

Abstract
Galacto-N-biose/lacto-N-biose I phosphorylase (GLNBP) from Bifidobacterium longum, a key enzyme for intestinal growth, phosphorolyses galacto-N-biose and lacto-N-biose I with anomeric inversion. GLNBP homologues are often found in human pathogenic and commensal bacteria, and their substrate specificities potentially define the nutritional acquisition ability of these microbes in their habitat. We report the crystal structures of GLNBP in five different ligand-binding forms. This is the first three-dimensional structure of glycoside hydrolase (GH) family 112. The GlcNAc- and GalNAc-bound forms provide structural insights into distinct substrate preferences of GLNBP and its homologues from pathogens. The catalytic domain consists of a partially broken TIM barrel fold that is structurally similar to a thermophilic β-galactosidase, strongly supporting the current classification of GLNBP homologues as one of the GH families. Anion binding induces a large conformational change by rotating a half-unit of the barrel. This is an unusual example of molecular adaptation of a TIM barrel scaffold to substrates.
A unique metabolic pathway specific for galacto-N-biose (Gal-β1,3-GalNAc; GNB)2 and lacto-N-biose I (Gal-β1,3-Glc-NAc; LNB) has been identified in several strains of bifidobacteria (1). A key intracellular enzyme in the GNB/LNB pathway is a β-1,3-galactosyl-N-acetylhexosamine phosphorylase (Gal-HexNAcP; EC 2.4.1.211). GalHexNAcP catalyzes the phosphorolysis of GNB and LNB into α-d-galactose 1-phosphate (Gal1P) and corresponding N-acetylhexosamines with anomeric inversion (Fig. 1) (1-3). GalHexNAcPs from bifidobacteria exhibit comparable activities against GNB and LNB (1) and are, therefore, named galacto-N-biose/lacto-N-biose I phosphorylase (GLNBP) (4). GLNBP and its homologues are classified into glycoside hydrolase (GH) family 112 in the CAZy Database (available on the World Wide Web) (5).
FIGURE 1.

Schematic representation of the reaction catalyzed by GalHexNAcP.
Recently, two GH112 GalHexNAcPs from human pathogenic bacteria were reported to exhibit substrate specificities distinct from those of GLNBP. GalHexNAcPs from Clostridium perfringens ATCC13124 and Vibrio vulnificus CMCP6 are specific for GNB and LNB, respectively (4, 6). Therefore, they are named galacto-N-biose phosphorylase (GNBP) and lacto-N-biose I phosphorylase (LNBP), respectively. C. perfringens is found in the gastrointestinal tract of humans, and some strains cause gas gangrene, septicemia, and food poisoning (7, 8). V. vulnificus is often detected in the marine environment (9), and infection by this bacteria causes severe symptoms and high mortality in humans (10). The distinct substrate specificity of GalHexNAcPs in these commensal or pathogenic bacteria can be related to nutritional acquisition in their habitat (4).
Interestingly, most of the members of GH112 (GLNBP gene homologues) are found in human commensal or pathogenic bacteria as hypothetical proteins through genome projects. GNB and LNB structures are often found in biologically functional glycoconjugates of animals. GNB exists in O-glycans of mucin glycoproteins as core 1 or T-antigen disaccharide and also in glycosphingolipids (11-13). LNB exists in glycolipids as blood type antigens and human milk oligosaccharides (14-16). These two similar disaccharides are important oligosaccharides in cell surface glycoconjugates, and cancer-associated antigens have structures related to them (17-19). The presence of the genes in commensal and pathogenic bacteria suggests that the enzymes play an important role in their growth in human tissues by utilizing GNB and/or LNB as nutrients.
Bifidobacteria naturally colonize the human intestinal tract and have health-promoting effects, such as prevention of diarrhea (20-22), especially in infants (23-26). The GNB/LNB pathway of Bifidobacterium longum JCM1217 involves a GNB- and LNB-specific ABC transporter (27). In addition, the pathway includes a set of intracellular enzymes to send the entire molecules of the reaction product of GLNBP (Gal1P, GalNAc, and GlcNAc) into the glycolytic and amino sugar metabolic pathways (28). Before the cellular uptake, GNB and LNB are liberated from their natural substrates by the bifidobacterial extracellular enzymes, endo-α-N-acetylgalactosaminidase (29), 1,2-α-l-fucosidase (30), and lacto-N-biosidase (31). We have hypothesized that a key role of this pathway is to metabolize LNB in human milk oligosaccharides (1, 28). In contrast to the gut contents of formula-fed infants, the guts of breast-fed infants generally contain microflora dominated by bifidobacteria (32-35). Therefore, the selective growth of bifidobacteria observed in breast-fed infants has been attributed to human milk oligosaccharides (32, 36-38), and our hypothesis explains its molecular basis.
The molecular scaffold and mechanisms of sugar phosphorylases are interesting, because they have been converged from various evolutionary origins. For example, pyridoxal phosphate-dependent glycogen phosphorylase shares structural and mechanistic similarities with typical NDP-dependent glycosyltransferases (GTs) (39) and is classified in the GT35 family. In contrast, some phosphorylases are very similar to standard GHs in their structures and reaction mechanisms (40). Therefore, sugar phosphorylases are classified in both the GH and GT classes in the CAZy Database. Another important aspect of phosphorylases is their ability to produce oligosaccharides due to the reversible nature of their reactions (41). GLNBP is actually used to prepare kilogram quantities of LNB (42). As a candidate for the bona fide bifidus factor, LNB attracts considerable interest in applications to improve infant health. In this study, we determined the crystal structures of GLNBP from B. longum JCM1217 in several ligand-binding forms and elucidated the molecular mechanism of the reaction and structural basis for substrate specificity of GalHexNAcPs. This is the first three-dimensional structure of a GH112 enzyme.
