

Abstract
β-Cell-type KATP channels are octamers assembled from Kir6.2/KCNJ11 and SUR1/ABCC8. Adenine nucleotides play a major role in their regulation. Nucleotide binding to Kir6.2 inhibits channel activity, whereas ATP binding/hydrolysis on sulfonylurea receptor 1 (SUR1) opposes inhibition. Segments of the Kir6.2 N terminus are important for open-to-closed transitions, form part of the Kir ATP, sulfonylurea, and phosphoinositide binding sites, and interact with L0, an SUR cytoplasmic loop. Inputs from these elements link to the pore via the interfacial helix, which forms an elbow with the outer pore helix. Mutations that destabilize the interfacial helix increase channel activity, reduce sensitivity to inhibitory ATP and channel inhibitors, glibenclamide and repaglinide, and cause neonatal diabetes. We compared Kir6.x/SUR1 channels carrying the V59G substitution, a cause of the developmental delay, epilepsy, and neonatal diabetes syndrome, with a V59A substitution and the equivalent I60G mutation in the related Kir6.1 subunit from vascular smooth muscle. The substituted channels have increased PO values, decreased sensitivity to inhibitors, and impaired stimulation by phosphoinositides but retain sensitivity to Ba2+-block. The V59G and V59A channels are either not, or poorly, stimulated by phosphoinositides, respectively. Inhibition by sequestrating phosphatidylinositol 4,5-bisphosphate with neomycin and polylysine is reduced in V59A, and abolished in V59G channels. Stimulation by SUR1 is intact, and increasing the concentration of inhibitory ATP restores the sensitivity of Val-59-substituted channels to glibenclamide. The I60G channels, strongly dependent on SUR stimulation, remain sensitive to sulfonylureas. The results suggest the interfacial helix dynamically links inhibitory inputs from the Kir N terminus to the gate and that sulfonylureas stabilize an inhibitory configuration.
ATP-sensitive K+ channels (KATP channels)2 consist of four pore-forming subunits Kir6.x and four sulfonylurea receptor (SURx) subunits, which are members of the ATP-binding cassette (ABC) protein superfamily (1). Adenine nucleotides have a balanced action on KATP channels: Mg2+-independent nucleotide binding to Kir6.x closes the channel (2–4), whereas MgADP binding to, or MgATP hydrolysis by, SURx stimulates channel openings (5–9). These results obtained with recombinant systems give an explanation for the older observation that KATP channels are sensitive to the ratio of ADP to ATP (10, 11). There are several subtypes of KATP channels (12) that subserve important functions in many tissues (13). The Kir6.2/SUR1 neuroendocrine channel in pancreatic β-cells couples insulin secretion to the plasma glucose level. KATP channels determine theβ-cell membrane potential and changes in the levels of ATP and ADP induced by changes in glucose metabolism determine channel activity. Antidiabetic sulfonyl-ureas such as glibenclamide (GBC) bind to SUR1 reducing its stimulatory action on the pore thus inducing channel closure that prompts insulin secretion (14, 15).
Mutations in either subunit can alter the balanced action of adenine nucleotides on KATP channels and result in disorders of insulin secretion (16, 17). Loss of channel function is a cause of hyperinsulinemic hypoglycemia (reviewed in Ref. 18), whereas mutations that increase channel activity are one cause of neonatal diabetes (reviewed in Ref. 17). Mutations in SUR1 have been identified that produce more active channels via increased Mg-nucleotide-dependent stimulation of the pore (6, 19, 20; reviewed in Ref. 17). Mutations in Kir6.2 have been identified in which the ability of ATP to close the channel is reduced due to a decreased affinity for inhibitory ATP or to an increased stability of the open state in the absence of ATP (16, 21, 22). In patients carrying one copy of these “gain of function” mutations the balanced action of adenine nucleotides is altered and the resulting increase in channel activity leads to β-cell hyperpolarization and the decrease in insulin secretion that causes (transient or permanent) neonatal diabetes. KATP channel subunits are found in neurons (Kir6.2 with SUR1), in striated muscle (Kir6.2 with SUR2A), and in some smooth muscle (Kir6.1 with SUR2B). Some Kir6.2 mutations result in hyperactive channels that produce more severe syndromic phenotypes that include muscle weakness, developmental delay, epilepsy, and neonatal diabetes, termed the DEND syndrome (16, 21, 23–25).
One DEND mutation, V59G, is in the slide or interfacial helix of Kir6.2 (21), an amphipathic stretch of 13 amino acids (residues 54–66) that lies at the membrane-cytosol interface and is assumed to be important in the mechanics of channel gating (26–28). In intact Xenopus oocytes, homozygous V59G channels are essentially open (29) and in isolated patches, 3 mm MgATP produces only a 10% block; in addition, the sensitivity of heterozygous channels to tolbutamide is strongly reduced (29, 30). In experiments at the single channel level in the absence of ATP, homozygous V59G channels exhibit a high open probability (PO = 0.83 versus 0.53 for the wild type (29)). The reduced sensitivity of V59G channels to inhibition by ATP and tolbutamide has been attributed to the higher open probability, because both compounds stabilize a long lived interburst closed state (31–34). In addition, the V59G substitution reduces the surface expression of the mutant channel to ∼20% of wild type (35). Two other substitutions of Val-59 occur, V59M (21) and V59A,3 which produce an “intermediate” DEND phenotype. The V59M channel has been extensively characterized (22, 29, 30), and the properties of the recently identified V59A channel have not been established.
Homology models imply the interfacial helix could serve to transmit conformational changes from the regulatory subunit to the outer transmembrane (M1) helices of Kir6.1 or 6.2 pores and thus affect gating. The nature of these conformational changes is uncertain, but the first N-terminal residues of Kir6.2 are known to be important for transitions from the open to the long lived closed state. Deletions of 5–35 residues from Kir6.1 and Kir6.2 produce highly active channels, PO > 0.9 (36, 37), with reduced sensitivity to sulfonylureas and ATP (3, 33, 37, 38), similar to those seen in the V59G channels. We suggest that the N terminus of Kir6.x is an inhibitory element that interacts with parts of SUR1, specifically with the L0 linker, to facilitate the transition to the closed state and thus to restrict channel openings (39). The observation that soluble, Kir N-terminal-like peptides can reduce the PO of ΔNKir6.1/SUR1 and ΔNKir6.2/SUR1 channels supports this idea (36). The interfacial helix is the physical connection between the proximal N terminus and the outer M1 helix. We propose that substitutions (e.g. glycine, methionine, and alanine) for valine at position 59 can introduce flexibility into the V59G helix that impairs the inhibitory action of the proximal N terminus.
To support this hypothesis, we sought to characterize the V59G mutation further, compare it with a V59A substitution and the analogous substitution, I60G, in Kir6.1 the pore-forming subunit of the vascular KATP channel. This channel is composed of Kir6.1 and SUR2B (40), however, for comparison we examined the I60G/SUR1 channel. Kir6.1 and 6.2 are 71% homologous overall, and the amino acids in the interfacial helix are identical except Val-59 is replaced by Ile-60 in Kir6.1. The Kir6.1 and Kir6.2 channels differ in their unitary conductance (∼36 versus 66 pS for Kir6.1 versus Kir6.2, respectively (41)). The channels also differ in their gating by nucleotides in isolated patches: Kir6.1/SUR1 channels require strong activating conditions, whereas Kir6.2/SUR1 channels are open in nucleotide-free solutions (42, 43). The Kir6.1-based channels have been reported either to have very low (41, 43) or equivalent (34) sensitivity to inhibitory ATP when compared with Kir6.2-based channels. In view of these differences in gating on one hand and of the similarity in amino acid sequence around the mutation on the other it was of interest to compare the V59G and I60G/SUR1 KATP channels.
