

Abstract
Thrombomodulin (TM) increases the catalytic efficiency of thrombin (IIa)-mediated activation of thrombin-activable fibrinolysis inhibitor (TAFI) 1250-fold. Negatively charged residues of the C-loop of TM-EGF-like domain 3 are required for TAFI activation. Molecular models suggested several positively charged residues of TAFI with which the C-loop residues could interact. Seven TAFI mutants were constructed to determine if these residues are required for efficient TAFI activation. TAFI wild-type or mutants were activated in the presence or absence of TM and the kinetic parameters of TAFI activation were determined. When the three consecutive lysine residues in the activation peptide of TAFI were substituted with alanine (K42/43/44A), the catalytic efficiencies for TAFI activation with TM decreased 8-fold. When other positively charged surface residues of TAFI (Lys-133, Lys-211, Lys-212, Arg-220, Lys-240, or Arg-275) were mutated to alanine, the catalytic efficiencies for TAFI activation with TM decreased by 1.7–2.7-fold. All decreases were highly statistically significant. In the absence of TM, catalytic efficiencies ranged from 2.8-fold lower to 1.24-fold higher than wild-type. None of these, except the 2.8-fold lower value, was statistically significant. The average half-life of the TAFIa mutants was 8.1 ± 0.6 min, and that of wild type was 8.4 ± 0.3 min at 37 °C. Our data show that these residues are important in the activation of TAFI by IIa, especially in the presence of TM. Whether the mutated residues promote a TAFI-TM or TAFI-IIa interaction remains to be determined. In addition, these residues do not influence spontaneous inactivation of TAFIa.
The endothelial membrane protein thrombomodulin (TM)2 is a central regulator in the balance between coagulation and fibrinolysis (1, 2). It serves as a molecular switch that changes the specificity of thrombin from a procoagulant enzyme to both an anticoagulant and an antifibrinolytic enzyme. The thrombin-thrombomodulin complex (IIa-TM) mediates its anticoagulant and antifibrinolytic effects through the activation of two zymogens, protein C (3) and thrombin-activable fibrinolysis inhibitor (TAFI) (4).
TAFI, also known as procarboxypeptidase U (pro-CPU) (5), plasma procarboxypeptidase B (6), and carboxypeptidase R (7), is a 60-kDa glycoprotein that circulates in plasma at concentration of about 75 nm (8). It can be activated to form TAFIa, a carboxypeptidase B-like enzyme, by proteolytic cleavage at arginine 92 (4, 6, 9). TAFIa catalyzes the removal of C-terminal lysine and arginine residues from partially degraded fibrin, thereby suppressing the cofactor activity of fibrin for plasminogen activation and attenuating fibrinolysis (10). TAFIa also can inactivate pro-inflammatory mediators such as the anaphylatoxins C3a and C5a, as well as bradykinin by hydrolysis of their C-terminal arginine, thereby reducing their pro-inflammatory effects (7, 11, 12). In addition, TAFIa can remove the C-terminal arginine of thrombin-cleaved osteopontin, thereby down-regulating its pro-inflammatory properties (13).
TAFI can be activated by the IIa-TM complex (14), free thrombin (4, 9), plasmin (4, 15), or trypsin (9). Although IIa at the high level generated after clotting via the factor XI-dependent pathway can activate sufficient TAFI to suppress fibrinolysis (16), IIa by itself is a relatively weak activator because this activation is very inefficient (Km = 2.14 ± 0.59 μm; kcat = 0.0021 ± 0.0004 s-1) (14). When IIa binds to TM and forms a complex, however, the catalytic efficiency of TAFI activation increases by 1250-fold, which occurs almost exclusively through an increase in kcat (Km = 1.01 ± 0.09 μm; kcat = 1.24 ± 0.06 s-1). Thus, the IIa-TM complex is probably the physiological activator of TAFI (14).
Eaton et al. (9) originally sequenced TAFI and showed that it consists of a 22-amino acid signal peptide, a 92-amino acid activation peptide, and a 309-amino acid catalytic domain (9). The sequence revealed that TAFI is homologous to tissue-type procarboxypeptidases A and B. TAFI, however, is unique in that it has triple lysine residues (Lys-42, Lys-43, and Lys-44) in its activation peptide (9).
TM consists of 10 structural elements: a N-terminal domain homologous to the family of C-type lectins (residues 1–226), six tandem epidermal growth factor (EGF)-like domains joined by small interdomain peptides (residues 227–462), a serine/threonine-rich domain (residues 463–497), a transmembrane domain (residues 498–521), and a cytoplasmic tail (residues 522–559) (1, 2, 17, 18). It is predominantly expressed on the luminal surface of endothelial cells lining normal blood vessels (19). The structure of the IIa-TM complex has been revealed by x-ray crystallography of human thrombin bound to EGF-like domains 4, 5, and 6 (20). EGF-like domains 5 and 6 are IIa-binding sites. EGF 5 and part of EGF 6 bind to a cluster of lysine and arginine residues of anion-binding exosite-I on IIa. This prevents binding between IIa and its procoagulant substrates, such as fibrinogen (20).
Although thrombin binds exclusively to the EGF-like domains 5 and 6, this is not sufficient to stimulate the activation of TAFI (16) or protein C (21, 22). The primary structure of TM required for efficient TAFI activation includes the 13 residues from the C terminus of EGF-like domain 3 extending through EGF-like domain 6 (16, 18). In addition, Wang et al. (16) demonstrated in alanine-scanning experiments that mutation of residues Val-340, Asp-341, or Glu-343 within the C-loop of EGF-like domain 3 resulted in a 90% or greater reduction in TAFI activation. They also demonstrated that the mutation D349A in the peptide connecting EGF-like domains 3 and 4 eliminated the cofactor activity of TM for TAFI activation (16). In addition, Schneider et al. (23) showed that the TM dependence of TAFI activation is not determined by residues occupying the P6-P3′ positions of TAFI.
To further understand the structural basis for the activity of the IIa-TM complex in TAFI activation, we generated a structure model of the TAFI-IIa-TM EGF-like domains 4–5-6 complex. From this model, two positively charged surface patches on TAFI which could complement the key negatively charged residues near the N terminus of EGF-like domain 4 (Asp-341, Glu-343, and Asp-349) were identified. One region consists of residues Lys-133, Lys-211, Lys-212, and Arg-220. Further away is another positively charged surface patch comprising residues Lys-240 and Arg-275. An additional positive surface patch consists of the three-lysine residues K42/43/44 unique to the TAFI activation peptide.
