

Abstract
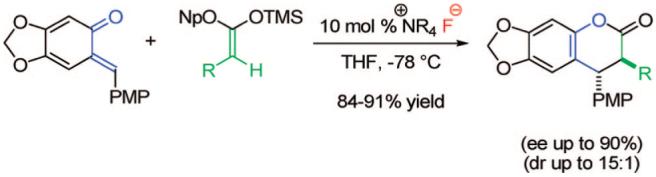
The catalytic, enantioselective, [4 + 2] cycloaddition reaction of ortho-quinone methides with silyl ketene acetals is described. This mechanistically interesting reaction, initiated by a chiral cinchona alkaloid-derived ammonium fluoride “precatalyst” complex, affords a variety of alkyl- and aryl-substituted 3,4-dihydrocoumarin products in excellent yield and with good enantioselectivity.
o-Quinone methides (o-QMs) are a valuable class of compounds with an illustrious chemical history.1 QMs serve as important intermediates in a variety of biological pathways,1a,2 and their reactions generate products of broad synthetic utility.1a Yet surprisingly, catalytic, asymmetric cycloadditions of o-quinone methides are virtually absent from the literature. Typically, o-QMs are generated under Lewis acidic,1a,3 thermal,1a,4 or photochemical conditions1a,5 and are used/trapped in situ. As a result, polymerization is a common problem, as is generation of a significant quantity of the desired compound at any given time. o-QMs that are isolable and whose reactivity is controllable would be ideal; their utilization in asymmetric cycloaddition reactions would provide access to a basic scaffold on which many therapeutic agents are based. For example, molecules containing the coumarin and dihydrocoumarin skeleton are prevalent in nature.6 Derivatives of these compounds have been shown to exhibit a variety of pharmacological properties, including antiherpetic activity,7a inhibition of protein kinases7b and aldose reductase,7c activity against several cancer lines,7d-f and selective inhibition of HIV-1 reverse transcriptase.7g Drawing from our successes with catalytic [4 + 2] cycloadditions,8 we sought to extend the scope of these highly enantioselective ketene enolate reactions to those of o-quinone methides, using cinchona alkaloid-derived precatalysts.9 Chiral ammonium halide catalysis is well documented,10 and it has found ubiquitous application of late. However, fluoride ion catalysis remains relatively underutilized.11 In this letter, we disclose the first example of a cycloaddition with o-quinone methides using silyl ketene acetals12 and chiral ammonium fluoride precatalysts.13 This methodology allows the production of 3,4-dihydrocoumarin derivatives in excellent yield with up to 90% ee and up to 15.4:1 dr.
The electron-rich o-quinone methide (1), first synthesized by Jurd in 1977, was chosen as a test substrate. This compound stood out as an excellent candidate because of its facile preparation and isolability. The o-QM (1) is easily synthesized in two steps from sesamol and 4-methoxybenzyl alcohol under mild Friedel-Crafts conditions, followed by Ag2O oxidation. Initially, we treated the o-QM (1) with butyryl chloride, thermodynamic Hünig’s base, and kinetic base/chiral catalyst benzoylquinidine (BQd) (Scheme 1, A). To our dismay, these conditions that worked beautifully in previous systems8a produced no product in the reaction. We reasoned that the high electron density of the o-QM was incompatible with the mildly nucleophilic, zwitterionic ketene enolate. Instead, we drew on silyl ketene acetal (2)asour ketene enolate precursor.
Scheme 1.
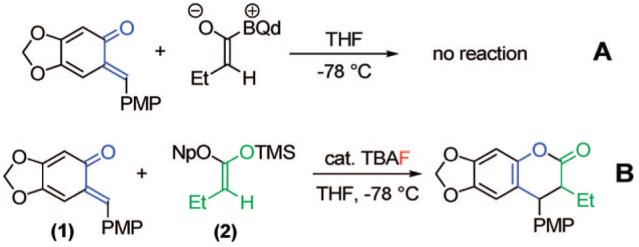
o-Quinone Methide Test Reactions
This substrate was then combined with the o-QM and catalytic tetrabutylammonium fluoride (TBAF) in THF14 at -78 °C (Scheme 1, B). We saw full consumption of the o-QM after only 1 h, to form the desired [4 + 2] cycloadduct in excellent yield after workup. To probe the potential enantioselectivity of the reaction, a number of chiral ammonium fluoride precatalysts were tested. The most successful precatalyst, N-(3-nitrobenzyl)quinidinium fluoride (Figure 1), when combined with ketene acetal (2), afforded cycloaddition products with 80% ee. We determined that the precatalyst’s effect on the enantioselectivity of the reaction was not dictated by steric bulk but rather by electronic effects.15
Figure 1.