EXPERIMENTAL PROCEDURES
Structure Prediction—To examine the effect of the mutations on the secondary structure of the interfacial helix of Kir6.x (Kir6.1: amino acids 55–67, Kir6.2: 54–66), the following programs were used: Predator (version 5a), JPRED, PSIPRED, nnpredict, and PhD (see Ref. 44 for references).
Molecular Biology, Cell Culture, and Transfection—Human Kir6.1, Kir6.2, and SUR1 cDNAs were subcloned into the pcDNA3.1 vector. A Myc epitope was introduced into human Kir6.2 after leucine 100 by ligating complementary oligonucleotides with appropriate overhangs; this modification did not affect the functional properties of the protein (39). The V59G, V59A, and I60G mutations were introduced into Kir6.2 and Kir6.1, respectively, using the QuikChange II XL site-directed mutagenesis kit (Stratagene). The mutations were verified by sequencing the relevant DNA region (V59G) or the whole coding region (I60G and V59A).
HEK 293 cells were cultured in minimum essential medium containing glutamine and supplemented with 10% fetal bovine serum and 20 μg/ml gentamycin as described (45). Cells were transfected with wild-type or mutant Kir6.x and SUR1 at a molar ratio of 1:1 using Lipofectamine 2000 and Opti-MEM (Invitrogen) according to the manufacturer's instructions (46). The pEGFP-C1 vector (Clontech, Palo Alto, CA), encoding green fluorescent protein, was added for identification of transfected cells.
Patch Clamp Experiments—Patch clamp experiments in the whole cell configuration were performed at 37 °C as described by Russ et al. (47). The bath was filled with (in mm): NaCl, 142; KCl, 2.8; MgCl2, 1; CaCl2, 1; d(+)-glucose, 11; HEPES, 10; pH 7.4. Patch pipettes were filled with (in mm) potassium glutamate, 132; NaCl, 10; MgCl2, 2; HEPES, 10; EGTA, 1; Na2ATP, 1; and Na2GDP, 0.3 at pH 7.2 such that [Mg2+]free was ∼0.85 mm and that there was a balance between inhibition and activation. Pipettes had a resistance of 3–5 MΩ. Cells were clamped at –60 mV. To determine the reversal potential of the currents, square pulses, 0.5-s duration, ranging from –110 to +10 mV in 20-mV steps were applied every 12 s. Recordings in which the reversal potential deviated from ∼–90 mV were rejected.
For experiments in the cell-attached and inside-out configuration, pipettes with a larger diameter were used (resistance of 1.0–1.5 MΩ). Experiments were performed at 22 °C. Pipette and bath were filled with a high K+-Ringer solution containing (in mm) KCl, 142; NaCl, 2.8; MgCl2, 1; CaCl2, 1; d(+)-glucose, 11; HEPES, 10; titrated to pH 7.4 with NaOH. In the inside-out mode and after patch excision, the pipette was moved in front of a pipe filled with a high K+ buffer containing (in mm) KCl, 142; MgCl2, 0.7–30.7 (according to the nucleotides added); d(+)-glucose, 11; Na2ATP, 0–30; EGTA, 0.1 (0 when adding Ba2+); HEPES, 10; titrated to pH 7.2 with NaOH at 22 °C and containing the channel modulators of interest. For experiments in Mg2+-free buffer, MgCl2 was omitted and EGTA was replaced by 1 mm EDTA. In a series of experiments using high ATP concentrations (10 and 30 mm), NaCl was added to the bath and pipe solutions with lower ATP concentration to minimize differences in osmolarity. Patches were generally clamped at –50 mV except for examination of the Ba2+-induced block in the inside-out configuration. Ba2+ applied to the inside of the patch produced only a slight block at –50 mV (because it was flushed out of the pore by the inward K+ current at negative voltage), whereas at +50 mV a complete block was achieved (as Ba2+ was dragged into the pore by the outward current). Some inhibitors (e.g. neomycin) were tested at both –50 and +50 mV. In experiments with the V59G/SUR1 channel in the inside-out configuration and at symmetrical high K+ solution, the leak current was determined as the outward current remaining in the presence of 1 mm Ba2+ at +50 mV, and it was assumed that, at –50 mV, the leak current was of the same magnitude (with inverted sign), thus determining the zero current level. Data were filtered at 0.2 kHz and sampled at 1 kHz.
The open probability (PO) of the V59A channel was determined in the inside-out configuration using Sylgard-coated pipettes. Recordings from patches with one channel were filtered at 2.5 kHz and sampled at 5 kHz. Amplitude histograms were generated using PulseTools (Heka, Lambrecht, Germany) and analyzed by fitting a superposition of 2 Gaussian distributions to the data.
[3H]GBC Competition Experiments—Binding experiments were performed in intact cells at 37 °C as described by Hambrock et al. (45) using an incubation buffer containing (in mm): NaCl, 129; KCl, 5; MgCl2, 1.2; CaCl2, 1.25; d(+)-glucose, 11; NaHCO3, 5; HEPES, 10 at pH 7.4. [3H]GBC was 1–2 nm; nonspecific binding was determined in the presence of 100 nm GBC and was ∼10% of total binding.
Data Analysis and Statistics—Channel inhibition and equilibrium binding inhibition curves were analyzed according to the Hill equation,
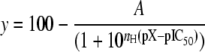 |
(Eq. 1) |
as described before (45) with y denoting the current or total binding, A the maximum inhibition (amplitude or extent of specific binding), nH the Hill coefficient, and x the inhibitor concentration with pX = –log X and pIC50 = –log IC50. In the text, the IC50 values with their 95% confidence interval are given. IC50 values are lognormally distributed (48); therefore the corresponding pIC50 values were compared by using the Student t-test after the data had passed the normality and equal variance tests using the program SigmaStat 3.1 (SPSS Science, Chicago, IL).
Materials—The reagents and media used for cell culture and transfection were from Invitrogen, the other chemicals, including nucleotides, were from Sigma. Glibenclamide, tolbutamide, and diazoxide were purchased from Sigma; repaglinide was a kind gift from Novo Nordisk (Bagsvaerd, Denmark). The KATP channel modulators were dissolved in DMSO/ethanol (50/50, v/v) and further diluted with the same solvent or with incubation buffer (final solvent concentration in the assays, <1%). [3H]GBC (specific activity, 1.85 TBq/mmol) was purchased from Perkin-Elmer Life Sciences. Poly-d-lysine-HBr (mean molecular weight 41,400; chain length ∼200) was from Sigma.
RESULTS
Structure Prediction—Glycine has a lower helix propensity than valine (49), and two, Predator and nnpredict, out of five protein secondary structure programs predicted that the V59G substitution will destabilize and thus break the interfacial helix. The PSIPRED and PhD programs predicted no structural change with the V59G mutation; JPRED did not recognize the helix structure. The same structural predictions were obtained for the I60G substitution in Kir6.1. Alanine and methionine have a greater helix propensity on the Chou-Fasman scale (49) than glycine, are not expected to interrupt the interfacial helix, but may change its flexibility.