EXPERIMENTAL PROCEDURES
Enzyme Preparation—The nonlabeled GLNBP protein was expressed in Escherichia coli strain BL21 (DE3) and purified as described previously (2). The selenomethionine-labeled enzyme was expressed in the methionine auxotroph E. coli strain B834 (DE3) (Novagen). The purification procedures for the selenomethionine-labeled enzyme were the same as those for the nonlabeled enzyme. The point mutations of R32E, N166A, R210E, R358E, Y362F, Y362N, and F364N were made using the QuikChange site-directed mutagenesis kit (Stratagene). The oligonucleotide primers are listed in Table S1. Preparation, purification, and activity measurements of the mutant enzymes were done as described previously (2).
Crystallization and Data Collection—Ligand-free and selenomethionine-labeled GLNBP crystals were obtained at 4 °C using the sitting drop vapor diffusion method by mixing 2 μl of a protein solution with 2 μl of a reservoir solution composed of 0.1 m sodium cacodylate (pH 6.5), 0.2 m Mg(NO3)2, and 15% (v/v) polyethylene glycol 4000. Rodlike crystals (0.1 × 0.1 × 0.2 mm) grew within 3-7 days. The GalNAc complex form crystals were obtained by co-crystallization using a reservoir solution containing 100 mm GalNAc. The GlcNAc complex and GlcNAc-NO3-ethylene glycol (EG) complex form crystals were obtained by co-crystallization using a reservoir solution containing 100 mm GlcNAc. Crystals of the GlcNAc-SO4 complex were obtained by co-crystallization using a reservoir solution containing 100 mm GlcNAc and 50 mm MgSO4. The crystals were transferred to a reservoir solution containing 15% (v/v) glycerol (selenomethionine, ligand-free, GalNAc or GlcNAc complex, and GlcNAc-SO4) or 15% (v/v) EG (GlcNAc-NO3-EG) and then flash-cooled in a stream of cold nitrogen gas at 95 K. The data set for the selenomethionine-labeled crystal was collected at wavelengths of 0.9645 Å (remote), 0.9793 Å (peak), and 0.9797 Å (edge). X-ray diffraction data sets were collected using synchrotron radiation (beamline BL-5A, BL-17A, and NW12A; Photon Factory, Tsukuba, Japan). The data sets were processed and scaled using HKL2000 (43). The statistics for data collection and processing are given in Table 1.
TABLE 1.
Data collection statistics
The numbers in parentheses correspond to the data shell at the highest resolution.
|
Crystals
|
Selenomethionine-labeled
|
Ligand-free
|
GalNAc
|
GlcNAc
|
GlcNAc-NO3-EG
|
GlcNAc-SO4
|
||
|---|---|---|---|---|---|---|---|---|
| Peak | Edge | Remote | ||||||
| Wavelength (Å) | 0.97934 | 0.97974 | 0.96450 | 1.00000 | 1.00000 | 1.00000 | 1.00000 | 1.00000 |
| Beamline | BL5A | BL5A | BL5A | BL5A | BL17A | NW12A | NW12A | NW12A |
| Space group | C2 | P1 | P1 | P1 | C2 | P1 | ||
| Unit cell parameters | ||||||||
| a (Å) | 212.2 | 67.8 | 67.9 | 67.8 | 204.0 | 67.9 | ||
| b (Å) | 67.8 | 111.5 | 111.7 | 111.5 | 68.0 | 112.0 | ||
| c (Å) | 118.2 | 118.8 | 118.7 | 118.4 | 116.0 | 118.7 | ||
| α (degrees) | 90.0 | 105.1 | 105.2 | 105.2 | 90.0 | 105.2 | ||
| β (degrees) | 106.6 | 90.2 | 90.5 | 90.5 | 108.8 | 90.4 | ||
| γ (degrees) | 90.0 | 107.8 | 107.3 | 107.3 | 90.0 | 107.3 | ||
| Resolution (Å) | 50.00-2.80 | 50.00-2.10 | 50.00-1.90 | 50.00-2.30 | 50.00-1.85 | 50.0-2.10 | ||
| (2.90-2.80) | (2.18-2.10) | (1.97-1.90) | (2.38-2.30) | (1.92-1.85) | (2.18-2.10) | |||
| Measured reflections | 283,825 | 279,509 | 274,172 | 344,949 | 640,661 | 269,456 | 529,437 | 713,861 |
| Unique reflections | 40,008 | 39,999 | 40,119 | 178,361 | 243,435 | 137,195 | 126,352 | 180,455 |
| Completeness (%) | 99.7 (98.5) | 99.7 (98.2) | 98.9 (90.9) | 97.7 (95.9) | 97.0 (96.0) | 97.8 (97.5) | 98.5 (97.7) | 98.2 (97.1) |
| Redundancy | 6.9 (4.5) | 7.0 (5.8) | 6.9 (4.5) | 1.9 (1.9) | 2.6 (2.6) | 2.0 (2.0) | 3.8 (3.9) | 4.0 (4.0) |
| Mean I/σ | 17.9 (3.0) | 21.1 (2.4) | 17.9 (2.2) | 15.8 (3.2) | 21.0 (3.5) | 15.3 (2.6) | 21.8 (2.7) | 15.9 (2.8) |
| Rmerge (%) | 8.3 (36.6) | 7.0 (30.3) | 8.3 (36.6) | 5.2 (21.2) | 5.7 (23.2) | 5.4 (24.6) | 6.8 (25.0) | 8.9 (32.7) |
Phase Calculation and Refinement—The autoSHARP (44, 45) and SOLVE/RESOLVE (46) programs were used for site detection of selenium, phase calculation, and initial model building of the multiple-wavelength anomalous dispersion data set. The resulting initial model was used as a search model for molecular replacement of the nonlabeled GLNBP data set with the MOLREP program (47). Visual inspection of the models, introduction of water molecules, and crystallographic refinement were achieved using Coot (48) and Refmac5 (49). The refinement statistics and contents in the asymmetric unit (ASU) are given in Table 2. The figures were prepared using PyMol (50), and the structural alignment was performed by LSQMAN (51). Pairwise structural comparisons were carried out using the whole structure, whereas the closed state and other structures were superimposed by excluding the half-barrel unit and C-terminal domain.
