinib with paclitaxel results in markedly enhanced antitumor activity of paclitaxel in an ABCB1-overexpressing tumor xenograft model (Fig. 4 A and B).
Our results suggest that lapatinib significantly potentiated the toxicity of established ABCB1 or ABCG2 substrates in ABCB1- or ABCG2-overexpressing MDR cells unrelated to its inhibitory action of tyrosine kinase. Several groups have published in vitro data that supports our findings. Coley et al. reported that the addition of GW282974A, an analogue of lapatinib, to paclitaxel resulted in a synergistic inhibition of cell survival in ABCB1-expressing human ovarian cancer cell line PEO1TaxR (36). The dual EGFR and Her-2 directed small molecule tyrosine kinase inhibitor CI1033 enhanced the uptake and cytotoxicity of SN-38 and topotecan in ABCG2-expressing glioblastoma T98G cells, colorectal carcinoma HCT8 cells and ABCG2-transfected MDA-MB-231 cells (37). Recently, Polli et al. (38) reported that lapatinib is a substrate of ABCB1 and ABCG2 and an inhibitor of ABCB1 and ABCG2 (IC50 values of 3.9 μM, and 0.025 μM, respectively). Their results are not only consistent with our findings that lapatinib is an inhibitor of ABCB1 and ABCG2, but their data are also agreement with our findings that low concentrations of lapatinib are able to stimulate the ATPase activity of ABCG2 (Fig. 2 A and B) and inhibit the photolabeling of ABCB1 and ABCG2 with IAAP (Fig. 2 C and D) indicating that lapatinib directly interacts with these transporters. Taken as a whole, these data suggest that the pharmacokinetics of conventional chemotherapeutic agents that are affected by ABC transporters may be altered in the presence of lapatinib.
Clinical studies have also hinted at interactions between lapatinib and ABC transporters. Lapatinib has been shown to have clinical benefit in patients with brain-metastasized breast cancer, increasing drug penetration across the blood-brain barrier, presumably via inhibition of ABCB1(37). The combination of lapatinib and tamoxifen an ABCB1 substrate or conventional chemotherapeutic agents, such as paclitaxel and docetaxel, may be active against hormone-refractory and chemotherapeutic drug-resistant metastasized breast cancer (14, 39-41). In the phase I study, when compared to irinotecan alone, the co-administration of lapatinib and irinotecan significantly increased the area under the plasma concentration-time curve of SN-38, the active metabolite of irinotecan, which is an ABCB1 and ABCG2 substrate (42). Despite the aforementioned promising findings, the authors of these papers did not propose any clear mechanisms to explain the synergy between lapatinib and chemotherapeutic agents. However, in human pharmacokinetic studies, the highest peak plasma lapatinib level was roughly 3 μmol/L, the half-life was approximately 17 hours and steady-state concentrations were achieved after six to seven days of once-daily dosing (43, 44). These data suggest that the in vitro concentrations of lapatinib used in our experiments are similar to those obtained in plasma after therapeutic treatment. Thus, it is possible that lapatinib affects chemosensitivity of refractory or resistant cancer cells through its interaction with ABC transporters.
Recently, Baker SD et al reported that one common functional single-nucleotide polymorphism in the ABCG2 gene, ABCG2 421C→A (Q141K), is associated with diarrhea, a gefitinib-induced adverse effect, and led to a high risk of diarrhea in patients treated with oral gefitinib (45). The same group also reported that this functional variant of ABCG2 was associated with a greater accumulation of gefitinib at steady-state and this may be relevant to toxicity and antitumor activity of EGFR TKIs (46). These findings suggest that the functional variants of ABCG2 in patients may affect the pharmacokinetics and pharmacodynamics of not only established ABCG2 substrates such as camptothecins and mitoxantrone, but also novel molecular target anticancer drugs such as gefitinib and lapatinib. Therefore, these functional single-nucleotide polymorphisms can cause alterations in the adverse events and therapeutic effects of chemotherapy. Similar to gefitinib, the most frequent adverse effects of lapatinib in patients are skin rash and diarrhea (47). Thus, these functional single-nucleotide polymorphisms of ABCG2 in patients may also affect the pharmacokinetics and pharmacodynamics of lapatinib, resulting in an attenuation of its adverse events and therapeutic effects. Indeed, Johnston et al. reported that the first-pass metabolism of lapatinib is mediated by the CYP3A4/5 enzymes that (47).