Basic Observations on Kir6.2 Channels—Fig. 1 shows basic characteristics of the wild-type and Val-59-substituted channels determined in the whole cell configuration. After breaking into a cell expressing wild-type channels, a current developed during cell dialysis with a nucleotide containing activating solution (Fig. 1a). The current was totally inhibited by glibenclamide (GBC, 0.1 μm), indicating it goes through KATP channels. In contrast, in cells expressing the Val-59-substituted channels, a current was present immediately upon breaking into the cell indicating channels were open prior to dialysis (Fig. 1, b and c). In agreement with their reported reduced sensitivity to sulfonylureas (29) the V59G currents were not affected by GBC (1 μm) (Fig. 1b; n = 12). The V59A channels display an intermediate sensitivity to 1 μm glibenclamide, ∼40% inhibition. Both of the Val-59-subsitituted channels are blocked by a high concentration of Ba2+ (1 mm). Fig. 2 shows there is no significant difference in Ba2+ sensitivity between the wild-type and V59G channels.
FIGURE 1.

Current recordings from cells transfected with Kir6.2/SUR1 (a), Kir6.2V59G/SUR1 (b), and Kir6.2V59A/SUR1 (c). Wild-type channels activated slowly upon cell dialysis with activating solution (MgATP, 1 mm, MgGDP, 0.3 mm, and [Mg2+]free, 0.7 mm), whereas both mutant channels gave a current immediately upon breaking into the cell. Addition of glibenclamide (GBC, 0.1 or 1 μm, black bars) to the bath-closed wild type (n = 9), did not affect V59G channels but inhibited V59A channels by ∼40%; mutant channels were completely blocked by Ba2+ (1 mm)(n = 12). Cells were clamped to –60 mV.
FIGURE 2.

Concentration-dependent inhibition of V59G and wild-type Kir6.2/SUR1 channels by Ba2+. Experiments were performed using the inside-out configuration of the patch clamp technique at +50 mV (see “Experimental Procedures”). Data are means of eight and six patches for the mutant and wild-type channels, respectively, and curve analysis with the Hill coefficient 1.0 gave pIC50 values of 5.38 ± 0.02 and 5.45 ± 0.07, respectively (not different). The inset shows a typical experiment with V59G channels. The zero current level was determined in the presence of Ba2+ (1 mm) and is given by the dotted line. There was a slight channel rundown; Ba2+ was applied via a pipe to the inside of the patch.
Inhibition by ATP, GBC, and Their Combination—As shown in Fig. 1b, 1 μm GBC had no significant effect on the V59G channel when cells were dialyzed with a standard activating solution containing 1 mm ATP and 0.3 mm GDP. Under these conditions MgATP will inhibit channel activity via binding to Kir and stimulate activity via binding and hydrolysis on SUR1 (2, 3, 6). The Kir inhibitory nucleotide-binding site is adenine-selective (34, 50), thus MgGDP primarily stimulates channel activity. Dialysis with 10 mm MgATP alters the nucleotide balance by adding an inhibitory component, and now 1 μm GBC inhibits the whole cell V59G currents by 20 ± 4% (n = 4; data not shown). This suggested that high concentrations of MgATP could “potentiate” channel inhibition by GBC. We used the inside-out membrane configuration to analyze this effect further (Fig. 3). Fig. 3a shows that the combined inhibitory and stimulatory actions of 10 mm MgATP produced a small, ∼7%, inhibition. In the absence of nucleotides, 1 μm GBC was inactive, but the co-application of 10 mm MgATP induced a more marked, ∼24%, inhibition. Under these conditions SUR1, whose affinity for GBC is >109 m–1, is saturated with the sulfonylurea, which strongly inhibits its Mg-nucleotide-dependent stimulatory action on Kir6.2. Thus the apparent “potentiation” of GBC by 10 mm MgATP reflects the combined inhibitory actions of ATP and GBC after disruption of nucleotide stimulation by the sulfonylurea. Fig. 3b provides support for this interpretation. MgATP (30 mm) has balanced inhibitory versus stimulatory effects on channel activity via binding to Kir6.2 and SUR1, respectively. Limiting the stimulatory action of SUR1 with 0.01 μm GBC unmasks the inhibitory effect of 30 mm MgATP producing ∼70% inhibition. The application of additional GBC (0.1–1 μm) did not result in further inhibition (n = 3–5). MgATP inhibition curves for the Val-59-substituted channels in the presence and absence of GBC (1 μm) are presented in Fig. 4 (upper panels); the resulting IC50 values are listed in Table 1. MgATP alone weakly inhibited V59G channels, but blocking the stimulatory action of SUR1 with 1 μm GBC shifted the inhibition curve ∼10× toward the left (Table 1). The V59A channels were ∼90 times more sensitive to inhibition by MgATP than the V59G channels. Similarly, blocking SUR1 stimulation with GBC potentiated inhibition of V59A channels by MgATP ∼6-fold (Fig. 4, upper panel, and Table 1).
FIGURE 3.

Recordings from inside-out patches showing inhibition of V59G channels by MgATP and GBC alone and in combination. The zero current level is represented by the dotted line. a, MgATP (10 mm) inhibited the current in the absence and presence of GBC (1 μm) and in the presence of [Mg2+]free = 0.7 mm by 7 and 24%, respectively. GBC (1 μm) alone did not affect the current. b, MgATP (30 mm) alone inhibited the current by 14%; in the presence of GBC (0.01–1 μm) inhibition was 70%. c, enhancement of the inhibition by ATP (3 mm, no Mg2+) by GBC.
FIGURE 4.

Concentration-dependent inhibition of the V59G and V59A channels by ATP: effect of GBC (1 μm) and Mg2+ ([Mg2+]free = 0.7 mm). Data were obtained as shown in Fig. 3 and are means ± S.E. of 3–10 patches. Inhibition curves were generated by fitting Equation 1 to the data; for V59G, curves were assumed to extrapolate to 100% inhibition with the Hill coefficient set to 1. Parameters are listed in Table 1.
TABLE 1.
Potency of ATP inhibition of V59G and V59A currents
Effects of Mg2+ and GBC (1 μm). IC50 values (followed by the 95% confidence interval) were determined as described in Fig. 4. Statistical tests were performed with the respective pKi values (which are normally distributed (49)).
|
Conditions
|
V59G
|
V59A
|
||
|---|---|---|---|---|
| Mg2+ (0.7 mm) | GBC (1 μm) | IC50 | IC50 | nHa |
| mm | mm | |||
| + | - | ∼200?b | 2.3 (0.9, 5.9) | 0.47 ± 0.13 |
| + | + | 20 (10, 38) | 0.36 (0.28, 0.48) | 0.85 ± 0.10 |
| - | - | 21 (15, 31)c | 0.32 (0.21, 0.49)c | 1.3 ± 0.2 |
| - | + | 3.1 (1.9, 5.1)d | 0.11 (0.077, 0.15)d | 1.3 ± 0.2 |
nH is the Hill coefficient; for V59G nH was set to 1.0.
Value uncertain (cf. Fig. 4).
Different from values in the presence of either Mg2+ or GBC (but not both), p < 0.05.
Different from the corresponding value in the presence of Mg2+, p < 0.05.