In this study, we constructed seven TAFI mutants in order to determine whether some or all of the positively charged residues referred to above are required in TAFI activation. We then determined the kinetics of activation by IIa for each mutant and wild-type TAFI in the presence or absence of TM. We also determined the thermal stability of TAFIa for each mutant and wild-type. The results indicate that all mutations decrease activation efficiency of TAFI, but none influence the thermal stability of TAFIa.
EXPERIMENTAL PROCEDURES
Materials—The synthetic carboxypeptidase substrate anisolylazoformylarginine (AAFR) was from Bachem Biosciences Inc. (King of Prussia, Pennsylvania), CNBr-activated Sepharose 4B was from Amersham Biosciences (Uppsala, Sweden), and the thrombin inhibitor d-Phe-Pro-Arg chloromethyl ketone (PPACK) was from Calbiochem (San Diego, CA). Bovine serum albumin, RIA Grade, Fraction V, minimum 96% was from Sigma-Aldrich Inc. Plasmid DNA Preparation kits were from Qiagen Inc. (Mississauga, Ontario). DNA restriction and modification enzymes were from New England BioLabs (Mississauga, Ontario) and Invitrogen (Burlington, Ontario). DNA Polymerase I, Large (Klenow) Fragment was from New England BioLabs (Mississauga, Ontario). QuikChange® Site-directed Mutagenesis kit was from Stratagene (La Jolla, CA). Baby hamster kidney (BHK) cells and the mammalian expression vector pNUT were kindly provided by Dr. Ross MacGillivray (University of British Columbia). Newborn calf serum, Dulbecco's modified Eagle's medium/F-12 nutrient mixture (1:1) (DMEM/F-12), Opti-MEM, penicillin/streptomycin/Fungizone mixture (PSF) were from Invitrogen. Methotrexate (Mayne Pharma Inc., Montreal, Canada) was purchased at Kingston General Hospital. Thrombin was prepared as described previously (24). Recombinant soluble thrombomodulin (Solulin) was a generous gift from Dr. Henning Brohmann (Paion, GmbH, Aachen, Germany). Antibodies for the TAFI ELISA were purchased from Affinity Biologicals Inc. (Ancaster, Ontario). The mouse-anti-human TAFI monoclonal antibody mAb16 was prepared as described previously (25). The mouse-anti-human TAFI monoclonal antibodies (MA-T12D11, MA-T3D8, and MA-T18A8) were prepared as described previously (26).
Structure Model of TAFI-IIa-TM-EGF 456 Complex—The homology model of TAFI was initially built based on the high resolution structure of human procarboxypeptidase B (CPB) (structure ID PDB1KWM, 1.6 Å), using Modeler (Accelrys, 2005). The loop involving the cleavage site between the activation peptide and catalytic domain was built based on the homologous loop of the bovine procarboxypeptidase A (CPA) structure that is in complex with chymotrypsinogen C (PDB1PYT, 2.35 Å). Based on this structural complex and the structure of IIa in complex with TM (PDB1DX5, 2.3 Å), the ternary complex of the TAFI, IIa, and the 4th to 6th EGF-like domains of TM was built. The backbone atoms of the TAFI model were initially superimposed onto those of the CPA structure from the complex, and then the catalytic triad of IIa was superimposed onto those of the chymotrypsinogen C. From this model, positively charged residues on the surface of TAFI that could complement the negatively charged residues on the C-loop of TM-EGF3 were identified for further study.
Construction of TAFI Variant Plasmids—The plasmid pBluescript (SK+) vector containing full-length human TAFI cDNA (27) was used to construct all of the TAFI mutants. Mutations were introduced into the plasmid coding for wild-type TAFI cDNA using a QuikChange Mutagenesis kit. A pair of primers was used to construct each of the 7 mutants: 1) Lys to Ala at position 133 (TAFI-K133A), 2) Lys to Ala at position 211 (TAFI-K211A), 3) Lys to Ala at position 212 (TAFI-K212A), 4) Arg to Ala at position 220 (TAFI-R220A), 5) Lys to Ala at position 240 (TAFI-K240A), 6) Arg to Ala at position 275 (TAFI-R275A), and 7) three consecutive Lys to Ala mutations at positions 42, 43, and 44 (TAFI-K42/43/44A). The triple mutant TAFI-K42/43/44A was constructed by three steps, in which single sequential mutations were made, using a new construct as the template for the next mutation. The plasmids for each mutant were verified by DNA sequencing. All of the numbers used to designate the various derivatives are based on the TAFI amino acid sequence numbering (9).
The newly constructed TAFI derivative cDNAs were ligated into the mammalian expression vector pNUT as described previously (27). The proper orientation of the construct was confirmed by EcoRI digestion, which yielded a fragment of ∼1850 bp for the forward orientation or a fragment of ∼650 bp for the reverse orientation.
Transfection and Expression of TAFI Variants—Baby hamster kidney (BHK) cells were cultured in DMEM/F-12 nutrient mixture (1:1) (DMEM/F-12) supplemented with 5% newborn calf serum (NCS) in a 37 °C incubator. TAFI-pNUT plasmids expressing mutants and wild-type were transfected into BHK cells using a calcium phosphate co-precipitation method (28, 29). Eight hours later, the cells were washed and fed with fresh DMEM/F-12 containing 5% NCS. The cells were allowed to recover overnight, and then the medium was replaced with DMEM/F-12 containing 5% NCS supplemented with 400 μm methotrexate.
After 2 weeks of selection, surviving colonies were transferred into individual wells in 6-well tissue culture plates (Becton Dickinson Labware, Franklin Lakes, New Jersey). Once the cells became confluent, the media were screened for TAFI protein production by ELISA. Clones with high TAFI expression levels were picked and seeded into triple flasks (500 cm2; Nunc, Roskilde, Denmark) in DMEM/F-12 with 5% NCS and 100 μm methotrexate. Once the cells became confluent, the medium was changed to serum-free Opti-MEM, supplemented with 1% (v/v) penicillin/streptomycin/Fungizone mixture (PSF) and 50 μm ZnCl2. Conditioned medium was collected at 48-h intervals and replaced with fresh Opti-MEM/PSF/ZnCl2. Collected medium was centrifuged at 1,200 × g for 20 min and then stored at -20 °C.