The expression of the Arg(482), Gly(482), and Thr(482) variant forms of ABCG2 has been shown to confer greater resistance to some substrates such as mitoxantrone and the sensitivity to some ABCG2 modulators has been shown to be decreased compared to the wild-type form (48-50). Our results showed that lapatinib significantly enhances the sensitivity of ABCG2 substrates not only in cells overexpressing wild-type but also the R482G/T variants of ABCG2. Mechanistically, similar to other MDR inhibitors, lapatinib may be able to reverse ABCB1- or ABCG2-mediated drug resistance by inhibiting drug efflux. Consistent with this hypothesis, we found that incubating MDR cells (such as MCF7/adr and S1-M1-80 cells) concomitantly with conventional chemotherapeutic drugs (substrates of ABC transporters) and lapatinib resulted in a higher intracellular drug accumulation in ABCB1 and ABCG2 expressing cells than cells incubated with drug alone (Fig. 1A and B). A similar result was obtained when we examined accumulation of rhodamine 123 in ABCB1-expressing cells (data not shown). Furthermore, the transport of E217βG and methotrexate inhibited by lapatinib in a concentration-dependent manner in membrane vesicles overexpressed wild-type ABCG2. However, the majority of substrates that interact with the ABC drug transporters stimulate ATP hydrolysis and the fact that lapatinib stimulated the ATP hydrolysis of both ABCB1 and ABCG2 suggested that it behaved similar to other known substrates of these transporters. These data led us to speculate that lapatinib interacts directly with the transporters. Indeed, this was confirmed by the finding that lapatinib significantly inhibited the binding of the compound IAAP, which photolabels the drug-substrate binding site of ABCB1 and ABCG2. Lapatinib, however, had no significant effect on the expression of ABCB1 in MCF-7/adr cells and ABCG2 in S1-M1-80 cells (Fig. 3C and D). These results suggested that lapatinib reverses ABCB1- and ABCG2-mediated MDR by inhibiting the function as opposed to expression of these two pumps. The expression of EGFR and Her2 (Fig. 3B) did not significantly alter lapatinib toxicity in MCF-7/adr, S1-M1-80 cells or parental MCF-7 and S1 cells (Fig. S1). Additional in vitro studies in cell lines expressing wild-type and mutant EGFR may be useful to determine if there is a difference in the efficacy between tumors expressing wild-type or mutant EGFR. The observed toxicity of lapatinib at such relatively high concentrations may be generated by a non-EGFR phosphorylation pathway. However, in this study, we did not examine the potential mechanisms of lapatinib toxicity in our cell lines.
In conclusion, lapatinib may inhibit cellular ABCB1 and ABCG2 functions at clinically relevant concentrations. The inhibition of drug efflux as a result of direct interaction of lapatinib with ABCB1 or ABCG2 may lead to increased clinical response when combined with conventional chemotherapeutic agents. Our analysis of the reversal effect of lapatinib in tumor xenograft model indicates that combination of lapatinib with other anticancer drugs may be important in surmounting clinical resistance in cancer chemotherapy.
Supplementary Material
Acknowledgments
We thank Dr. Susan E. Bates (NCI, National Institute of Health) for S1 and S1-M1-80 cells, and FTC, Dr. Somnath Pal (St. John’s University) for assistance with statistical analyses, Dr. Jian-je Zhang (Sun Yat-Sen University) for technical assistance, Tong Shen (St. John’s University) and Yangmin (Mimi) Chen (Montgomery High School, New Jersey) for the editorial assistance of the manuscript. This work was supported by funds from China National Natural Sciences Foundation No.30672407 (L Fu) and 863 Project Foundation No.2006AA09Z419 (L Fu); St. John’s University Tenure Track Faculty Position Start-Up Funding No.C-0531 and St. John’s University Seed Grant No.579-1110 (ZS Chen). CP Wu, RW Robey, and SV Ambudkar were supported by the Intramural Research Program, Center for Cancer Research, National Cancer Institute, NIH.