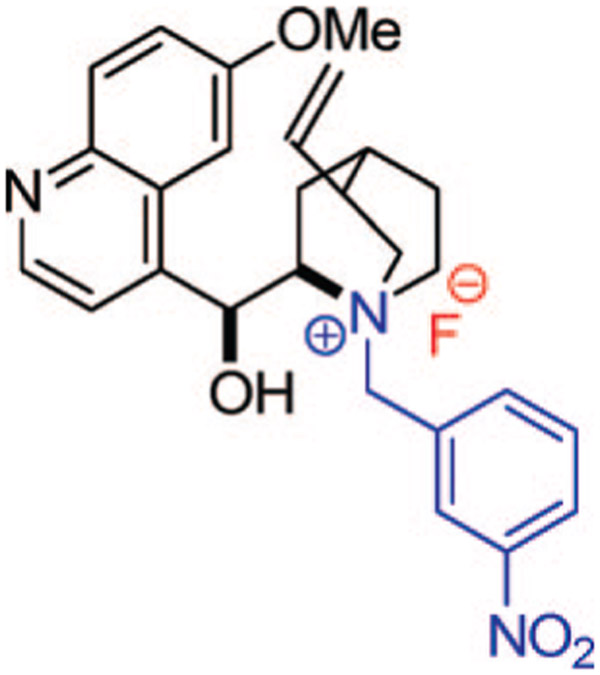
Chiral cinchona alkaloid-derived ammonium fluoride precatalyst.
Various precatalyst optimization experiments led us to the strongly withdrawing nitro group, which afforded the best results. The substitution pattern of the nitro group also proved to be important, with the 4-nitro versus 3-nitro precatalysts giving a difference of 10% ee (70 versus 80%, respectively).
In addition, we found that the hydroxyl group of the cinchona alkaloid moiety was critical to high optical induction. In all cases, O-alkylated and acylated precatalysts gave lower % ee than their respective precatalysts bearing a free -OH. The necessity for a hydrogen bond donation source led us to explore chiral H-bond donors as additives in the reaction. An assortment of chiral additives was screened, and we found that 10 mol % of the bis(diphenylphosphoryl)pyrrolidine16 (Figure 2, right), in conjunction with 4-nitrobenzylquinidinium fluoride precatalyst, o-QM (1), and ketene acetal (2) in THF at -78 °C, increased the % ee from 70 to 80 (Figure 2). Unfortunately, no such increase in % ee was afforded when combined with the N-3-nitrobenzyl precatalyst.
Figure 2.

Chiral additive (N-Boc-protected and free -NH).
With optimized conditions in hand, we illustrated the versatility of the methodology by screening a selection of ketene acetals (Table 1). As illustrated by the diverse array of products produced, the system is tolerant of both aliphatic and aryl substituents and those with sterically hindered, small, or heavyatom groups. The use of quinidine-derived precatalyst produces products with the 7S,8R absolute configuration as the predominant enantiomer,17 and the analogous quinine-based precatalysts afford cycloadducts with equal and opposite selectivity. Of note, by replacing the 2-naphthyl group of the ketene acetal with a phenyl group, the ee decreased about 5% for each ketene acetal tested. It is also interesting to mention that, by replacing the 2-naphthyl group of the ketene acetal with a methyl group, the reaction afforded only alkylated product (5, after hydrolytic workup). In this case, by halting lactonization (4→6) and the release of the aryloxide to perpetuate the catalytic cycle, an autocatalytic mechanism is implied. These data have led us to propose a mechanism for the reaction, which is initiated by fluoride ion desilylation of the ketene acetal (2), forming chiral ion-paired ketene enolate (3). The ketene enolate then regioselectively alkylates the o-QM at the methide carbon, with restoration of aromaticity acting as the driving force. At this point, two operative pathways are envisioned. Lactonization forms the desired cycloadduct (6) and releases 2-naphthoxide, which then replaces the catalyst counterion and, eventually, desilylates the next molecule of ketene acetal (2). Alternatively, the phenoxide of the alkylated product (4) can desilylate the ketene acetal (2), forming O-silylated, alkylated product (5) and ketene enolate (3). After full consumption of the o-quinone methide, a sodium fluoride wash in the workup ensures complete conversion to the lactone product (6).