Purification of Recombinant TAFI—The isolation of recombinant TAFI was carried out as described previously (30). Conditioned medium (2 liters) was filtered and loaded onto a 3.0 ml of anti-TAFI mAb-Sepharose column that had been equilibrated at 4 °C in 0.02 m HEPES, 0.15 m NaCl, pH 7.4 (HBS). The column was then washed extensively with HBS. TAFI was eluted with 0.2 m glycine (pH 3.0), and 1-ml fractions were collected into tubes containing 1-ml aliquots of 1 m Tris-HCl (pH 8.0), for a final volume of 2 ml per fraction. The mixture was pipetted up and down immediately to neutralize the eluate. All the fractions were quantified for TAFI levels by ELISA and fractions containing high TAFI were pooled, dialyzed against HBS, and concentrated 10-fold using an Amicon Ultra Centrifugal Filter (Millipore Corporation, Billerica, MA). Purified TAFI was quantified by measurement of absorbances at 280 nm and 320 nm (ε1%,280 = 26.4; Mr = 48,442) (4). The protein was then aliquoted, snap-frozen, and stored at -80 °C. The isolated TAFI derivatives were then analyzed using 5–15% gradient SDS-PAGE under reducing conditions, with the Neville system (31). The antibodies used to isolate TAFI were coupled to Sepharose 4B as described previously (25). Antibody mAb16 was used to isolate wild-type TAFI and all of the mutants, except the triple mutant K42/43/44A, which did not bind the column. For its isolation, antibody MA-T12D11 (30) was used.
Kinetics of Activation of TAFI Variants by the IIa-TM Complex—TAFI activation was performed as described previously (30). Briefly, each TAFI variant at various concentrations (0–2.0 μm) was titrated with TM at concentrations ranging from 0 to 50 nm in the presence of IIa (0.5 nm), CaCl2 (5 mm) and HBS with 0.01% Tween 80. Reaction mixtures (40 μl) were incubated at 25 °C for 10 min. The reactions then were stopped by adding a solution containing PPACK (1 μm) and synthetic substrate AAFR (120 μm) to achieve a final volume of 200 μl. The samples then were monitored at 349 nm in a SpectraMax Plus Plate Reader (Molecular Devices, Sunnyvale, CA), and the initial rates of hydrolysis of AAFR were determined from the slopes of the absorbance versus time relationships.
TAFIa standard curves were generated as described previously (30). Briefly, each TAFI variant (1 μm) was completely activated by incubating it with IIa (25 nm), TM (100 nm) and CaCl2 (5 mm) at 25 °C for 15 min and placing it on ice. TAFIa at varying concentrations was added into AAFR (120 μm) and PPACK (1 μm). Because TAFI zymogen also shows small activity toward AAFR (30), a TAFI standard also was made with it (1 μm). The standard curves were used to calculate two rate constants: K1, for the activity of TAFI zymogen toward AAFR and K2, for the activity of TAFIa toward AAFR. The initial rate of AAFR hydrolysis (r = dAbs/dt) is described by Equation 1.
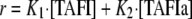 |
(Eq. 1) |
Because the total concentration of TAFI, [TAFI]0 = [TAFI] + [TAFIa], Equation 2 can be derived from Equation 1.
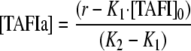 |
(Eq. 2) |
The TAFIa concentrations at 10 min were used to calculate the initial rates of TAFI activation, v (mol TAFIa formed/liter/s). In no instance did the TAFIa concentration at 10 min exceed 10% of the input concentration of TAFI. The activation kinetics then were analyzed by the enzyme central, parallel assembly model described previously (14, 23, 32). In this model, the enzyme (T) can interact with TM to form the T-TM complex with dissociation constant Kd1 or with TAFI to form the T-TAFI complex with dissociation constant Km1. The T-TM complex can interact with TAFI to form the T-TM-TAFI complex with Km2, or the T-TAFI complex can interact with TM to form the T-TM-TAFI complex with Kd2. The ternary T-TM-TAFI complex turns over to TAFIa with first order rate constant, kcat. The rate of the reaction is given by Equation 3.
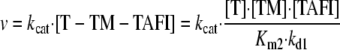 |
(Eq. 3) |
The total concentration of thrombin, [T]0, is given by Equation 4, which can be rearranged to give Equations 5 and 6.
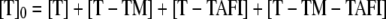 |
(Eq. 4) |
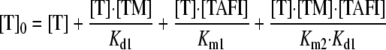 |
(Eq. 5) |
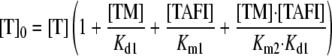 |
(Eq. 6) |
The TAFI activation rate divided by the total concentration of thrombin is obtained by dividing Equation 3 by Equation 6. The result, after algebraic rearrangement, is given by Equation 7.
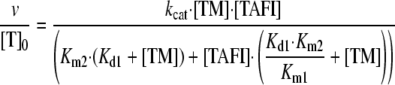 |
(Eq. 7) |
All of the rate data were fit simultaneously by non-linear regression to Equation 7 with the NONLIN module of SYSTAT (SPSS Inc., Chicago, IL). The best fit parameters were kcat, Km1, and Km2 for wild-type and each variant. Kd1 (the dissociation constant for the IIa-TM interaction) was a best fit parameter also, but its value was assumed to be the same for wild-type TAFI and the variants, because all reactions have this interaction in common. The regression analysis returned best fit values along with their asymptotic standard errors (A.S.E.). The analyses were applied to no less than four and no more than five independent experiments with wild-type TAFI and each of the variants. The mean and standard deviations for kcat, Km2, and kcat/Km2 values were calculated. Statistical analysis on the variance of catalytic efficiencies (kcat/Km2) of activation of the TAFI derivatives with respect to the wild-type was performed using independent samples t test in SPSS (SPSS Inc., Chicago, IL).
Kinetics of Activation of TAFI Variants by IIa in the Absence of Thrombomodulin—Each TAFI variant at various concentrations (0–2.0 μm) was incubated with IIa (50 nm) and CaCl2 (5 mm) in HBS with 0.01% Tween 80 at 25 °C for 20 or 30 min. All reactions were carried out, and the initial rates of hydrolysis of AAFR were measured as described above.