In the absence of Mg2+, ATP inhibits channel activity by binding to Kir6.2, but the Mg2+-nucleotide dependent stimulatory action of SUR1 is absent. Fig. 3c shows that ATP4– (3 mm) inhibited KATP currents by 25%, and the addition of GBC (0.01 μm) increased this inhibition to ∼60%. Higher concentrations of GBC (up to 1 μm) had no further effect. Thus in the absence of the Mg2+-dependent stimulatory action of SUR1, ATP, and GBC both stabilize the interburst closed state. GBC (1 μm) alone did not affect the V59G channel, inhibited the V59A channels ∼15% and the wild-type channels ∼70% (Table 2), the maximum inhibition attainable in inside-out patches. The ATP inhibition curves in the absence of Mg2+ are shown in Fig. 4 (lower panels). ATP inhibited V59G channels with an IC50 of ∼21 mm; GBC (1 μm) shifted the ATP inhibition curve to ∼7× lower concentrations (Table 1). The V59A channels were ∼65-times more sensitive to inhibition by ATP4– than the V59G channels; blocking SUR1 stimulation with GBC potentiated the inhibitory action of ATP4– ∼3-fold (Fig. 4, lower panel, and Table 1).
TABLE 2.
Properties of Kir6.2 wild type, V59G, V59A, and ΔN10 channels formed with SUR1
Experiments in inside/outside patches –Mg2+; number of experiments are in parentheses.
|
Kir6.2
|
POa
|
GBC (1 μm)
|
PIP2 (10 μm)
IPIP2/Icontrol
|
Neomycin
|
||
|---|---|---|---|---|---|---|
| 30 μm | 1000 μm | |||||
| % Inhibition | % | % Inhibition | ||||
| Wild type | 0.32 ± 0.10 (5)b | 71 ± 10 (4) | 900 ± 390 (12) | 57 ± 5 (9) | 96 ± 2 (7) | |
| 0.53 ± 0.02 (8)c | ||||||
| V59G | 0.83 ± 0.01 (8)c | 0 (10) | 0 (4) | 3 ± 2 (5) | 16 ± 2 (5) | |
| V59A | 0.86 ± 0.01 (4) | 15 ± 8 (7) | 5 ± 3 (5) | 17 ± 6 (6) | 50 ± 11 (6) | |
| ΔN10 | 0.61 ± 0.08 (4) (ΔN9)b | 9 ± 5 (12) | 270 ± 40 (4) | 17 ± 4 (10) | 60 ± 8 (7) | |
To examine further the effect of an increased open probability (PO) on the sensitivity of the channel toward GBC, we used an engineered subunit lacking N-terminal amino acids 2–10, Kir6.2ΔN10. Deletion of the N terminus impairs the transition to the interburst closed state thus increasing the open probability, PO (33, 37, 38). Table 2 shows that this mutant has an impaired sensitivity to GBC (1 μm), which resembles that observed in the Val-59-substituted channels.
Lipids and Polycations—The effect of channel-activating lipids and inhibitory polycations on Kir6.2/SUR1 channels was studied in the inside-out configuration; sample traces for the wild-type versus V59G channels are shown in Fig. 5; the results are summarized in Table 2. Application of PIP2 (10 μm) or oleoyl-CoA (1 μm) increased the current through wild-type channels by 900 ± 390% (n = 13) and 300 ± 90% (n = 11, data not shown), respectively. The effect of the lipids was greater when applied after considerable rundown had occurred. Activation by PIP2 was poorly reversible upon washout, whereas the effect of oleoyl-CoA was rapidly reversed. In contrast, PIP2 and oleoyl-CoA were unable to further increase the POmax of V59G channels (n = 4–7) and had only small stimulatory effects on V59A channels (Table 2). To determine the contribution of PIP2 to the elevated POmax of the Val-59-substituted channels, we assessed the effects of agents that sequester phosphoinositides. The polycations, neomycin, gentamycin, and polylysine, inhibited wild-type channels with IC50 values of ∼16 and 82 μm and <1 μg/ml, respectively. Inhibition of wild-type channels by neomycin and gentamycin was reversible upon washout; inhibition by polylysine was not. In contrast, the Val-59-substituted channels were only weakly affected by these polycations. Inhibition of the V59G channels by neomycin and gentamycin (1 mm) was <20% (n = 5) and by polylysine was 27 ± 3, 29 ± 3, and 34 ± 6% at 1, 10, and 100 μg/ml, indicating that a full block was not achievable (Fig. 5). Inhibition at +50 mV was only slightly stronger. The inhibitory actions of neomycin on the V59A and ΔN10Kir6.2 channels were comparably attenuated (Table 2).
FIGURE 5.

Traces showing the effects of lipids and polycations on wild-type and V59G KATP channels at –50 mV. The zero current level is indicated by the dotted line. a, PIP2 (10μm); b, neomycin (30 and 1000μm); and c, polylysine (100 μg/ml).
V59G Channels Have a Reduced Efficacy, but Not Affinity for the KATP Channel Antagonist, Repaglinide—Previous work showed that coexpression of wild-type Kir6.2 increased the affinity of SUR1 for glibenclamide ∼3-fold and for repaglinide ∼130- to 200-fold (51, 52). It was therefore of interest to determine whether the V59G channels had an altered affinity for repaglinide. Binding experiments on intact cells gave a Ki value of 0.70 (0.55 and 0.87) nm (n = 3) for the V59G channels comparable to that determined for wild-type channels (0.72 (0.60 and 0.87) nm (51)). Functionally, repaglinide had an effect similar to GBC. In experiments similar to those in Fig. 3a repaglinide (1 μm) did not inhibit the mutant channel by itself but increased the inhibition by MgATP (10 mm) from 5 ± 2 to 50 ± 7% (n = 4, data not shown).
Response of Kir6.1/SUR1 Channels to Nucleotides and Sulfonylureas—Fig. 6 compares the responses of cells expressing wild-type versus I60G/SUR1 channels to stimulation with 1 mm MgATP/0.3 mm MgGDP. Similar to Kir6.2/SUR1 channels, wild-type Kir6.1/SUR1 channels activated upon dialysis, whereas the Kir6.1I60G/SUR1 channels were open immediately upon seal formation and showed little rundown (n = 15) thus resembling the V59G channels. An immediate, stable current was also obtained when cells expressing the I60G channels were dialyzed with 10 mm MgATP indicating that 10 mm MgATP did not close the mutant channel (n = 5). Dialysis with Mg2+- or nucleotide-free solutions led to a steady loss of current over 2–10 min indicating that the I60G channel required activation to remain in the open state (n = 12). GBC (10 nm) inhibited both the wild-type and I60G channels ≥90% (Fig. 6). This inhibition of the I60G channels is in sharp contrast to the V59G Kir6.2 channels (Fig. 1b), which were insensitive to GBC up to 1 μm under equivalent activating conditions.
FIGURE 6.

Inhibition of wild-type and Kir6.1I60G/SUR1 channels by GBC. a, wild-type channels activated slowly upon cell dialysis and closed in response to GBC (10 nm). b, mutant channels were open prior to cell dialysis and closed in response to GBC (10 nm). Cells were clamped to –60 mV.