Table 1.
Catalytic, Asymmetric [4 + 2] Cycloadditions of o-Quinone Methides with Silyl Ketene Acetalsa
| entry | product | ee % | yield %c | drd |
|---|---|---|---|---|
| 1 |  |
72 | 85 | 7.5:1 |
| 2 | 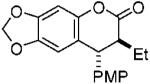 |
80(95)b | 87 | 15.4:1 |
| 3 |  |
79 | 88 | 10.2:1 |
| 4 |  |
81 | 91 | 11.3:1 |
| 5 | 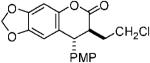 |
90 | 88 | 15.2:1 |
| 6 | 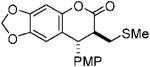 |
85 | 84 | 9.5:1 |
| 7 | 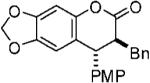 |
80 | 86 | 13.7:1 |
Reaction conditions: N-(3-nitrobenzyl)quinidinium fluoride (0.l equiv), o-QM (1.0 equiv), ketene acetal (1.1 equiv) in THF at -78 °C for 1-2 h.
The % ee after single recrystalization.
Yield of analytically pure product.
Determined by NMR prior to purification.
In conclusion, we have demonstrated an effective entry into catalytic, asymmetric o-quinone methide cycloaddition chemistry. The [4 + 2] cycloadducts were formed in excellent yield and with very good enantioselectivity. This system provides access to products having potentially broad therapeutic application. Efforts to expand the scope of the system by introducing additional o-quinone methide cores are forthcoming.
Supplementary Material
Scheme 2.

Proposed Mechanism of Our Enantioselective, Chiral Ammonium Fluoride Initiated [4 + 2] Cycloaddition Reaction
Acknowledgment
T.L. thanks the NIH (Grant GM064559) for support.
References
- (1) (a).Van De Water RW, Pettus TRR. Tetrahedron. 2002;58:5367–5405. [Google Scholar]; (b) Jurd L. Tetrahedron. 1977;33:163–168. [Google Scholar]; (c) Jurd L, Roitman JN. Tetrahedron. 1978;34:57–62. [Google Scholar]; (d) Jurd L. Aust. J. Chem. 1978;31:347–352. [Google Scholar]
- (2) (a).Peter MG. Angew. Chem., Int. Ed. Engl. 1989;28:555–570. [Google Scholar]; (b) Wang QP, Dechert U, Jirik F, Withers SG. Biochem. Biophys. Res. Commun. 1994;200:577–583. doi: 10.1006/bbrc.1994.1487. [DOI] [PubMed] [Google Scholar]; (c) Buchan G, Turner AB. J. Chem. Soc., Perkin Trans. 1. 1979:1326–1332. [Google Scholar]
- (3).Chiba K, Hirano T, Kitano Y, Tada M. Chem. Commun. 1999:691–692. [Google Scholar]
- (4) (a).Arduini A, Bosi A, Pochini A, Ungaro R. Tetrahedron. 1985;41:3095–3103. [Google Scholar]; (b) Chambers JD, Crawford J, Williams HWR, Dufresne C, Scheigetz J, Bernstein MA, Lau CK. Can. J. Chem. 1992;70:1717–1732. [Google Scholar]
- (5) (a).Flegel M, Lukeman M, Huck L, Wan P. J. Am. Chem. Soc. 2004;126:7890–7897. doi: 10.1021/ja039078g. [DOI] [PubMed] [Google Scholar]; (b) Chen Y, Steinmetz MG. J. Org. Chem. 2006;71:6053–6060. doi: 10.1021/jo060790g. [DOI] [PubMed] [Google Scholar]
- (6).Murray RDH, Mendez J, Brown SA. The Natural Coumarins: Occurrence, Chemistry, and Biochemistry. Wiley; New York: 1982. [Google Scholar]; (b) O’Kennedy R, Thornes RD. Coumarins: Biology, Applications, and Mode of Action. 1st ed. Wiley; Chichester, UK: 1997. [Google Scholar]