In the absence of TM, the relationships between rates and the concentration of TAFI were linear indicating high Km values. Thus, the slopes of rate versus the concentration of TAFI were measured by linear regression. The catalytic efficiencies (kcat/Km1) were then equated with the slopes according to Equation 8.
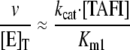 |
(Eq. 8) |
The catalytic efficiency was calculated for each TAFI mutant and compared with that of TAFI wild-type. Statistical analysis on the variance of catalytic efficiencies of activation of the TAFI derivatives with respect to the wild-type was performed using independent samples t test in SPSS (SPSS Inc., Chicago, IL).
Thermal Stability Assay of TAFIa Variants—Half lives of various recombinant TAFI were determined as described previously (30). Briefly, TAFI (1 μm) was fully activated to TAFIa by incubation with IIa (25 nm), TM (100 nm) and CaCl2 (5 mm) in HBS containing 0.01% (v/v) Tween 80 for 15 min at 25 °C. Thrombin activity was then quenched by the addition of 1 μm PPACK, and the mixture was placed on ice until use.
TAFIa was then diluted 8-fold using HBS with 0.01% Tween 80 and 1 mg/ml bovine serum albumin. The mixture was then transferred to a 37 °C water bath. Samples (50 μl) were removed at various times (0, 2, 5, 10, 15, 20, 30, and 45 min) into a solution containing 50 μl of 240 μm AAFR in a microtiter plate. The initial rates of AAFR hydrolysis were measured at 349 nm in a microtiter plate reader. Residual activity over time was fit to the exponential decay equation [TAFIa] = [TAFIa]0 · e-k·t by non-linear regression using SigmaPlot 8 (SigmaPlot 2002 for Windows Version 8.02). The first-order decay constant k was obtained and used to calculate the half-life of each TAFI variant.
RESULTS
The Use of Prior Biochemical Data and Structure Modeling of the TAFI-IIa-TM Complex to Identify Residues of TAFI for Mutagenesis—Previous work using alanine-scanning mutagenesis of TM indicated that residues Asp-341 and Glu-343 from the C-loop of EGF-like domain 3 and Asp-349 in the interdomain peptide joining EGF domains 3 and 4 are very important in TAFI activation, but not protein C activation (16). Other work showed that residues occupying the P6-P3′ positions of TAFI do not determine the TM dependence of its activation (23). In addition, the interaction of IIa and TM does not appreciably alter the active site structure of IIa (20, 33). These observations suggest the hypothesis that an interaction between the negatively charged residues of C-loop of EGF-like domain 3 of TM and positively charged residues of TAFI in the ternary IIa-TM-TAFI complex promote efficient activation of TAFI by IIa. To identify positively charged residues of TAFI that might be involved in this putative interaction, a model structure of the IIa-TM-TAFI complex was built (Fig. 1).
FIGURE 1.

Structure model of the TAFI-IIa-TM-EGF 456 complex. This complex was built based on the structure of bovine procarboxypeptidase A (CPA) in complex with chymotrypsinogen C (44) and the structure of IIa (secondary structures shown in green) in complex with TM-EGF 456 (red) (20). The TAFI model (yellow) and the catalytic triad of IIa were superimposed on CPA and the catalytic triad of chymotrypsinogen C complex, respectively. TM-EGF 456 structure retains its original orientation with respect to IIa (20). The positive residues on TAFI (side chains indicated in dark and light blue) may complement the negative residues on the C-loop of TM EGF 3, where the predicted position is indicated with the arrow.
The model was evaluated with Profiles-3D (Accelrys, 2005), and no putative misfolded regions were identified. The backbone root-mean-squared deviation between the structurally conserved regions of CPB and the TAFI model is 0.6 Å, and between the catalytic triads of the chymotrypsinogen C and IIa is 0.35 Å. In the model of the ternary complex, the orientation between TM-EGF 456 and IIa is identical to that in the original structure complex (20).
Although EGF3 of TM is not in this structure because it was not part of the structure determined by Fuentes-Prior et al. (20), it would be expected to be located at the N terminus of TM-EGF 456 as indicated in Fig. 1. Thus, residues deemed to be “close” to this region were selected for mutagenesis. These included the triple lysine residues K42/43/44 of the activation peptide of TAFI, and two positively charged patches, one defined by residues Lys-133, Lys-211, Lys-212, and Arg-220, and the other by residues Lys-240 and Arg-275. Therefore, site-directed mutagenesis was used to construct the mutants K42/43/44A, K133A, K211A, K212A, R220A, K240A, and R275A.
Construction, Expression, and Isolation of TAFI Variants— Seven mutants of human TAFI with alanine substituted for lysine or arginine were constructed. The sequence of primers used for introduction of the mutations are listed in supplemental Table S2. The mutants were constructed in the pBlueScript (SK+) vector. They were subsequently subcloned into the mammalian expression vector pNUT, which was stably transfected into BHK cells. TAFI variants were expressed at concentrations ranging from 0.6 to 1.1 μg/ml.
All recombinant TAFI variants, except for the triple mutant, TAFI-K42/43/44A, were isolated with the anti-TAFI mAb16-Sepharose column with a yield of 50–60%, as determined by ELISA. Antibody MA-T12D11 was used for the isolation of the TAFI-K42/43/44A derivative. The yield was 50%. Purified proteins were quantified by spectrophotometry and analyzed by SDS-PAGE. All of the TAFI variants migrated as a single band at ∼60 kDa in SDS-PAGE (Fig. 2).
FIGURE 2.

SDS-PAGE analysis of TAFI wild-type and derivatives. The purified proteins (2 μg) were resolved on 5–15% polyacrylamide gradient gel under reducing conditions. Protein bands were visualized with Coomassie Blue. The lanes are: M, marker; 1, TAFI-WT; 2, TAFI-K42/43/44A; 3, TAFI-K133A; 4, TAFI-K211A; 5, TAFI-K212A; 6, TAFI-R220A; 7, TAFI-K240A; 8, TAFI-R275A.