The inhibition of SUR1 containing channels by glibenclamide is essentially irreversible on the time scale of electrophysiological experiments. Tolbutamide, which inhibits the pancreatic KATP channel with ∼1000-fold lower potency and is rapidly reversible, was also tested. In the whole cell configuration, wild-type Kir6.1/SUR1 channels were completely and reversibly inhibited by tolbutamide (100 μm), whereas the I60G channels were 89 ± 6 and 91 ± 4% inhibited by 100 and 300 μm tolbutamide (n = 4 and 8), respectively. Assuming complete inhibition, an IC50 value of ∼10 μm was estimated for the I60G channel.
The experiments in the whole cell configuration (Fig. 6) suggest that, in the cellular environment, wild-type Kir6.1 channels are essentially closed, whereas the I60G channels are open. These observations were confirmed using the cell-attached configuration of the patch clamp technique. Fig. 7a shows activity was low when wild-type Kir6.1 channels are exposed to cellular nucleotide levels. Application of 100 μm diazoxide stimulates the activity of channels with bursting behavior characteristic of KATP channels and 1.8 pA currents expected for a single channel conductance of 36 pS in symmetrical high K+ solution (41) at a patch potential of –50 mV (n = 13). The diazoxide-induced activity of wild-type Kir6.1 channels was blocked reversibly by tolbutamide (300 μm). As shown in Fig. 7b, the I60G channels were spontaneously active in on-cell patches and exhibited transitions between long lived states that differed by ∼1.8 pA. Tolbutamide (300 μm) reversibly inhibited the I60G channels (Fig. 7b), whereas diazoxide had no effect (n = 9). Diazoxide and tolbutamide had no effect on non-transfected cells (n = 7).
FIGURE 7.

Recordings of the wild-type and mutant Kir6.1I60G/SUR1 channels in the cell-attached mode. Pipette and bath were filled with a high K+ buffer at room temperature, and cells were clamped to –50 mV. a, wild-type channel: effects of diazoxide and tolbutamide. Prior to drug application, there was almost no channel activity. Diazoxide (100 μm) greatly enhanced channel activity. Tolbutamide (300 μm) blocked activation by diazoxide. The effects of both drugs were easily washed out. The inset shows the opening and closing of a single 1.8-pA channel opened by diazoxide. b, Kir6.1I60G/SUR1. Channels were spontaneously open; tolbutamide (300 μm) induced the sequential closure of channels (1.8 pA, long open times); one channel opened again. After washout, channels reappeared. The inset shows closing and opening of a 1.8-pA channel in the presence of tolbutamide.
DISCUSSION
Structural Hypothesis—We compared Val-59-substituted Kir6.2/SUR1 and analogous I60G Kir6.1/SUR1 channels with their respective wild-type channels. The mutations are located in the middle of a 13-amino acid helix, termed the interfacial or slide helix, that lies at the membrane-cytosol interface and forms an elbow with the outer transmembrane helix, M1, via a short loop (26, 27). In Kir6.2, the interfacial helix is flanked by basic residues, Arg-54 and Lys-67, proposed to be part of the PIP2 binding site (53, 54) and is C-terminal of Arg-50 and Arg-54 proposed to be part of the inhibitory adenine nucleotide binding site (39, 53, 55). The interfacial helix is adjacent to a peptide segment, residues 37–44, which specifies major gating differences between Kir6.1 and Kir6.2 (56) and has two residues, Lys-39 and Asn-41, which also participate in PIP2 binding (53, 54). Additionally, the interfacial helix is proposed to interact with a submembrane helix in the L0 loop of SUR1 (Ref. 39; reviewed in Ref. 57). The interfacial helix is thus positioned to transduce conformational changes resulting from interactions with inhibitory nucleotides, phosphoinositides, and the SUR regulatory subunits directly to the pore and thus to affect slow channel gating. Efficient transduction presumably depends on the integrity of the interfacial helix. Structure prediction programs suggest that substitution of a glycine for valine at position 59 will introduce a break, a short extended sheet, in the interfacial helix that would introduce flexibility and impair signal transmission. Substitution of methionine or alanine for valine is not predicted to have as drastic an effect on helix structure and does perturb signal transduction to a lesser, albeit physiologically significant degree because both mutations produce a severe phenotype.
There is strong evidence for the view that the proximal N termini of Kir6.2 (and Kir6.1) are “inhibitory” segments critical for the transition of these channels to the long lived interburst closed state. This idea is based on the observations that deletion of the Kir N terminus increases the channel PO by impairing the transition to the closed state (3, 33, 38) and that a synthetic Kir6.2 N-terminal-like peptide can reduce the PO of Kir6.2ΔN32/SUR1 channels (36). In addition, application of the synthetic Kir6.2 N-terminal-like peptide to intact SUR1/Kir6.2 channels increases their PO, presumably by displacing the endogenous N terminus (36). We hypothesize that an increased flexibility of the interfacial helix would impair transmission of the inhibitory N-terminal signal and thus impede transitions to the interburst closed state. This will reduce the potency of inhibitory ATP (and GBC) and thus produce more active channels that, interestingly, have a reduced requirement for PIP2 to maintain their activity.
Sensitivity to ATP—Experiments in the inside-out configuration showed that in the absence of Mg2+, the V59G channel had an ∼4000-fold lower sensitivity toward inhibition by ATP4– than the wild-type (21 mm versus 5 μm). For comparison, deletion of up to 44 N-terminal amino acids from Kir6.2 led to a 10- to 20-fold reduction in the sensitivity to inhibitory ATP (3, 33, 38). At least two explanations could rationalize this difference. The V59G mutation could reduce the affinity for inhibitory ATP more strongly than deletion of the N terminus in the ΔNKir channels. Alternatively, signal transduction may be more seriously impaired by the V59G mutation. ATP is suggested to stabilize the interburst closed state, and thus the reduced sensitivity to inhibitory ATP has been hypothesized to reflect the reduced time the V59G and ΔNKir channels spend in the closed state. Both of the Val-59 substitutions and N-terminal truncation produce channels with PO values > 0.8, but the Val-59-substituted channels are far less sensitive to inhibition by ATP. We suggest that both types of “mutation” affect the same inhibitory linkage, but in somewhat different ways. The results are consistent with a “sequential transduction model” in which the interfacial helix acts as the final common element that transduces multiple signals to the pore, including inputs from the proximal N-terminal segment, L0, and the ATP and PIP2 binding sites. The increase in PO attributable to N-terminal truncation reflects removal of one inhibitory input, whereas destabilization of the interfacial helix by the Val-59 substitutions weakens or abolishes multiple inputs, including that from the N terminus. Destabilization is greatest for substitution with glycine, which can adopt the widest range of psi-phi angles. We presume that both the Val-59-substituted and ΔNKir subunits retain ATP binding, perhaps with reduced affinity. We suggest that the inhibitory effect of ATP on a ΔNKir channel is greater, only a 10- to 20-fold reduction, because the interfacial helices are intact and able to transduce the effect of ATP binding, whereas the link is destabilized in the V59A and V59M channels and largely absent in V59G channels.
In the presence of Mg2+, inhibition of the Val-59-substituted channels by MgATP was ∼7–10 times weaker than for ATP4–, consistent with effective Mg-nucleotide-dependent stimulation via SUR1. Assuming that MgATP and ATP interact with the Val-59-substituted channels with equal affinity, the results indicate the “pathway” for the stimulatory action of MgATP via SUR1 is intact in the Val-59-substituted channels. Babenko (6) has reported a comparable (∼10-fold) MgATP-dependent net stimulation of wild-type channels. This implies the increased mutant channel activity is primarily due to reduced nucleotide inhibition (see however, Ref. 30, which suggests the stimulatory action of MgATP via SUR1 is enhanced in Val-59-substituted channels).