- (7) (a).Takechi M, Tanaka Y, Takehara M, Nonaka G, Nishioka I. Phytochemistry. 1985;24:2245–2250. [Google Scholar]; (b) Hsu F-L, Nonaka G-I, Nishioka I. Chem. Pharm. Bull. 1985;33:3142–3152. [Google Scholar]; (c) Iinuma M, Tanaka T, Mizuno M, Katsuzaki T, Ogawa H. Chem. Pharm. Bull. 1989;37:1813–1815. doi: 10.1248/cpb.37.1813. [DOI] [PubMed] [Google Scholar]; (d) Jurd L. J. Heterocycl. Chem. 1996;33:1227–1232. [Google Scholar]; (e) Jurd L. J. Heterocycl. Chem. 1988;25:89–96. [Google Scholar]; (f) Jurd L. J. Heterocycl. Chem. 1997;34:601–604. [Google Scholar]; (g) Tillekeratne LMV, Sherette A, Grossman P, Hupe L, Hupe D, Hudson RA. Bioorg. Med. Chem. Lett. 2001;11:2763–2767. doi: 10.1016/s0960-894x(01)00577-7. [DOI] [PubMed] [Google Scholar]
- (8) (a).Bekele T, Shah M, Wolfer J, Abraham CJ, Weatherwax A, Lectka T. J. Am. Chem. Soc. 2006;128:1810–1811. doi: 10.1021/ja058077g. [DOI] [PubMed] [Google Scholar]; (b) Wolfer J, Bekele T, Abraham CJ, Dogo-Isonagie C, Lectka T. Angew. Chem., Int. Ed. 2006;45:7398–7400. doi: 10.1002/anie.200602801. [DOI] [PubMed] [Google Scholar]; (c) Abraham CJ, Paull DH, Scerba MT, Grebinski JW, Lectka T. J. Am. Chem. Soc. 2006;128:13370–13371. doi: 10.1021/ja065754d. [DOI] [PubMed] [Google Scholar]
- (9).The term “precatalyst” signifies that the fluoride ion is sacrificed to initiate the reaction. Thus, the ammonium ion of the catalyst presumably forms ion pairs with the various anionic species throughout the course of the reaction.
- (10) (a).Ooi T, Maruoka K. Angew. Chem., Int. Ed. 2007;46:4222–4266. doi: 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]; (b) Hashimoto T, Maruoka K. Chem. Rev. 2007;107:5656–5682. doi: 10.1021/cr068368n. [DOI] [PubMed] [Google Scholar]
- (11).Kruger J, Carreira EM. J. Am. Chem. Soc. 1998;120:837–838. [Google Scholar]
- (12).Kobayashi S, Sano T, Mukaiyama T. Chem. Lett. 1989:1319–1322. [Google Scholar]
- (13) (a).Colonna S, Hiemstra H, Wynberg H. J. Chem. Soc., Chem. Commun. 1978:238–239. [Google Scholar]; (b) Ando A, Miura T, Tatematsu T, Shiori T. Tetrahedron Lett. 1993;34:1507–1510. [Google Scholar]; (c) Drew MD, Lawrence NJ. Tetrahedron Lett. 1997;38:5857–5860. [Google Scholar]
- (14).THF proved to be the superior solvent in the reaction, affording products with higher yields and selectivities than toluene, diethyl ether and CH2Cl2.
- (15).N-Benzylquinidinium fluoride and N-(9-anthracenylmethyl)quinidinium fluoride precatalysts afforded products with similar enantioselectivities (50% ee).
- (16).The bis(diphenylphosphoryl)pyrrolidine is synthesized in six steps from trans-4-hydroxy-l-proline. See:Ojima I, Kogure T, Yoda N. J. Org. Chem. 1980;45:4728–4739.
- (17).Absolute stereochemical configuration of the product was determined by X-ray crystallography.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.