TABLE 2.
Refinement statistics and contents in the asymmetric unit
| Crystal | Ligand-free | GalNAc | GlcNAc | GlcNAc-NO3-EG | GlcNAc-SO4 |
|---|---|---|---|---|---|
| Refinement statistics | |||||
| Protein Data Bank code | 2ZUS | 2ZUT | 2ZUU | 2ZUV | 2ZUW |
| Resolution range (Å) | 32.1-2.11 | 44.8-1.90 | 40.7-2.30 | 35.1-1.85 | 41.1-2.11 |
| R-factor/Rfreea (%) | 17.0/22.5 | 16.1/20.4 | 17.0/23.1 | 14.9/19.2 | 16.9/22.5 |
| r.m.s. deviation bond lengths (Å) | 0.014 | 0.014 | 0.010 | 0.014 | 0.013 |
| r.m.s. deviation bond angles (degrees) | 1.3 | 1.4 | 1.2 | 1.3 | 1.3 |
| Average B-factor (Å2) | |||||
| Protein (chain A/B/C/D) | 25.4/30.9/26.8/27.5 | 24.7/30.4/24.8/28.4 | 32.1/39.0/31.9/37.2 | 20.6/20.2 | 22.3/28.7/22.9/27.6 |
| Ligands in each active site | - | 22.2/24.0/21.3/23.0 | 26.2/32.7/29.3/30.5 | 20.0/14.6 | 19.2/22.6/20.0/34.2 |
| Water | 31.5 | 36.0 | 37.1 | 33.6 | 32.4 |
| Ramachandran plot (% in favored/allowed/disallowed regions) | |||||
| Chain A | 97.1/2.3/0.7 | 96.6/3.0/0.4 | 96.5/3.1/0.4 | 97.3/2.6/0.1 | 96.8/3.2/0 |
| Chain B | 96.2/3.4/0.4 | 96.4/3.6/0 | 94.9/4.6/0.6 | 96.6/3.3/0.1 | 95.9/3.7/0.4 |
| Chain C | 96.5/3.0/0.5 | 97.0/2.8/0.1 | 96.6/3.4/0 | 96.5/3.4/0.1 | |
| Chain D | 96.6/3.4/0 | 96.5/3.4/0.1 | 95.4/4.5/0.1 | 95.1/4.3/0.6 | |
| ASU content | |||||
| No. of protein atoms | 23,724 | 23,573 | 23,549 | 11,787 | 23,395 |
| No. of solvent atoms | 1,898 | 3,114 | 1,638 | 1,826 | 2,281 |
| No. of heteroatoms | 2 | 124 | 85 | 94 | 106 |
| No. of ligands | |||||
| Mg2+ | 2 | 6 | 1 | 8 | 1 |
| GalNAc/GlcNAc | 4 (GalNAc) | 4 (GlcNAc) | 2 (GlcNAc) | 4 (GlcNAc) | |
| Glycerol/EG | 9 (glycerol) | 4 (glycerol) | 9 (EG) | 6 (glycerol) | |
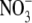 / /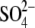
|
1 (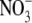 ) )
|
1 (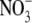 ) )
|
1 (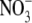 ) + 1
( ) + 1
(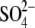 ) )
|
||
| Model built/Conformationb/Ligands in active site | |||||
| Chain A | 2-750/open/NAc | 3-34, 43-754/semiclosed/GalNAc | 3-34, 41-754/semiclosed/GlcNAc | 3-35, 42-749/closed/GlcNAc, EG, 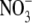
|
3-33, 40-753/semiclosed/GlcNAc |
| Chain B | 3-34, 37-41, 43,44, 47-751/open/NA | 3-34, 47-749/semiclosed/GalNAc | 3-33, 47-367, 371-749/semiclosed/GlcNAc | 3-33, 42-750/semiclosed/GlcNAc | 3-32, 40-366, 371-751/semiclosed/GlcNAc |
| Chain C | 3-35, 38-40, 43-750/open/NA | 3-33, 43-754/semiclosed/GalNAc | 3-34, 43-754/semiclosed/GlcNAc | 3-35, 40-754/semiclosed/GlcNAc | |
| Chain D | 3-35, 39-750/open/NA | 334, 43750/semiclosed/GalNAc | 3-33, 43-750/semiclosed/GlcNAc |
5-12, 22-33, 46-372, 375-400, 407-427, 433-751/closed/GlcNAc,
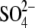
|
Rfree = ∑hkl||Fo| - |Fc||/∑hkl|Fo|, where the crystallographic R-factor was calculated including and excluding refinement reflections. The free reflections constituted 5% of the total number of reflections.