Information on the Kir6.1I60G/SUR1 channels is more limited and their reduced expression impeded experiments in the inside-out configuration. Whole cell and cell-attached recordings show that the I60G channels are more active than wild-type in intact cells (Fig. 7). This suggests that under physiologic conditions the I60G channels either have a reduced sensitivity to inhibitory ATP versus wild-type or are more efficiently activated by MgATP. We did not observe I60G channel activity in the absence of Mg2+ and activating nucleotides consistent with earlier studies on the Kir6.1/SUR1 wild-type (34, 40, 42, 43), whereas both the Val-59-substituted and wild-type Kir6.2 channels are open in the absence of activating nucleotides (e.g. Figs. 3 and 5). Kondo et al. (56) have shown that amino acids 37–44 are the major determinant of the requirement for nucleotide stimulation in Kir6.1 channels, while a C-terminal sequence, 243–248, has a lesser role. The mechanism by which these two short segments in Kir6.1 prevent opening of the channel in the absence of SUR activation is unknown. We note that deletion of the Kir6.1 N terminus, ΔN33Kir6.1, which would remove the putative inhibitory segment produces ΔN33Kir6.1/SUR1 channels that are spontaneously active in the absence of activating nucleotides and whose PO is reduced by the Kir6.2 synthetic peptide (36). One interpretation of these results is that the putative N-terminal inhibitory segment of Kir6.1 is more firmly tethered to its binding site than its Kir6.2 counterpart and thus exhibits an increased dependence on SUR activation for activity. In support of this idea, we note that, although the application of the Kir6.2 synthetic peptide to intact Kir6.1/SUR1 channels does increase their activity, the effect is smaller than that observed with intact Kir6.2/SUR1 channels (36).
Sensitivity to GBC—GBC (1 μm) was without effect on V59G channel activity under standard activating conditions (1 mm MgATP, 0.3 mm MgGDP) and had a markedly reduced inhibitory action on the V59A channels. Interestingly, although the GBC binding site is composed of parts of SUR1 and the N terminus of Kir6.2 (58, 59), and deletion of the Kir6.2 N terminus reduces sensitivity to sulfonylureas (33, 37, 38), the reduced inhibition of Val-59-substituted channels is not the result of altered GBC or repaglinide binding to SUR1. With a KD value of 0.5 nm (51), at 1 μm GBC, binding to both wild-type and mutant channels was 99.95% saturated. The reduced effect of GBC on the Val-59-substituted channels implies that the mechanisms by which sulfonylurea binding to SUR1 closes the channel are markedly less effective when the inhibitory transduction pathway is disrupted (Fig. 1). GBC limits the activating effect of Mg-nucleotides; however the hyper-activation observed in the Val-59-substituted channels also requires that the normal counterbalancing nucleotide inhibition is reduced. GBC stabilizes long lived closed states, but these are reduced dramatically, because the inhibitory linkage is disrupted and nucleotide inhibition is impaired in the Val-59-substituted and ΔNKir6.2 channels. Two observations support this interpretation. First, partially restoring the balance between activation and inhibition by significantly increasing the concentration of ATP confirms that the activation pathway is intact and is inhibited by GBC (Fig. 3 and see “Discussion” below). Second, the I60G channels, whose activity is strongly dependent on Mg-nucleotide-dependent stimulation by SUR1, are nearly as sensitive to GBC as the wild-type (potency difference ∼ 4×). GBC limits the stimulatory input from SUR1 thereby inducing closure of the I60G channels.
Effect of ATP and GBC in Combination—We observed synergistic effects when ATP and GBC were co-applied to Val-59-substituted channels. We suggest these effects reflect the altered balance of inhibitory versus stimulatory actions of nucleotides on KATP channels and support a role for the Kir N termini in GBC inhibition. In the inside-out configuration of the patch clamp technique, concentrations of sulfonylureas that saturate SUR1 are reported to reduce the activity of wild-type channels ∼50% when tested in the absence of nucleotide activation (–Mg2+) and to show an increased efficacy under activating conditions (60, 61). Deletion of 10–20 N-terminal residues from Kir6.2 reduces the potency of ATP4– ∼20× (IC50 increase from ∼5 to 100 μm) and eliminates sensitivity to a saturating concentration (200 μm) of tolbutamide without affecting the Mg-nucleotide-dependent stimulatory pathway (33, 37, 38). In the absence of stimulation (–Mg2+), the V59G mutation increases the IC50 for ATP from ∼5 μm to ∼20 mm and 1 μm GBC, ineffective when applied alone, shifts the ATP4– inhibition curve to ∼7× lower concentrations (Fig. 4 and Table 1). The V59A results were broadly similar. These observations emphasize the interplay between the proposed inhibitory segments in the Kir N terminus, the interfacial helix, and GBC binding to SUR1. The observations suggest a speculative hypothesis: We assume that the interactions of the Kir N terminus with SUR1 are dynamic, as suggested by experiments with a synthetic N-terminal peptide (36), and we hypothesize that GBC binding to SUR1 stabilizes a non-stimulatory configuration. In this case, deletion of the N terminus, in the ΔNKir6.2 channels, eliminates the inhibitory segments, abolishes GBC inhibition and, as discussed above, impairs inhibition by nucleotides.
Similarly, although the Kir N terminus is intact in the Val-59-substituted channels, destabilizing the interfacial helix impairs dynamic signaling to the gate. The stabilization of an inhibitory conformation of the Kir N terminus by ATP and/or GBC can partially overcome the disruption of the interfacial helix. When Mg2+ is present the strong stimulatory action of SUR1 is apparent; the IC50(MgATP) of the V59A channels is increased, ∼5 μm to 2.3 mm, whereas the IC50 for the V59G channels can only be estimated at ∼200 mm. The combined effect of ATP inhibition and GBC reducing the stimulatory action of SUR1 and stabilizing an inhibitory conformation of the Kir N terminus shifts the inhibition curve for the V59G channels to ∼10-fold, and the V59A to ∼6-fold, lower concentrations, similar to the effect of ATP4– in the absence of Mg2+ and GBC (∼20 mm for V59G and 0.32 mm for V59A, respectively, Table 1). These quantitative effects are clinically relevant; patients with the V59G mutation are not responsive to sulfonylureas, whereas the V59A patient has responded to GBC.4
The inhibitory potency of GBC was explored using a synergism protocol with the V59G channels (Fig. 3). At high ATP concentrations, in the presence and absence of activation (±Mg2+), the maximum level of inhibition was reached at <10 nm GBC, consistent with an affinity of GBC ≤1 nm. The result is in agreement with the lack of effect of the V59G mutation on GBC binding.
PIP2 and Polycations—The V59G channels are totally insensitive to activation by PIP2 (Fig. 5) and oleoyl-CoA. This may simply reflect the fact that they are maximally active. However, the polycations, gentamycin, neomycin, and polylysine, produced only a partial block at high concentrations. The V59A channels showed only a small activation by PIP2 and a markedly reduced sensitivity to neomycin. Similarly, the ΔNKir6.2 channels were activated by PIP2, albeit to a lesser degree than wild-type, and the inhibitory effect of phosphoinositide sequestration by neomycin was reduced (Table 2) consistent with a reduced requirement for PIP2. We cannot eliminate the possibility that alteration of the N termini actually greatly increases the affinity of the Kirs for PIP2, but favor the idea that the affinities are reduced and that the reduction of Val-59-substituted channel activity by polycations does not involve sequestration of PIP2, but rather that they partially block the channel pore at the high concentrations used.