Activity of TAFIa and TAFI Zymogen Toward the Synthetic Substrate AAFR—The activities toward AAFR of both TAFIa and TAFI zymogen at known concentrations determined by absorbance at 280 nm were measured for wild type and all of the mutant proteins. The data are summarized in Table 1. Wild type TAFIa yielded a value of 124 (mOD/min/nm). All mutants yielded activities similar to wild-type TAFIa, except for K212A and K240A with respective values of 177 and 245. All zymogens also showed some activity toward the small substrate, which is typical for the carboxypeptidase family of enzymes and zymogens. The wild-type zymogen shows 1.72% of the activity of the enzyme, which is similar to the 2% found previously (30). The zymogen activities of the mutants, relative to the corresponding enzymes, ranged from 0.89 to 2.64%. These data show that none of the mutations grossly affected the activities of either the zymogen or the enzyme.
TABLE 1.
Comparison of zymogen and enzyme activities for TAFI wild-type and derivatives Various recombinant TAFI zymogens (1 μm) and TAFIa at known concentrations (0–200 nm) was added into AAFR and PPACK, and rates were measured as described under “Experimental Procedures.” The values are the means ± S.D. of at least two and at most five independent experiments.
| K1a(Zymogen) | K2a(Enzyme) | K1/K2 | |
|---|---|---|---|
| WT | 2.13 ± 0.55 | 124 ± 23 | 0.0172 |
| K42/43/44A | 2.03 ± 0.47 | 116 ± 2 | 0.0175 |
| K133A | 3.68 ± 0.22 | 140 ± 23 | 0.0264 |
| K211A | 1.42 ± 0.28 | 135 ± 3 | 0.0105 |
| K212A | 1.58 ± 0.16 | 177 ± 6 | 0.0089 |
| R220A | 1.26 ± 0.04 | 134 ± 22 | 0.0094 |
| K240A | 2.93 ± 0.18 | 245 ± 17 | 0.0120 |
| R275A | 1.75 ± 0.22 | 135 ± 27 | 0.0130 |
Unit (mOD/min/nm, A349), activity measured with 120 μm AAFR, 22 °C, pH 7.4
Activation of TAFI Variants by the IIa-TM Complex—Initial rates of TAFIa formation were measured as the concentrations of TAFI and TM were systematically varied (Fig. 3). The data were analyzed according to the enzyme central, parallel assembly equilibrium model of Boskovic et al. (32) by fitting them to Equation 7 through non-linear regression. Best values for the kinetic parameters, Km1, kcat, Km2 and Kd1 thus were obtained. The results are presented in Table 2. All data were regressed together with a single value of the Kd for the binding of IIa to TM (Kd1 in Equation 7). The regression analysis yielded Kd = 28.3 ± 8.3 nm, which agrees well with the value of 22 nm measured directly and reported previously (14). The lines in Fig. 3 are regression lines and they indicate a good fit of the model to the data.
FIGURE 3.

Kinetics of activation of TAFI variants by the IIa-TM complex. The zymogens TAFI-WT (a), TAFI-K42/43/44A (b), TAFI-K133A (c), TAFI-K211A (d), TAFI-K212A (e), TAFI-R220A (f), TAFI-K240A (g), and TAFI-R275A (h), each at various concentrations, were incubated with IIa (0.5 nm) and TM at concentrations of 1.56 nm (•), 3.13 nm (○), 6.25 nm (▾), 12.5 nm (▿), 25 nm (▪), and 50 nm (□). TAFIa formation rates (mol of TAFIa formed/mol of thrombin/second) were calculated, and the complete data set was fit globally to Equation 7 as described previously (32). The lines are best-fit regression lines.
TABLE 2.
Comparison of kinetic parameters for TAFI wild-type and mutants The Km1, kcat, Km2, and Kd values were estimated by non-linear regression of the data presented in Fig. 3 to the rate Equation 7. The rate constant values listed here are given as the mean ± S.D. of the estimates of at least four and at most five independent experiments. p values were calculated using independent samples t test for the catalytic efficiencies (kcat/Km2) of each derivative in comparison to the wild-type.
| Km1 | Kcat | Km2 | Kcat/Km2 | p value | Relative binding energy | |
|---|---|---|---|---|---|---|
| μm | s–1 | μm | μm–1 s–1 | |||
| WT | 40.6 ± 12.9 | 1.18 ± 0.24 | 0.25 ± 0.02 | 4.80 ± 0.70 | — | 1.000 |
| K42/43/44A | 60.5 ± 15.6 | 3.27 ± 0.37 | 5.49 ± 1.31 | 0.61 ± 0.10 | <0.001 | 0.797 |
| K133A | 8.8 ± 15.6 | 1.24 ± 0.19 | 0.59 ± 0.12 | 2.20 ± 0.55 | <0.001 | 0.944 |
| K211A | 32.4 ± 24.2 | 1.56 ± 0.15 | 0.89 ± 0.15 | 1.77 ± 0.16 | <0.001 | 0.916 |
| K212A | 2.0 ± 0.5 | 0.91 ± 0.61 | 0.49 ± 0.57 | 2.63 ± 0.86 | 0.005 | 0.956 |
| R220A | 41.5 ± 18.9 | 1.89 ± 0.45 | 0.86 ± 0.25 | 2.27 ± 0.44 | <0.001 | 0.919 |
| K240A | 38.3 ± 23.5 | 0.86 ± 0.28 | 0.32 ± 0.15 | 2.79 ± 0.29 | 0.002 | 0.984 |
| R275A | 16.3 ± 22.8 | 1.49 ± 0.60 | 0.66 ± 0.27 | 2.26 ± 0.38 | 0.001 | 0.936 |
When the triple lysine residues, Lys-42, Lys-43, and Lys-44 that are unique to the activation peptide of TAFI (9) were mutated to alanine, the catalytic efficiency (kcat/Km2) of TAFI activation by IIa-TM complex decreased by about 8-fold compared with the wild-type (Table 2). When the residues Lys-133, Lys-211, Lys-212, and Arg-220 of one surface patch of TAFI were substituted by alanine individually, the catalytic efficiencies of TAFI activation by the IIa-TM complex decreased from 1.8–2.7-fold. When residues Lys-240 and Arg-275 on another surface patch were individually substituted by alanine, the catalytic efficiencies of TAFI-K240A and TAFI-R275A activation decreased 1.7- and 2.1-fold, respectively. Statistical analysis shows that the catalytic efficiency of wild-type TAFI is significantly higher than those of all of the mutants, with p values ranging from less than 0.001 to 0.005 (Table 2).