See “Results” for designation of conformations.
NA, not applicable.
Docking Studies—The AUTO-DOCK program version 4.0
(52) was used for the
automated docking of ligands to the GLNBP active site. The LNB, GNB, and Gal1P
ligand models were prepared with the PCModel program (Serena Software,
Bloomington, IN) and optimized using the MMX force field. Rotatable ligand
bonds (13 in LNB and GNB and five in Gal1P) were defined using the
AutoDockTools interface. The closed state subunit of the
GlcNAc-NO3-EG form was prepared for docking by removing the water
molecules except for those included in the GlcNAc/GalNAc interactions.
Retaining these water molecules (shown in
Fig. 3) was essential for
GlcNAc docking into the subsite (+1). EG and GlcNAc were removed for LNB/GNB
docking, and EG and 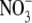 were removed
for Gal1P docking, respectively. One of the two alternative conformations of
the Arg-32 side chain in the closed state subunit of the
GlcNAc-NO3-EG form was selected based on its conformation in the
GlcNAc-SO4 form. After adding polar hydrogens, Gasteiger charges
were calculated for the ligand and protein. Grid maps were prepared with 40
× 40 × 40 points for LNB and GNB covering subsites -1 and +1 and
ranges at 40 × 30 × 30 points for Gal1P covering subsite -1 and
the anion binding site with a point spacing of 0.375 Å. The Lamarckian
genetic algorithm of the AutoDock 4.0 package performed 200 iterations with a
maximum of 27,000 generations per iteration, a population size of 50
individuals, and a maximum of 2.5 × 106 energy evaluations
per generation. The results of the iterations were clustered so that no
cluster member deviated >1.0 Å from other cluster members. After
docking, all structures generated for a single compound were assigned to
clusters based on a tolerance of 1.0 Å for all atom root mean square
(r.m.s.) deviations from the lowest-energy structure. The results of cluster
analyses are shown in Table S2. The best docking results with the lowest
Einter values are shown in
Fig. 3C and Fig.
S2C.
were removed
for Gal1P docking, respectively. One of the two alternative conformations of
the Arg-32 side chain in the closed state subunit of the
GlcNAc-NO3-EG form was selected based on its conformation in the
GlcNAc-SO4 form. After adding polar hydrogens, Gasteiger charges
were calculated for the ligand and protein. Grid maps were prepared with 40
× 40 × 40 points for LNB and GNB covering subsites -1 and +1 and
ranges at 40 × 30 × 30 points for Gal1P covering subsite -1 and
the anion binding site with a point spacing of 0.375 Å. The Lamarckian
genetic algorithm of the AutoDock 4.0 package performed 200 iterations with a
maximum of 27,000 generations per iteration, a population size of 50
individuals, and a maximum of 2.5 × 106 energy evaluations
per generation. The results of the iterations were clustered so that no
cluster member deviated >1.0 Å from other cluster members. After
docking, all structures generated for a single compound were assigned to
clusters based on a tolerance of 1.0 Å for all atom root mean square
(r.m.s.) deviations from the lowest-energy structure. The results of cluster
analyses are shown in Table S2. The best docking results with the lowest
Einter values are shown in
Fig. 3C and Fig.
S2C.
FIGURE 3.

Stereoviews of the interactions with the crystallographic (A and B) and docked (C) ligand molecules. The ligand molecules are shown in the ball-and-stick model with the |Fo| - |Fc| electron density maps (4.0σ in A and 3.5σ in B). Water molecules involved in ligand interactions are shown as spheres (cyan). The broken lines indicate hydrogen bonds. The labels of residues subjected to mutational analysis and from the other subunit are colored red and blue, respectively. A, GalNAc complex in the semiclosed state. B, GlcNAc-NO3-EG in the closed state. C, the best docking result of LNB into the closed subunit of GlcNAc-NO3-EG.
RESULTS
Crystallography—Five refined crystal structures are
presented here, and they are hereafter designated according to the ligands
found in the active site: ligand-free form; GalNAc complex; GlcNAc complex;
quaternary complex with GlcNAc, nitrate, and ethylene glycol
(GlcNAc-NO3-EG); and ternary complex with GlcNAc and
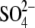 (GlcNAc-SO4)
(Table 2). These structures
also contain crystallization reagents (Mg2+ and
(GlcNAc-SO4)
(Table 2). These structures
also contain crystallization reagents (Mg2+ and
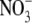 ) and cryoprotectants (glycerol or
EG) at their molecular surfaces. Ligand-free, GalNAc complex, GlcNAc complex,
and GlcNAc-SO4 crystals belong to space group P1 and
contain four subunits per ASU, whereas GlcNAc-NO3-EG belongs to
space group C2 and contains two subunits per ASU. The ASU contents in
these crystal structures are summarized in
Table 2. In all crystal forms,
each of the two molecules forms a dimer, as shown in
Fig. 2A. This should
correspond to the dimeric form of this enzyme in solution
(1).
) and cryoprotectants (glycerol or
EG) at their molecular surfaces. Ligand-free, GalNAc complex, GlcNAc complex,
and GlcNAc-SO4 crystals belong to space group P1 and
contain four subunits per ASU, whereas GlcNAc-NO3-EG belongs to
space group C2 and contains two subunits per ASU. The ASU contents in
these crystal structures are summarized in
Table 2. In all crystal forms,
each of the two molecules forms a dimer, as shown in
Fig. 2A. This should
correspond to the dimeric form of this enzyme in solution
(1).