References
- 1.Dean M, Rzhetsky A, Allikmets R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res. 2001;11:1156–66. doi: 10.1101/gr.184901. [DOI] [PubMed] [Google Scholar]
- 2.Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. J Clin Pharmacol. 2005;45:872–7. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 3.Gillet JP, Efferth T, Remacle J. Chemotherapy-induced resistance by ATP-binding cassette transporter genes. Biochim Biophys Acta. 2007;1775:237–62. doi: 10.1016/j.bbcan.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 4.Glavinas H, Krajcsi P, Cserepes J, Sarkadi B. The role of ABC transporters in drug resistance, metabolism and toxicity. Curr Drug Deliv. 2004;1:27–42. doi: 10.2174/1567201043480036. [DOI] [PubMed] [Google Scholar]
- 5.Ambudkar SV, Kimchi-Sarfaty C, Sauna ZE, Gottesman MM. P-glycoprotein: from genomics to mechanism. Oncogene. 2003;22:7468–85. doi: 10.1038/sj.onc.1206948. [DOI] [PubMed] [Google Scholar]
- 6.Kruh GD, Belinsky MG. The MRP family of drug efflux pumps. Oncogene. 2003;22:7537–52. doi: 10.1038/sj.onc.1206953. [DOI] [PubMed] [Google Scholar]
- 7.Doyle LA, Ross DD. Multidrug resistance mediated by the breast cancer resistance protein BCRP (ABCG2) Oncogene. 2003;22:7340–58. doi: 10.1038/sj.onc.1206938. [DOI] [PubMed] [Google Scholar]
- 8.Normanno N, De Luca A, Bianco C, et al. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 9.Grant S, Qiao L, Dent P. Roles of ERBB family receptor tyrosine kinases, and downstream signaling pathways, in the control of cell growth and survival. Front Biosci. 2002;7:d376–89. doi: 10.2741/grant. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee A, Dhadda AS, Shehata M, Chan S. Lapatinib: a tyrosine kinase inhibitor with a clinical role in breast cancer. Expert Opin Pharmacother. 2007;8:2189–204. doi: 10.1517/14656566.8.13.2189. [DOI] [PubMed] [Google Scholar]
- 11.Nahta R, Yuan LX, Du Y, Esteva FJ. Lapatinib induces apoptosis in trastuzumab-resistant breast cancer cells: effects on insulin-like growth factor I signaling. Mol Cancer Ther. 2007;6:667–74. doi: 10.1158/1535-7163.MCT-06-0423. [DOI] [PubMed] [Google Scholar]
- 12.Geyer CE, Forster J, Lindquist D, et al. Lapatinib plus capecitabine for HER2-positive advanced breast cancer. N Engl J Med. 2006;355:2733–43. doi: 10.1056/NEJMoa064320. [DOI] [PubMed] [Google Scholar]
- 13.Johnston SR. Clinical trials of intracellular signal transductions inhibitors for breast cancer--a strategy to overcome endocrine resistance. Endocr Relat Cancer. 2005;12:S145–57. doi: 10.1677/erc.1.00992. [DOI] [PubMed] [Google Scholar]
- 14.Chu I, Blackwell K, Chen S, Slingerland J. The dual ErbB1/ErbB2 inhibitor, lapatinib ( GW572016), cooperates with tamoxifen to inhibit both cell proliferation- and estrogen-dependent gene expression in antiestrogen-resistant breast cancer. Cancer Res. 2005;65:18–25. [PubMed] [Google Scholar]
- 15.Elkind NB, Szentpetery Z, Apati A, et al. Multidrug transporter ABCG2 prevents tumor cell death induced by the epidermal growth factor receptor inhibitor Iressa (ZD1839, Gefitinib) Cancer Res. 2005;65:1770–7. doi: 10.1158/0008-5472.CAN-04-3303. [DOI] [PubMed] [Google Scholar]
- 16.Yang CH, Huang CJ, Yang CS. Gefitinib reverses chemotherapy resistance in gefitinib-insensitive multidrug resistant cancer cells expressing ATP-binding cassette family protein. Cancer Res. 2005;65:6943–9. doi: 10.1158/0008-5472.CAN-05-0641. [DOI] [PubMed] [Google Scholar]
- 17.Shi Z, Peng XX, Kim IW, et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007;67:11012–20. doi: 10.1158/0008-5472.CAN-07-2686. [DOI] [PubMed] [Google Scholar]
- 18.Fu L, Liang Y, Deng L, et al. Characterization of tetrandrine, a potent inhibitor of P-glycoprotein-mediated multidrug resistance. Cancer Chemother Pharmacol. 2004;53:349–56. doi: 10.1007/s00280-003-0742-5. [DOI] [PubMed] [Google Scholar]