The Km2 values were interpreted as reflecting the binding of the TAFI mutants to the IIa-TM complex. This interpretation, along with the thermodynamic relationship ΔG° = -RTlnKd (and therefore ΔG° = -RTlnKm2), allowed for the calculation of the binding energies for the interactions between the various forms of TAFI and the IIa-TM complex. The results are presented in Table 2, each expressed relative to the binding energy for the interaction of wild-type TAFI with the IIa-TM complex. The losses of binding energy ranged from a low of 1.6% (K240A) to a high of 20.3% (K42/43/44A). The sum of losses of all of the mutants is 54.8%. Thus, no single mutation eliminated binding of TAFI to the IIa-TM complex, but the nine residues together account for 54.8% of the binding energy.
The Km1 values were interpreted as reflecting the binding of the TAFI mutants to IIa alone. The values for Km1, as shown in Table 2, were typically very high (8.8–60.5 μm), except for the K212A mutant (2.0 μm). The high values were accompanied by very high standard deviations, indicating that these binding interactions are relatively weak. This is consistent with the apparently high Km values suggested by the linear rate versus TAFI concentrations observed experimentally in the absence of TM (Fig. 4). Because the IIa and TAFI binding was weak and thus standard deviations for Km1 were high, an alternative analysis was carried out where the data were regressed to a one pathway model (supplemental Eq. S1), which does not include Km1 because it does not include the IIa/TAFI interaction. The results of this analysis are found in the supplemental material (supplemental Fig. S1). The kcat/Km values were very similar to the kcat/Km2 values found in Table 2. The fit of the one pathway model to the data, however, was marginally poorer than the fit of the parallel pathway model, as can be seen by comparing Figs. 3 and supplemental S1.
FIGURE 4.

Kinetics of activation of TAFI variants by thrombin alone. TAFI-WT (•), TAFI-K42/43/44A (○), TAFI-K133A (▾), TAFI-K211A (▿), TAFI-K212A (▪), TAFI-R220A (□), TAFI-K240A (♦), and TAFI-R275A (⋄) were incubated at various concentrations with IIa. The rate of TAFIa formation (mol of TAFIa formed/mol of thrombin/second) was calculated. The slope of the relationship between the rate and TAFI concentration was measured to determine the catalytic efficiency (kcat/Km1) of TAFI activation.
Activation of TAFI Variants by IIa Alone—TAFIa formation rates were calculated from the initial rates of AAFR hydrolysis and plotted as a function of the TAFI concentration (Fig. 4). The catalytic efficiencies (kcat/Km1) of TAFI activation were calculated from the slopes of the lines.
In the absence of TM, wild-type TAFI has a catalytic efficiency of 0.00095 ± 0.00025 μm-1 s-1 for its activation (Table 3). The catalytic efficiency value of TAFI-K42/43/44A is 2.8-fold lower (0.00034 ± 0.00004 μm-1 s-1). The catalytic efficiencies of the other mutants range from a low of 64% of wild-type (K212A) to a high of 124% (R275A). None of these values, however, are significantly different from that of wild-type TAFI, with p values ranging from 0.067 to 0.572 (Table 3).
TABLE 3.
Catalytic efficiencies for TAFI activations by IIa in the absence of TM The catalytic efficiencies of TAFI activation by IIa alone are given as the mean ± S.D. For TAFI wild-type, n = 4. For TAFI-K42/43/44A, n = 2. For all other TAFI mutants, n = 3. p values were calculated using independent samples t test for each derivative in comparison to the wild-type.
| kcat/Km1 | p value | |
|---|---|---|
| μm–1 s–1 | ||
| WT | 0.00095 ± 0.00025 | — |
| K42/43/44A | 0.00034 ± 0.00004 | 0.014 |
| K133A | 0.00062 ± 0.00016 | 0.086 |
| K211A | 0.00073 ± 0.00004 | 0.171 |
| K212A | 0.00061 ± 0.00005 | 0.067 |
| R220A | 0.00071 ± 0.00013 | 0.163 |
| K240A | 0.00104 ± 0.00015 | 0.572 |
| R275A | 0.00118 ± 0.00019 | 0.221 |
Thermal Stabilities of the TAFI Variants—TAFIa is intrinsically unstable and this is likely the main mechanism for its down-regulation in vivo (34). Thus, we measured and compared the half lives at 37 °C of the various TAFIa derivatives to that of wild-type TAFIa (Fig. 5). Wild-type TAFI has a half-life of 8.4 ± 0.3 min, while the derivatives TAFI-K42/43/44A, TAFI-K133A, TAFI-K211A, TAFI-K212A, TAFI-R220A, TAFI-K240A, and TAFI-R275A have half lives of 8.5 ± 0.2, 7.0 ± 0.2, 8.4 ± 0.2, 8.7 ± 0.3, 8.7 ± 0.2, 7.3 ± 0.2, and 8.3 ± 0.3 min, respectively. Thus, mutations of these arginine or lysine residues do not appreciably affect the stabilities of the activated forms of these TAFI derivatives.
FIGURE 5.

Comparison of thermal stabilities of TAFIa variants at 37 °C. The residual TAFIa activities were measured over time. The lines represent the nonlinear regression of the data to the exponential decay function.
DISCUSSION
Previous studies revealed some of the elements of the structures of IIa, TAFI, and TM needed for efficient TAFI activation (16, 18, 20, 23). To further understand the mechanism by which TM stimulates TAFI activation, we replaced several positively charged residues on TAFI that are likely to interact with TM, and potentially IIa, with alanine. Our data support a model, which posits that acidic residues of the C-loop of the TM-EGF-like domain 3 interacts with these basic residues on TAFI and aligns it to present its activation peptide to the IIa active site.
Monoclonal antibody mAb16, which does not recognize the triple mutant K42/43/44A, has been shown to inhibit TAFI activation by the IIa-TM complex but not by IIa alone (25). This loss of antigenicity due to mutations of these residues on the activation peptide are consistent with the epitope of mAb16 overlapping a region of TAFI important for a TM-dependent interaction. The antibody MA-T12D11, which does bind the triple mutant, recognizes an epitope on TAFI at Gly-66 (35), which is presumably away from the K42/43/44 location on the activation peptide.
In addition to the triple lysine residues in the activation peptide of TAFI, we also describe two positive surface patches on the catalytic domain of TAFI which may be important in expressing the TM dependence of TAFI activation. These two positive patches on TAFI might interact with two separate sites that are required for its efficient activation. Although the two patches seem to be on opposite ends of the TAFI molecule (Fig. 1), all single mutations show comparable catalytic efficiency decreases. Perhaps the positively charged residues studied here, both on the catalytic domain and the activation peptide, interact simultaneously with both the negatively charged C-loop of TM-EGF-like domain 3 and the negatively charged rim of IIa.