FIGURE 2.

Overall structure of GLNBP. The TIM barrel fold domain
(blue), Ig-like fold domain (green), α/β fold
domain (yellow), and C-terminal domain (red) are shown.
A, dimeric structure of GlcNAc-NO3-EG crystal form.
Subunits in the closed and semiclosed states are shown in a ribbon
model. The semiclosed state subunit is shown with a different color
code (TIM barrel domain in light blue, Ig-like domain in
light green, α/β domain in brown, and C-terminal
domain in pink). The ligand-free form in the open state (subunit A)
(B) and GlcNAc-NO3-EG in the closed state (subunit A)
(C) are shown from the same view as A. Ligands in the active
site (GlcNAc, ethylene glycol, and 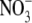 )
are shown as a space-filling model. Asp-313 and Trp-233 are shown as
a stick model (cyan), and the Cα atom of Gly-371 is
shown as a red sphere.
)
are shown as a space-filling model. Asp-313 and Trp-233 are shown as
a stick model (cyan), and the Cα atom of Gly-371 is
shown as a red sphere.
Overall Structure of Ligand-free Form—Fig. 2B shows a ribbon diagram of the monomer structure of ligand-free form. The GLNBP monomer consists of four domains. The first is the (β/α)8 barrel (TIM barrel) domain (residues 3-70 and 181-437; blue). The second domain, the Ig-like fold domain (residues 71-180; green), is inserted between β-3 and α-3 of a TIM barrel (Fig. S1). The remaining two regions form the α/β fold (residues 438-695; yellow) and the C-terminal β-sheet (residues 696-750; red) domains. The dimer interface is mainly formed by Ig-like and α/β fold domains (Fig. 2A), and the buried molecular surface area is about 2,060 Å2/subunit. Structures of the four subunits in the ASU are almost identical; the r.m.s. deviations for Cα atoms between all pairs are within 0.5 Å, except for the relatively flexible half-barrel unit and the C-terminal domain (for discussion, see below).
GalNAc and GlcNAc Complex—In the GalNAc complex form, each subunit in the ASU holds a GalNAc molecule (Table 2). It is bound at the center of the C-terminal loop side of the TIM barrel domain. All of the four GalNAc molecules in the ASU are in an α-anomeric state and take a standard 4C1 conformation (Fig. 3A). In addition to the hydrophobic interactions that recognize the pyranose moiety of GalNAc, two direct and several water-mediated hydrogen bonds are present. One direct hydrogen bond is formed between the O3 atom of GalNAc and the side chain of Asp-313. This residue has been proposed to be the catalytic proton donor residue based on a mutational analysis (2). Another direct hydrogen bond is formed between the carbonyl oxygen atom of the N-acetyl group and the Nε atom of the Trp-233 side chain. Most of the water-mediated hydrogen bonds are formed at one side of the GalNAc molecule (O1, O5, and O6). The water molecules are held by the side chains of Tyr-165, Glu-228, and Ser-612 (from the other subunit), and by the main chain carbonyl oxygen atom of His-460.
The overall structure of the GalNAc complex is very similar to that of the ligand-free form (Fig. S2B). In the complex form, however, an α-helix region in the Ig-like domain (residues 160-170) shifts toward the ligand (Fig. 4C, green), and the Cα displacement is 1.5 Å at Val-162 (Fig. 4A). This region participates in the active site formation and partially covers the substrate-binding pocket. Hereafter, this region is referred to as “160-170 helix.” The structural change is accompanied by a side chain switch of Pro-161 and Gln-217 at the root of the 160-170 helix (Fig. 4C). Because this conformation exhibits slight but clear active site closure compared with an “open” state of the ligand-free form, we designate it as the “semiclosed” state.
FIGURE 4.

Conformational differences in the three states of GLNBP. A, Cα difference plot between open and semiclosed states (blue) and between open and closed states (red). The red bars indicate the highly flexible regions (half-barrel unit and C-terminal domain colored as in Fig. 1A). B, overall structural changes of TIM barrel domain between open (ligand-free form; black) and closed (GlcNAc-NO3-EG form; light gray and red) states. A half-barrel unit in the closed state is colored red. Side chains of Tyr-362, Phe-364, and Asp-313 (general acid) are shown. The Cα atom of Gly-371 is shown as a sphere. C, stereoview of the active site of the semiclosed state of the GalNAc complex (yellow and green) superimposed on the open conformation subunit (black). The 160-170 helix is colored green. The residues labeled with red characters were subjected to mutational analysis. D, stereoview of the active site of GlcNAc-NO3-EG in the closed state (yellow and orange) superimposed on the subunit in the open conformation (black). The mobile half-barrel unit is colored orange.
We have also determined the complex structure of GlcNAc, which is the O4 epimer of GalNAc. The binding interactions of the GlcNAc molecule as well as the overall structure are almost identical with those of the GalNAc complex (Fig. S3A).
GlcNAc-NO3-EG Complex—In the
GlcNAc-NO3-EG complex form, the two subunits in the ASU form a
dimer (Fig. 2A). One
of the two subunits takes a semiclosed state whose active site pocket contains
a GlcNAc molecule, as in the GalNAc and GlcNAc complex forms. The other
subunit contains EG and 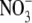 as well as
a GlcNAc molecule in the active site pocket
(Fig. 3B). In
comparison with the ligand-free form, this subunit exhibits a large domain
closure at the substrate binding site (Figs.