Ba2+—The Val-59-substituted channels were completely blocked by Ba2+ applied from the outside. Experiments in the inside-out mode showed there was no difference in the blockade of V59G versus wild-type channels. The site of Ba2+ block is located at the inner side of the selectivity filter (62); the data imply the V59G substitution in the interfacial helix does not affect the Ba2+ site.
In conclusion, we have compared Kir6.x/SUR1 channels with Kir substitutions predicted to increase the flexibility of, or disrupt, the interfacial helix and thus impair signal transduction within the channel. The substituted channels have strongly increased PO values in the cellular environment. Disruption of the interfacial helix in the V59G channel effectively uncouples the pore from nucleotide inhibition and phosphoinositide activation, actions that require direct binding to the pore, without affecting the binding of GBC or repaglinide to SUR1. The V59A results are consistent with this substitution partially destabilizing the interfacial helix. Destabilization of the interfacial helix does not significantly affect the Mg-nucleotide-dependent stimulatory action of SUR1, and understanding the synergistic actions of ATP and sulfonylureas on the mutant channels underscores the idea of a balance between nucleotide inhibition and stimulation.
The altered inhibitory effects of ATP4– and sulfonylureas on the channel, in the absence of Mg2+, are interpretable in terms of a simple hypothesis in which the interfacial helix is part of a dynamic link between inhibitory elements in the Kir N terminus and the gate and GBC binding stabilizes a non-stimulatory configuration of SUR1. The Val-59 substitutions in the interfacial helix leave the stimulatory transduction pathway from SUR1, and the differential responses of the Kir6.x subtypes to these signals, intact, thus explaining the difference in the sensitivity of the Kir6.1- and Kir6.2-based channels to sulfonylureas.
Acknowledgments
We gratefully acknowledge Drs. Louis Philipson and Graeme Bell for sharing their finding that the V59A substitution can produce the intermediate DEND syndrome.
This work was supported, in whole or in part, by National Institutes of Health Grant DK044311 (to J. B.). This work was also supported by the Deutsche Forschungsgemeinschaft Grant Qu100/4-1 (to U. Q.) and a grant from the Dr. Karl Kuhn-Stiftung (to U. R.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: KATP channel, ATP-sensitive K+ channel; DEND syndrome, developmental delay, epilepsy, and neonatal diabetes syndrome; GBC, glibenclamide; Kir, inwardly rectifying potassium channel; PIP2, phosphatidylinositol 4,5-bisphosphate; PO, open probability of single channels; SUR, sulfonylurea receptor.
L. Philipson and G. Bell, personal communication.
L. Philipson, personal communication.
References
- 1.Clement IV, J. P., Kunjilwar, K., Gonzalez, G., Schwanstecher, M., Panten, U., Aguilar-Bryan, L., and Bryan, J. (1997) Neuron 18 827–838 [DOI] [PubMed] [Google Scholar]
- 2.Tucker, S. J., Gribble, F. M., Zhao, C., Trapp, S., and Ashcroft, F. M. (1997) Nature 387 179–183 [DOI] [PubMed] [Google Scholar]
- 3.Babenko, A. P., Gonzalez, G., Aguilar-Bryan, L., and Bryan, J. (1999) FEBS Lett. 445 131–136 [DOI] [PubMed] [Google Scholar]
- 4.Drain, P., Li, L., and Wang, J. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 13953–13958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nichols, C. G., Shyng, S. L., Nestorowicz, A., Glaser, B., Clement IV, J. P., Gonzalez, G., Aguilar-Bryan, L., Permutt, M. A., and Bryan, J. (1996) Science 272 1785–1787 [DOI] [PubMed] [Google Scholar]
- 6.Babenko, A. P. (2008) J. Biol. Chem. 283 8778–8782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsuo, M., Kimura, Y., and Ueda, K. (2005) J. Mol. Cell. Cardiol. 38 907–916 [DOI] [PubMed] [Google Scholar]
- 8.Zingman, L. V., Alekseev, A. E., Bienengraeber, M., Hodgson, D., Karger, A. B., Dzeja, P. P., and Terzic, A. (2001) Neuron 31 233–245 [DOI] [PubMed] [Google Scholar]
- 9.Bienengraeber, M., Olson, T. M., Selivanov, V. A., Kathmann, E. C., O'Cochlain, F., Gao, F., Karger, A. B., Ballew, J. D., Hodgson, D. M., Zingman, L. V., Pang, Y. P., Alekseev, A. E., and Terzic, A. (2004) Nat. Genet. 36 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dunne, M. J., and Petersen, O. H. (1986) FEBS Lett. 208 59–62 [DOI] [PubMed] [Google Scholar]
- 11.Misler, S., Falke, L. C., Gillis, K., and McDaniel, M. L. (1986) Proc. Natl. Acad. Sci. U. S. A. 83 7119–7123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Babenko, A. P., Aguilar-Bryan, L., and Bryan, J. (1998) Annu. Rev. Physiol. 60 667–687 [DOI] [PubMed] [Google Scholar]
- 13.Seino, S., and Miki, T. (2003) Prog. Biophys. Mol. Biol. 81 133–176 [DOI] [PubMed] [Google Scholar]
- 14.Aguilar-Bryan, L., Nichols, C. G., Wechsler, S. W., Clement IV, J. P., Boyd III, A. E., Gonzalez, G., Herrera-Sosa, H., Nguy, K., Bryan, J., and Nelson, D. A. (1995) Science 268 423–426 [DOI] [PubMed] [Google Scholar]
- 15.Proks, P., Reimann, F., Green, N., Gribble, F., and Ashcroft, F. M. (2002) Diabetes 51 S368–S376 [DOI] [PubMed] [Google Scholar]
- 16.Ashcroft, F. M. (2005) J. Clin. Investig. 115 2047–2058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aguilar-Bryan, L., and Bryan, J. (2008) Endocr. Rev. 29 265–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aguilar-Bryan, L., and Bryan, J. (1999) Endocr. Rev. 20 101–135 [DOI] [PubMed] [Google Scholar]
- 19.Babenko, A. P., Polak, M., Cave, H., Busiah, K., Czernichow, P., Scharfmann, R., Bryan, J., Aguilar-Bryan, L., Vaxillaire, M., and Froguel, P. (2006) N. Engl. J. Med. 355 456–466 [DOI] [PubMed] [Google Scholar]