The major hypothesis of our studies was based on a computer structure model of TAFI generated using the sequence homology between TAFI and procarboxypeptidase B. Recent studies have shown that the structure of TAFI is indeed similar to that of procarboxypeptidase B, thus supporting the model (36, 37).
To date, no physiological inhibitor for TAFIa has been identified. Instead, it is intrinsically unstable, with a half-life that is highly temperature-dependent (30, 38). Mutagenesis studies by others have shown that inactivation of TAFIa is the result of its conformational instability (39), which is in agreement with the finding that the decay of TAFIa activity is associated with a substantial decrease in the intrinsic fluorescence (27). A region of TAFIa comprising amino acids 302–330 has been shown to be important in its intrinsic instability, and TAFI variants with altered thermal stabilities show corresponding alterations in their antifibrinolytic abilities (38, 39). In our study, TAFIa variants were tested at 37 °C to determine whether the present mutations alter their thermal stabilities. As expected, TAFI-K42/43/44A, which has the triple alanine mutation in its activation peptide and no other changes in its enzyme moiety shows a very similar half-life compared with the wild-type. The other six individual mutations in catalytic domain of TAFI also resulted in minimal changes with respect to the wild-type, indicating that these arginine and lysine residues are not involved in the intrinsic instability of TAFIa.
In general, our studies show that replacement of any one of the six positively charged residues on TAFI catalytic domain (Lys-133, Lys-211, Lys-212, Arg-220, Lys-240, Arg-275) or the triple lysine residues of the activation peptide (K42/43/44) with alanine, reduces the efficiency of activation of TAFI by thrombin, especially in the presence of TM. Because none of the mutations resulted in total loss of activation efficiency, the mutated residues together may play a role, whereby each contributes partially to the interactions of TAFI with IIa or TM that are needed for efficient activation.
Because TAFI is a key protein in the fibrinolytic system, inhibition of TAFIa is considered as a therapeutic strategy to prevent thrombosis. In animal models, inhibitors of TAFIa suppressed experimental thrombosis and enhanced thrombolysis (40–43). Further elucidation of TAFI structure-function relationships may help to develop better TAFIa inhibitors as novel anti-thrombotic agents.
Supplementary Material
Acknowledgments
We thank Tom Abbott for his help with tissue culture preparations and Lauren Lazowski for statistical advice.
This work was supported, in whole or in part, by National Institutes of Health Grant 5 P01 HL046703 (to M. E. N.). This work was also supported by CIHR Grant MT-9781 (to M. E. N.), as well as Heart and Stroke Foundation of Ontario Grant T5575 (to M. E. N.). This study was also supported by the Heart and Stroke Foundation of Canada Doctoral Research Award (to P. Y. K.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Equations S1–S6, Tables S1 and S2, and Fig. S1.
Footnotes
The abbreviations used are: TM, thrombomodulin; IIa, thrombin; TAFI, thrombin-activatable fibrinolysis inhibitor; TAFIa, activated TAFI; EGF-like domain, epidermal growth factor-like domain; AAFR, anisolylazoformylarginine; PPACK, d-Phe-Pro-Arg chloromethyl ketone; BHK cells, baby hamster kidney cells; CPB, procarboxypeptidase B; CPA, procarboxypeptidase A; mAb 16, mouse-anti-human TAFI monoclonal antibody; MA-T12D11, mouse anti-human TAFI monoclonal antibody; DMEM, Dulbecco's modified Eagle's medium; pro-CPU, procarboxypeptidase U.
References
- 1.Esmon, C. T., Johnson, A. E., and Esmon, N. L. (1991) Ann. N. Y. Acad. Sci. 614 30-43 [DOI] [PubMed] [Google Scholar]
- 2.Sadler, J. E., Lentz, S. R., Sheehan, J. P., Tsiang, M., and Wu, Q. (1993) Haemostasis 23 183-193 [DOI] [PubMed] [Google Scholar]
- 3.Esmon, C. T., Esmon, N. L., and Harris, K. W. (1982) J. Biol. Chem. 257 7944-7947 [PubMed] [Google Scholar]
- 4.Bajzar, L., Manuel, R., and Nesheim, M. E. (1995) J. Biol. Chem. 270 14477-14484 [DOI] [PubMed] [Google Scholar]
- 5.Wang, W., Hendriks, D. F., and Scharpe, S. S. (1994) J. Biol. Chem. 269 15937-15944 [PubMed] [Google Scholar]
- 6.Tan, A. K., and Eaton, D. L. (1995) Biochemistry 34 5811-5816 [DOI] [PubMed] [Google Scholar]
- 7.Campbell, W., and Okada, H. (1989) Biochem. Biophys. Res. Commun. 162 933-939 [DOI] [PubMed] [Google Scholar]
- 8.Nesheim, M. E. (1999) Fibrinol. Proteol. 13 72-77 [Google Scholar]
- 9.Eaton, D. L., Malloy, B. E., Tsai, S. P., Henzel, W., and Drayna, D. (1991) J. Biol. Chem. 266 21833-21838 [PubMed] [Google Scholar]
- 10.Wang, W., Boffa, M. B., Bajzar, L., Walker, J. B., and Nesheim, M. E. (1998) J. Biol. Chem. 273 27176-27181 [DOI] [PubMed] [Google Scholar]