- 19.Dai CL, Xiong HY, Tang LF, et al. Tetrandrine achieved plasma concentrations capable of reversing MDR in vitro and had no apparent effect on doxorubicin pharmacokinetics in mice. Cancer Chemother Pharmacol. 2007;60:741–50. doi: 10.1007/s00280-007-0420-0. [DOI] [PubMed] [Google Scholar]
- 20.Litman T, Brangi M, Hudson E, et al. The multidrug-resistant phenotype associated with overexpression of the new ABC half-transporter, MXR (ABCG2) J Cell Sci. 2000;113(Pt 11):2011–21. doi: 10.1242/jcs.113.11.2011. [DOI] [PubMed] [Google Scholar]
- 21.Robey RW, Honjo Y, Morisaki K, et al. Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity. Br J Cancer. 2003;89:1971–8. doi: 10.1038/sj.bjc.6601370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu LW, Zhang YM, Liang YJ, Yang XP, Pan QC. The multidrug resistance of tumour cells was reversed by tetrandrine in vitro and in xenografts derived from human breast adenocarcinoma MCF-7/adr cells. Eur J Cancer. 2002;38:418–26. doi: 10.1016/s0959-8049(01)00356-2. [DOI] [PubMed] [Google Scholar]
- 23.Chen LM, Wu XP, Ruan JW, et al. Screening novel, potent multidrug-resistant modulators from imidazole derivatives. Oncology Res. 2004;14:355–62. doi: 10.3727/0965040041292378. [DOI] [PubMed] [Google Scholar]
- 24.Shi Z, Liang YJ, Chen ZS, et al. Reversal of MDR1/P-glycoprotein-mediated multidrug resistance by vector-based RNA interference in vitro and in vivo. Cancer Biol Ther. 2006;5:39–47. doi: 10.4161/cbt.5.1.2236. [DOI] [PubMed] [Google Scholar]
- 25.Chen LM, Liang YJ, Ruan JW, et al. Reversal of P-gp mediated multidrug resistance in-vitro and in-vivo by FG020318. J Pharm Pharmacol. 2004;56:1061–6. doi: 10.1211/0022357043879. [DOI] [PubMed] [Google Scholar]
- 26.Venne A, Li S, Mandeville R, Kabanov A, Alakhov V. Hypersensitizing effect of pluronic L61 on cytotoxic activity, transport, and subcellular distribution of doxorubicin in multiple drug-resistant cells. Cancer Res. 1996;56:3626–9. [PubMed] [Google Scholar]
- 27.Cornwell MM, Gottesman MM, Pastan IH. Increased vinblastine binding to membrane vesicles from multidrug-resistant KB cells. J Biol Chem. 1986;261:7921–8. [PubMed] [Google Scholar]
- 28.Kerr KM, Sauna ZE, Ambudkar SV. Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in Human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates. J Biol Chem. 2001;276:8657–64. doi: 10.1074/jbc.M010044200. [DOI] [PubMed] [Google Scholar]
- 29.Chen ZS, Robey RW, Belinsky MG, et al. Transport of methotrexate, methotrexate polyglutamates, and 17beta-estradiol 17-(beta-D-glucuronide) by ABCG2: effects of acquired mutations at R482 on methotrexate transport. Cancer Res. 2003;63:4048–54. [PubMed] [Google Scholar]
- 30.Ambudkar SV. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 1998;292:504–514. doi: 10.1016/s0076-6879(98)92039-0. [DOI] [PubMed] [Google Scholar]
- 31.Sauna ZE, Ambudkar SV. Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein. Proc Natl Acad Sci U S A. 2000;97:2515–20. doi: 10.1073/pnas.97.6.2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shukla S, Robey RW, Bates SE, Ambudkar SV. The calcium channel blockers, 1,4-dihydropyridines, are substrates of the multidrug resistance-linked ABC drug transporter, ABCG2. Biochemistry. 2006;45:8940–51. doi: 10.1021/bi060552f. [DOI] [PubMed] [Google Scholar]
- 33.Shi Z, Liang YJ, Chen ZS, et al. Overexpression of Survivin and XIAP in MDR cancer cells unrelated to P-glycoprotein. Oncol Rep. 2007;17:969–76. [PubMed] [Google Scholar]
- 34.Lostumbo A, Mehta D, Setty S, Nunez R. Flow cytometry: a new approach for the molecular profiling of breast cancer. Exp Mol Pathol. 2006;80:46–53. doi: 10.1016/j.yexmp.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 35.Honjo Y, Hrycyna CA, Yan QW, et al. Acquired mutations in the MXR/BCRP/ABCP gene alter substrate specificity in MXR/BCRP/ABCP-overexpressing cells. Cancer Res. 2001;61:6635–9. [PubMed] [Google Scholar]