2C and
4B). The
conformational change is prominent, because about 50% of the TIM barrel
elements (half-barrel unit, β1-α1-β2-α2-β3, and
β7-α7-β8-α8 formed by residues 3-50 and 355-437) and the
C-terminal domain (residues 696-750) shift toward the ligand molecules (r.m.s.
deviation = 2.7 Å; Fig.
4A). The half-barrel unit rotates about 10° around a
pivot point located at the bottom of the TIM barrel
(Fig. 4B). The maximal
difference is present at Gly-371 (6.8 Å), which is located at the
C-terminal loop of the half-barrel unit. Hereafter, this conformation is
referred to as the “closed” state. Note that the barrel structure
of the closed state is highly deformed
(Fig. 2C), whereas the
β-strands in the “open” state form an almost perfect circle
(Fig. 2B). In this
crystal form, the dimer is clearly asymmetric, since the two subunits are in a
distinct conformational state. Moreover, the two active sites are clearly
separated, and there is no apparent interaction between them (Fig.
S2A). These results are in agreement with the nonallosteric behavior
of this enzyme.
as well as
a GlcNAc molecule in the active site pocket
(Fig. 3B). In
comparison with the ligand-free form, this subunit exhibits a large domain
closure at the substrate binding site (Figs.
2C and
4B). The
conformational change is prominent, because about 50% of the TIM barrel
elements (half-barrel unit, β1-α1-β2-α2-β3, and
β7-α7-β8-α8 formed by residues 3-50 and 355-437) and the
C-terminal domain (residues 696-750) shift toward the ligand molecules (r.m.s.
deviation = 2.7 Å; Fig.
4A). The half-barrel unit rotates about 10° around a
pivot point located at the bottom of the TIM barrel
(Fig. 4B). The maximal
difference is present at Gly-371 (6.8 Å), which is located at the
C-terminal loop of the half-barrel unit. Hereafter, this conformation is
referred to as the “closed” state. Note that the barrel structure
of the closed state is highly deformed
(Fig. 2C), whereas the
β-strands in the “open” state form an almost perfect circle
(Fig. 2B). In this
crystal form, the dimer is clearly asymmetric, since the two subunits are in a
distinct conformational state. Moreover, the two active sites are clearly
separated, and there is no apparent interaction between them (Fig.
S2A). These results are in agreement with the nonallosteric behavior
of this enzyme.
Interactions with the GlcNAc molecule are virtually the same as in the
semiclosed state (Fig.
3B). One of the hydroxyl groups in EG forms a hydrogen
bond with the side chain of Asn-166, which is located in the 160-170 helix.
The hydrophobic part of EG interacts with the Tyr-362 and Phe-364 side chains
at the C terminus of β7 in the TIM barrel. The movement of the
half-barrel unit allows these hydrophobic residues to approach the EG binding
site. The binding site of 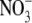 is
surrounded by three arginine residues (Arg-32, Arg-210, and Arg-358), and they
form direct hydrogen bonds with
is
surrounded by three arginine residues (Arg-32, Arg-210, and Arg-358), and they
form direct hydrogen bonds with 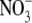 .
Arg-32 have two alternative conformations
(Fig. 4D), one of
which directly interacts with
.
Arg-32 have two alternative conformations
(Fig. 4D), one of
which directly interacts with 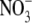 .
.
GlcNAc-SO4 Complex—The crystal of the
GlcNAc-SO4 complex is obtained by co-crystallization with 50
mm MgSO4 and 100 mm
Mg(NO3)2. Three of the four subunits in the ASU are in
the semiclosed state with a GlcNAc molecule in the active site
(Table 2), and one subunit is
in the closed state. However, a part of the electron density corresponding to
the half-barrel unit is disordered (Fig. S2C). A strong electron
density peak is present near the GlcNAc molecule in the active site of the
subunit in the closed state (Fig. S3B). Considering the shape and
height of the peak, the electron density peak definitely comes from
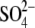 . The
. The
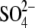 binding site corresponds to the
binding site corresponds to the
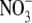 binding site in the
GlcNAc-NO3-EG complex form. The
binding site in the
GlcNAc-NO3-EG complex form. The
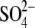 forms hydrogen bonds with the side
chain hydroxyl group of Tyr-362 as well as with the three arginine residues.
In contrast to the GlcNAc-NO3-EG complex form, Arg-32 in this form
is clearly in a single conformation. The tetrahedral structure of
forms hydrogen bonds with the side
chain hydroxyl group of Tyr-362 as well as with the three arginine residues.
In contrast to the GlcNAc-NO3-EG complex form, Arg-32 in this form
is clearly in a single conformation. The tetrahedral structure of
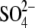 seems to be preferable to the
planar triangular structure of
seems to be preferable to the
planar triangular structure of 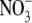 for
this binding site. These results suggest that the
for
this binding site. These results suggest that the
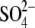 binding site is also suitable for
phosphate binding.
binding site is also suitable for
phosphate binding.