- 11.Shinohara, T., Sakurada, C., Suzuki, T., Takeuchi, O., Campbell, W., Ikeda, S., Okada, N., and Okada, H. (1994) Int. Arch. Allergy Immunol. 103 400-404 [DOI] [PubMed] [Google Scholar]
- 12.Campbell, W. D., Lazoura, E., Okada, N., and Okada, H. (2002) Microbiol. Immunol. 46 131-134 [DOI] [PubMed] [Google Scholar]
- 13.Myles, T., Nishimura, T., Yun, T. H., Nagashima, M., Morser, J., Patterson, A. J., Pearl, R. G., and Leung, L. L. (2003) J. Biol. Chem. 278 51059-51067 [DOI] [PubMed] [Google Scholar]
- 14.Bajzar, L., Morser, J., and Nesheim, M. (1996) J. Biol. Chem. 271 16603-16608 [DOI] [PubMed] [Google Scholar]
- 15.Mao, S. S., Cooper, C. M., Wood, T., Shafer, J. A., and Gardell, S. J. (1999) J. Biol. Chem. 274 35046-35052 [DOI] [PubMed] [Google Scholar]
- 16.Wang, W., Nagashima, M., Schneider, M., Morser, J., and Nesheim, M. (2000) J. Biol. Chem. 275 22942-22947 [DOI] [PubMed] [Google Scholar]
- 17.Esmon, N. L., Owen, W. G., and Esmon, C. T. (1982) J. Biol. Chem. 257 859-864 [PubMed] [Google Scholar]
- 18.Kokame, K., Zheng, X., and Sadler, J. E. (1998) J. Biol. Chem. 273 12135-12139 [DOI] [PubMed] [Google Scholar]
- 19.Esmon, C. T. (1995) FASEB J. 9 946-955 [DOI] [PubMed] [Google Scholar]
- 20.Fuentes-Prior, P., Iwanaga, Y., Huber, R., Pagila, R., Rumennik, G., Seto, M., Morser, J., Light, D. R., and Bode, W. (2000) Nature 404 518-525 [DOI] [PubMed] [Google Scholar]
- 21.Parkinson, J. F., Nagashima, M., Kuhn, I., Leonard, J., and Morser, J. (1992) Biochem. Biophys. Res. Commun. 185 567-576 [DOI] [PubMed] [Google Scholar]
- 22.Tsiang, M., Lentz, S. R., and Sadler, J. E. (1992) J. Biol. Chem. 267 6164-6170 [PubMed] [Google Scholar]
- 23.Schneider, M., Nagashima, M., Knappe, S., Zhao, L., Morser, J., and Nesheim, M. (2002) J. Biol. Chem. 277 9944-9951 [DOI] [PubMed] [Google Scholar]
- 24.Walker, J. B., and Nesheim, M. E. (1999) J. Biol. Chem. 274 5201-5212 [DOI] [PubMed] [Google Scholar]
- 25.Bajzar, L., Nesheim, M. E., and Tracy, P. B. (1996) Blood 88 2093-2100 [PubMed] [Google Scholar]
- 26.Gils, A., Alessi, M. C., Brouwers, E., Peeters, M., Marx, P., Leurs, J., Bouma, B., Hendriks, D., Juhan-Vague, I., and Declerck, P. J. (2003) Arterioscler. Thromb. Vasc. Biol. 23 1122-1127 [DOI] [PubMed] [Google Scholar]
- 27.Boffa, M. B., Wang, W., Bajzar, L., and Nesheim, M. E. (1998) J. Biol. Chem. 273 2127-2135 [DOI] [PubMed] [Google Scholar]
- 28.Chen, C., and Okayama, H. (1987) Mol. Cell. Biol. 7 2745-2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu, C., Wu, F., Pan, J., Morser, J., and Wu, Q. (2003) J. Biol. Chem. 278 25847-25852 [DOI] [PubMed] [Google Scholar]
- 30.Schneider, M., Boffa, M., Stewart, R., Rahman, M., Koschinsky, M., and Nesheim, M. (2002) J. Biol. Chem. 277 1021-1030 [DOI] [PubMed] [Google Scholar]
- 31.Neville, D. M. (1971) J. Biol. Chem. 246 6328-6334 [PubMed] [Google Scholar]
- 32.Boskovic, D. S., Giles, A. R., and Nesheim, M. E. (1990) J. Biol. Chem. 265 10497-10505 [PubMed] [Google Scholar]
- 33.Hall, S. C., Nagashima, M., Zhao, L., Morser, J., and Leung, L. L. K. (1999) J. Biol. Chem. 274 25510-25516 [DOI] [PubMed] [Google Scholar]
- 34.Fan, B., Crews, B. C., Turko, I. V., Choay, J., Zettlmeissl, G., and Gettins, P. (1993) J. Biol. Chem. 268 17588-17596 [PubMed] [Google Scholar]
- 35.Gils, A., Ceresa, E., Macovei, A. M., Marx, P. F., Peeters, M., Compernolle, G., and Declerck, P. J. (2005) J. Thromb. Haemost. 3 2745-2753 [DOI] [PubMed] [Google Scholar]
- 36.Anand, K., Pallares, I., Valnickova, Z., Christensen, T., Vendrell, J., Wendt, K. U., Schreuder, H. A., Enghild, J. J., and Aviles, F. X. (2008) J. Biol. Chem. 283 29416-29423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marx, P. F., Brondijk, T. H., Plug, T., Romijn, R. A., Hemrika, W., Meijers, J. C., and Huizinga, E. G. (2008) Blood. 112 2803-2809 [DOI] [PubMed] [Google Scholar]
- 38.Boffa, M. B., Bell, R., Stevens, W. K., and Nesheim, M. E. (2000) J. Biol. Chem. 275 12868-12878 [DOI] [PubMed] [Google Scholar]
- 39.Marx, P. F., Hackeng, T. M., Dawson, P. E., Griffin, J. H., Meijers, J. C., and Bouma, B. N. (2000) J. Biol. Chem. 275 12410-12415 [DOI] [PubMed] [Google Scholar]
- 40.Wu, C., Dong, N., da, C, V., Martin-McNulty, B., Tran, K., Nagashima, M., Wu, Q., Morser, J., and Wang, Y. X. (2003) Thromb. Haemost. 90 414-421 [DOI] [PubMed] [Google Scholar]
- 41.Nagashima, M., Werner, M., Wang, M., Zhao, L., Light, D. R., Pagila, R., Morser, J., and Verhallen, P. (2000) Thromb. Res. 98 333-342 [DOI] [PubMed] [Google Scholar]
- 42.Refino, C. J., DeGuzman, L., Schmitt, D., Smyth, R., Jeet, S., Lipari, M. T., Eaton, D., and Bunting, S. (2000) Fibrinol. Proteol. 14 305-314 [Google Scholar]
- 43.Klement, P., Liao, P., and Bajzar, L. (1999) Blood 94 2735-2743 [PubMed] [Google Scholar]
- 44.Gomis-Ruth, F. X., Gomez, M., Bode, W., Huber, R., and Aviles, F. X. (1995) EMBO J. 14 4387-4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.