- 36.Coley HM, Shotton CF, Ajose-Adeogun A, Modjtahedi H, Thomas H. Receptor tyrosine kinase (RTK) inhibition is effective in chemosensitising EGFR-expressing drug resistant human ovarian cancer cell lines when used in combination with cytotoxic agents. Biochem Pharmacol. 2006;72:941–8. doi: 10.1016/j.bcp.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 37.Erlichman C, Boerner SA, Hallgren CG, et al. The HER tyrosine kinase inhibitor CI1033 enhances cytotoxicity of 7-ethyl-10-hydroxycamptothecin and topotecan by inhibiting breast cancer resistance protein-mediated drug efflux. Cancer Res. 2001;61:739–48. [PubMed] [Google Scholar]
- 38.Polli JW, Humphreys JE, Harmon KA, et al. The role of efflux and uptake transporters in N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl ]amino}methyl)-2-furyl]-4-quinazolinamine ( GW572016, lapatinib) disposition and drug interactions. Drug Metab Dispos. 2008;36:695–701. doi: 10.1124/dmd.107.018374. [DOI] [PubMed] [Google Scholar]
- 39.Johnston SR, Martin LA, Leary A, Head J, Dowsett M. Clinical strategies for rationale combinations of aromatase inhibitors with novel therapies for breast cancer. J Steroid Biochem Mol Biol. 2007;106:180–6. doi: 10.1016/j.jsbmb.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 40.Higa GM, Abraham J. Lapatinib in the treatment of breast cancer. Expert Rev Anticancer Ther. 2007;7:1183–92. doi: 10.1586/14737140.7.9.1183. [DOI] [PubMed] [Google Scholar]
- 41.Agulnik M, Cohen EW, Cohen RB, et al. Phase II study of lapatinib in recurrent or metastatic epidermal growth factor receptor and/or erbB2 expressing adenoid cystic carcinoma and non adenoid cystic carcinoma malignant tumors of the salivary glands. J Clin Oncol. 2007;25:3978–84. doi: 10.1200/JCO.2007.11.8612. [DOI] [PubMed] [Google Scholar]
- 42.Midgley RS, Kerr DJ, Flaherty KT, et al. A phase I and pharmacokinetic study of lapatinib in combination with infusional 5-fluorouracil, leucovorin and irinotecan. Ann Oncol. 2007;18:2025–9. doi: 10.1093/annonc/mdm366. [DOI] [PubMed] [Google Scholar]
- 43.Bence AK, Anderson EB, Halepota MA, et al. Phase I pharmacokinetic studies evaluating single and multiple doses of oral GW572016, a dual EGFR-ErbB2 inhibitor, in healthy subjects. Invest New Drugs. 2005;23:39–49. doi: 10.1023/B:DRUG.0000047104.45929.ea. [DOI] [PubMed] [Google Scholar]
- 44.Burris HA, 3rd, Hurwitz HI, Dees EC, et al. Phase I safety, pharmacokinetics, and clinical activity study of lapatinib ( GW572016), a reversible dual inhibitor of epidermal growth factor receptor tyrosine kinases, in heavily pretreated patients with metastatic carcinomas. J Clin Oncol. 2005;23:5305–13. doi: 10.1200/JCO.2005.16.584. [DOI] [PubMed] [Google Scholar]
- 45.Cusatis G, Gregorc V, Li J, et al. Pharmacogenetics of ABCG2 and adverse reactions to gefitinib. J Natl Cancer Inst. 2006;98:1739–42. doi: 10.1093/jnci/djj469. [DOI] [PubMed] [Google Scholar]
- 46.Li J, Cusatis G, Brahmer J, et al. Association of Variant ABCG2 and the Pharmacokinetics of Epidermal Growth factor Receptor Tyrosine Kinase Inhibitors in Cancer Patients. Cancer Biol Ther. 2007:6. doi: 10.4161/cbt.6.3.3763. [DOI] [PubMed] [Google Scholar]
- 47.Johnston SR, Leary A. Lapatinib: a novel EGFR/HER2 tyrosine kinase inhibitor for cancer. Drugs Today (Barc) 2006;42:441–53. doi: 10.1358/dot.2006.42.7.985637. [DOI] [PubMed] [Google Scholar]
- 48.Mitomo H, Kato R, Ito A, et al. A functional study on polymorphism of the ATP-binding cassette transporter ABCG2: critical role of arginine-482 in methotrexate transport. Biochem J. 2003;373:767–74. doi: 10.1042/BJ20030150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shafran A, Ifergan I, Bram E, et al. ABCG2 harboring the Gly482 mutation confers high-level resistance to various hydrophilic antifolates. Cancer Res. 2005;65:8414–22. doi: 10.1158/0008-5472.CAN-04-4547. [DOI] [PubMed] [Google Scholar]
- 50.Ozvegy-Laczka C, Koblos G, Sarkadi B, Varadi A. Single amino acid (482) variants of the ABCG2 multidrug transporter: major differences in transport capacity and substrate recognition. Biochim Biophys Acta. 2005;1668:53–63. doi: 10.1016/j.bbamem.2004.11.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.