Docking Analysis—To investigate the possible interactions
with the galactose moiety of natural substrates or products, we performed
automated docking analysis of LNB and Gal1P in the closed state structure of
the GlcNAc-NO3-EG complex form. Several water molecules in the
substrate binding pocket are not removed to maintain the interactions around
the ligands. The best results of the docking analysis with LNB and Gal1P are
shown in Fig. 3C and
Fig. S3C. The GlcNAc moiety of the docked LNB almost entirely
overlaps with the crystallographic GlcNAc molecule (r.m.s. deviation = 0.43
Å). The galactose moiety of LNB also overlaps with the EG molecule in
the crystal structure, suggesting that this site is subsite -1. This moiety is
recognized by the side chains of Asn-166, Asp-313, Tyr-362, and Phe-364. The
former two residues form bifurcated hydrogen bonds with the equatorial O3 and
axial O4 atoms of the galactose moiety, whereas the latter two create
hydrophobic interactions. When Gal1P is docked, the galactose and phosphate
moieties are positioned at the binding sites for EG and
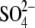 , respectively. The side chain of
Asn-166 again forms bifurcated hydrogen bonds, suggesting that this residue is
important for recognizing the galactose moiety. The docking analysis with GNB
produced a very similar result with LNB under the same conditions (data not
shown). We could not obtain any reliable models when the open and semiclosed
state structures were subjected to the docking analysis (data not shown).
, respectively. The side chain of
Asn-166 again forms bifurcated hydrogen bonds, suggesting that this residue is
important for recognizing the galactose moiety. The docking analysis with GNB
produced a very similar result with LNB under the same conditions (data not
shown). We could not obtain any reliable models when the open and semiclosed
state structures were subjected to the docking analysis (data not shown).
Mutational Analysis—Kinetic parameters of the synthetic reaction of mutant enzymes were measured to confirm the importance of the residues in the ligand binding sites (Table 3). Mutants at the three arginine residues in the anion binding site (R32E, R210E, and R358E) showed no detectable activity, indicating that they are all critical for the reaction. Mutations at the putative galactose binding site (N166A, Y362F, Y362N, and F364N) also severely impaired the activity. In all of the mutant enzymes, the Km values against Gal1P increased, and the kcat values significantly decreased. Elimination of the side chain of Tyr-362 (Y362N) resulted in the complete loss of the activity, and the loss of the hydroxyl group (Y362F) resulted in about 1,000-fold reduction of the catalytic efficiency. These results coincide with the docking results, since Tyr-362 is suggested to hydrophobically interact with the galactose pyranose ring as well as a hydrogen bond with phosphate. The activity of a mutant at the putative proton donor (D313N) was undetectable (2). All of these residues (Arg-32, Asn-166, Arg-210, Asp-313, Arg-358, Tyr-362, and Phe-364) are fully conserved in GH112 GalHexNAcPs (Fig. S1).
TABLE 3.
Kinetic parameters of the wild-type and mutant enzymes in the synthesis reaction with GIcNAc as an acceptor
| kcat | Km(Gal1P) | kcat/Km(Gal1P) | |
|---|---|---|---|
| s−1 | mm | mm·s−1 | |
| Wild type | 10.7 | 1.72 | 6.21 |
| R32E | NDa | ||
| N166A | 0.30 (0.02)b | 24 (13) | 0.012 (0.002) |
| R210E | ND | ||
| R358E | ND | ||
| Y362F | 0.09 (0.008) | 17 (10) | 0.005 (0.0008) |
| Y362N | ND | ||
| F364N | 4.83 (0.4) | 19 (10) | 0.260 (0.04) |
ND, activity not detected.
Numbers in parentheses correspond to the ratio compared with the wild type enzyme.
DISCUSSION
Reaction Mechanism of GLNBP—GLNBP forms a ternary complex
with two substrates (i.e. phosphate and GNB/LNB) or two products
(i.e. Gal1P and GalNAc/GlcNAc) during the reaction, because its
phosphorolytic reaction follows the sequential bi-bi mechanism
(6). Our results strongly
suggest that the EG and anion binding sites correspond to those of galactose
and phosphate, respectively. The anion binding site is in close proximity to
the anomeric carbon of the galactose moiety of the docked LNB. In addition, a
hydrogen bond is formed between the glycosidic bond oxygen and the Asp-313
side chain (Fig. 3), which has
been suggested to be the catalytic proton donor
(2). These structural features
suggest that the inverting phosphorolytic reaction begins with a direct
nucleophilic attack by phosphate on the anomeric carbon, but the evidence for
the mechanism has yet to be obtained. This type of mechanism has also been
proposed for cellobiose phosphorylase, the other inverting sugar phosphorylase
in GH94
(53-55).
In the crystal structures of cellobiose phosphorylase, subsite +1 is occupied
by glucose, and the adjacent subsite -1 and phosphate binding site are
occupied by glycerol and 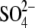 or
phosphate, respectively.
or
phosphate, respectively.
Efficient catalysis on the hydrolytic or phosphorolytic cleavage of β-glucosidic bonds is considered to require distortion of the glycon sugar ring (55-58). In this study, we tried to dock LNB with several distorted conformers at its glycon (e.g. 1S3, 1S5, and 2So at the galactosyl sugar ring), but we could not obtain any reasonably docked models. The docking procedures used here may not be sensitive enough to determine the most probable pretransition state conformer, because the structural differences are small between these conformers, or some rearrangements of the GLNBP active site may be required to accept a distorted galactose moiety. Alternatively, the conformational itinerary of β-galactosidases (or phosphorylases acting on β-galactosyl bonds) may be different from those of β-glucosidases because of potential crashes between the axial C4 hydroxyl and β-anomeric substituents. Espinosa et al. (59) conducted an NMR study of a nonhydrolyzable C-glycoside analogue of lactose bound to E. coli GH2 β-galactosidase. They suggested that the galactopyranose ring of the bound analog is not in a distorted conformation (e.g. a half-chair or sofa), because the intensities of the H-1′/H-3′ or H-1′/H-5′ intraresidue nuclear Overhauser effects were strong. Conformational changes on the β-galactosidic bond cleavage reactions need to be further studied.