- 20.Flanagan, S. E., Patch, A. M., Mackay, D. J., Edghill, E. L., Gloyn, A. L., Robinson, D., Shield, J. P., Temple, K., Ellard, S., and Hattersley, A. T. (2007) Diabetes 56 1930–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gloyn, A. L., Pearson, E. R., Antcliff, J. F., Proks, P., Bruining, G. J., Slingerland, A. S., Howard, N., Srinivasan, S., Silva, J. M., Molnes, J., Edghill, E. L., Frayling, T. M., Temple, I. K., Mackay, D., Shield, J. P., Sumnik, Z., van Rhijn, A., Wales, J. K., Clark, P., Gorman, S., Aisenberg, J., Ellard, S., Njolstad, P. R., Ashcroft, F. M., and Hattersley, A. T. (2004) N. Engl. J. Med. 350 1838–1849 [DOI] [PubMed] [Google Scholar]
- 22.Koster, J. C., Remedi, M. S., Dao, C., and Nichols, C. G. (2005) Diabetes 54 2645–2654 [DOI] [PubMed] [Google Scholar]
- 23.Hattersley, A. T., and Ashcroft, F. M. (2005) Diabetes 54 2503–2513 [DOI] [PubMed] [Google Scholar]
- 24.Koster, J. C., Permutt, M. A., and Nichols, C. G. (2005) Diabetes 54 3065–3072 [DOI] [PubMed] [Google Scholar]
- 25.Flanagan, S. E., Edghill, E. L., Gloyn, A. L., Ellard, S., and Hattersley, A. T. (2006) Diabetologia 49 1190–1197 [DOI] [PubMed] [Google Scholar]
- 26.Kuo, A., Gulbis, J. M., Antcliff, J. F., Rahman, T., Lowe, E. D., Zimmer, J., Cuthbertson, J., Ashcroft, F. M., Ezaki, T., and Doyle, D. A. (2003) Science 300 1922–1926 [DOI] [PubMed] [Google Scholar]
- 27.Nishida, M., Cadene, M., Chait, B. T., and MacKinnon, R. (2007) EMBO J. 26 4005–4015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Antcliff, J. F., Haider, S., Proks, P., Sansom, M. S., and Ashcroft, F. M. (2005) EMBO J. 24 229–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Proks, P., Antcliff, J. F., Lippiat, J., Gloyn, A. L., Hattersley, A. T., and Ashcroft, F. M. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 17539–17544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Proks, P., Girard, C., and Ashcroft, F. M. (2005) Hum. Mol. Genet. 14 2717–2726 [DOI] [PubMed] [Google Scholar]
- 31.Trapp, S., Proks, P., Tucker, S. J., and Ashcroft, F. M. (1998) J. Gen. Physiol. 112 333–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Enkvetchakul, D., Loussouarn, G., Makhina, E., Shyng, S. L., and Nichols, C. G. (2000) Biophys. J. 78 2334–2348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koster, J. C., Sha, Q., Shyng, S., and Nichols, C. G. (1999) J. Physiol. 515 19–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Babenko, A. P., and Bryan, J. (2001) J. Biol. Chem. 276 49083–49092 [DOI] [PubMed] [Google Scholar]
- 35.Lin, C. W., Lin, Y. W., Yan, F. F., Casey, J., Kochhar, M., Pratt, E. B., and Shyng, S. L. (2006) Diabetes 55 1738–1746 [DOI] [PubMed] [Google Scholar]
- 36.Babenko, A. P., and Bryan, J. (2002) J. Biol. Chem. 277 43997–44004 [DOI] [PubMed] [Google Scholar]
- 37.Babenko, A. P., Gonzalez, G., and Bryan, J. (1999) Biochem. Biophys. Res. Commun. 255 231–238 [DOI] [PubMed] [Google Scholar]
- 38.Reimann, F., Tucker, S. J., Proks, P., and Ashcroft, F. M. (1999) J. Physiol. 518 325–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Babenko, A. P., and Bryan, J. (2003) J. Biol. Chem. 278 41577–41580 [DOI] [PubMed] [Google Scholar]
- 40.Yamada, M., Isomoto, S., Matsumoto, S., Kondo, C., Shindo, T., Horio, Y., and Kurachi, Y. (1997) J. Physiol. 499 715–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Takano, M., Xie, L. H., Otani, H., and Horie, M. (1998) J. Physiol. 512 395–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beech, D. J., Zhang, H., Nakao, K., and Bolton, T. B. (1993) Br. J. Pharmacol. 110 573–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Satoh, E., Yamada, M., Kondo, C., Repunte, V. P., Horio, Y., Iijima, T., and Kurachi, Y. (1998) J. Physiol. 511 663–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rost, B. (2003) Methods Biochem. Anal. 44 559–587 [PubMed] [Google Scholar]
- 45.Hambrock, A., Loffler-Walz, C., Russ, U., Lange, U., and Quast, U. (2001) Mol. Pharmacol. 60 190–199 [DOI] [PubMed] [Google Scholar]
- 46.Hambrock, A., Loffler-Walz, C., Kurachi, Y., and Quast, U. (1998) Br. J. Pharmacol 125 577–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Russ, U., Hambrock, A., Artunc, F., Loffler-Walz, C., Horio, Y., Kurachi, Y., and Quast, U. (1999) Mol. Pharmacol. 56 955–961 [PubMed] [Google Scholar]
- 48.Christopoulos, A. (1998) Trends Pharmacol. Sci. 19 351–357 [DOI] [PubMed] [Google Scholar]
- 49.Chou, P. Y., and Fasman, G. D. (1978) Annu. Rev. Biochem. 47 251–276 [DOI] [PubMed] [Google Scholar]
- 50.MacGregor, G. G., Dong, K., Vanoye, C. G., Tang, L., Giebisch, G., and Hebert, S. C. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 2726–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stephan, D., Winkler, M., Kühner, P., Russ, U., and Quast, U. (2006) Diabetologia 49 2039–2048 [DOI] [PubMed] [Google Scholar]
- 52.Hansen, A. M. K., Hansen, J. B., Carr, R. D., Ashcroft, F. M., and Wahl, P. (2005) Br. J. Pharmacol. 144 551–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haider, S., Tarasov, A. I., Craig, T. J., Sansom, M. S., and Ashcroft, F. M. (2007) EMBO J. 26 3749–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schulze, D., Krauter, T., Fritzenschaft, H., Soom, M., and Baukrowitz, T. (2003) J. Biol. Chem. 278 10500–10505 [DOI] [PubMed] [Google Scholar]
- 55.Cukras, C. A., Jeliazkova, I., and Nichols, C. G. (2002) J. Gen. Physiol. 120 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kondo, C., Repunte, V. P., Satoh, E., Yamada, M., Horio, Y., Matsuzawa, Y., Pott, L., and Kurachi, Y. (1998) Receptors Channels 6 129–140 [PubMed] [Google Scholar]
- 57.Babenko, A. P. (2005) J. Mol. Cell Cardiol. 39 79–98 [DOI] [PubMed] [Google Scholar]
- 58.Vila-Carriles, W. H., Zhao, G., and Bryan, J. (2007) FASEB. J. 21 18–25 [DOI] [PubMed] [Google Scholar]
- 59.Winkler, M., Stephan, D., Bieger, S., Kuhner, P., Wolff, F., and Quast, U. (2007) J. Pharmacol. Exp. Ther. 322 701–708 [DOI] [PubMed] [Google Scholar]
- 60.Babenko, A. P., Gonzalez, G., and Bryan, J. (1999) FEBS Lett. 459 367–376 [DOI] [PubMed] [Google Scholar]
- 61.Ashfield, R., Gribble, F. M., Ashcroft, S. J., and Ashcroft, F. M. (1999) Diabetes 48 1341–1347 [DOI] [PubMed] [Google Scholar]
- 62.Jiang, Y., and MacKinnon, R. (2000) J. Gen. Physiol. 115 269–272 [DOI] [PMC free article] [PubMed] [Google Scholar]