Substrate Specificity Determinants of GalHexNAcPs—Major determinants of the substrate specificities of GalHex-NAcPs are envisaged to be located around the GalNAc/GlcNAc binding site (subsite +1). In particular, the Val-162 residue of GLNBP, which is located very close to the axial O4 atom of GalNAc (Fig. 3A), is the primary candidate for the determinants. GNBP shows strong preference for GNB (Km = 1.9 mm) compared with LNB (Km = 26 mm) (6). It has a threonine residue at the corresponding position of Val-162 in GLNBP (Fig. S1). Therefore, there would be a favorable polar interaction between the side chain of the threonine and the GalNAc axial O4 atom in GNBP. The factors for LNB preferences in LNBP are more complicated, because both GLNBP and LNBP retain a valine residue at this position. One possibility is that large insertions and deletions in the α/β domain affect the substrate specificity of LNBP (Fig. S1). His-460 and Ser-612 of GLNBP, which are involved in subsite +1 recognition (Figs. 3 and 4, C and D), are both located in the α/β domain. Perhaps the insertions and deletions alter the tertiary and/or quaternary structures around the sugar binding site and prevent GNB binding to LNBP. In the future, many more putative GalHexNAcP (GH112 GLNBP homologue) genes will be found, since a number of genomic and metagenomic projects on human-related microbes are now in progress worldwide (e.g. through the International Human Microbiome Project) (60). Our study will provide useful information on the possible substrate preferences for those putative GalHexNAcPs retained by microbes living around human bodies.
Structural Neighbor of GLNBP—A structural homology search
using the Secondary Structure Matching server
(61) indicated that GLNBP is
most similar to a GH42 β-galactosidase from Thermus thermophilus
A4, A4-β-galactosidase (Protein Data Bank code 1KWK, Q score =
0.110, r.m.s. deviation = 3.73 Å for 370 residues)
(62). A4-β-galactosidase
consists of three domains: a TIM barrel, an α/β fold, and
β-sheet domains. The structural similarity between GLNBP and
A4-β-galactosidase ranges from the TIM barrel domain to the subsequent
α/β fold domain (Fig. S4). Superimposition of the TIM barrel domain
alone is shown in Fig.
5A (Q score = 0.150, r.m.s. deviation = 2.90
Å for 242 Cα atoms). Their TIM barrel domains have a similar
“comma-like” shape with a long α-4 helix. A close-up view of
the superimposition at the active site is shown in
Fig. 5B.
A4-β-galactosidase is a retaining GH, and the two catalytic residues,
acid/base (Glu-141) and nucleophile (Glu-312), are located on the opposite
side of the bound galactose. Interestingly, the catalytic components of
A4-β-galactosidase overlap well with the general acid residue (Asp-313),
the phosphate binding site, and the subsite -1 (EG binding site) of GLNBP. The
side chain positions of Asp-313 in GLNBP and Glu-141 in
A4-β-galactosidase coincide, although they come from neighboring
β-strands in the barrel scaffold; the former is from β5, and the
latter is from β4. On the opposite side, the bound anion
(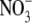 ) of GLNBP overlaps with the side
chain carboxyl group of the nucleophile residue of A4-β-galactosidase
(Glu-312). An arginine residue involved in anion binding (Arg-358) and Glu-312
of A4-β-galactosidase are both located in the same position at the
β7 strand of the barrel scaffold. Another interesting overlap was
observed between Arg-32 of GLNBP (anion binding) and Arg-32 in
A4-β-galactosidase, both being located at the same position in the barrel
(β2 strand). Arg-32 in A4-β-galactosidase is a highly conserved
residue and is considered to control the pKa of the
nucleophile residue (62,
63).
) of GLNBP overlaps with the side
chain carboxyl group of the nucleophile residue of A4-β-galactosidase
(Glu-312). An arginine residue involved in anion binding (Arg-358) and Glu-312
of A4-β-galactosidase are both located in the same position at the
β7 strand of the barrel scaffold. Another interesting overlap was
observed between Arg-32 of GLNBP (anion binding) and Arg-32 in
A4-β-galactosidase, both being located at the same position in the barrel
(β2 strand). Arg-32 in A4-β-galactosidase is a highly conserved
residue and is considered to control the pKa of the
nucleophile residue (62,
63).
FIGURE 5.

Structural similarity with GH42 thermophilic β-galactosidase. A, superimposition of TIM barrel domains of GLNBP (yellow) and A4-β-galactosidase (black). B, close-up view of A at the active site. Bound ligands and catalytically important residues in both structures are shown in ball-and-stick and wire frame models, respectively.
In summary, the positions of the nucleophile (phosphate in GLNBP and Glu-312 in A4-β-galactosidase), subsite -1 (galactoside moiety in both enzymes), and the proton donor (Asp-313 and Glu-141) are located at the same positions in GLNBP and A4-β-galactosidase. Although there is no clear evidence, the structural overlaps of the catalytic components as well as overall structural similarity suggest a possible evolutionary relationship between the inverting phosphorylase (GLNBP) and the retaining GH (A4-β-galactosidase). Moreover, the catalytic mechanism of GLNBP seems to be similar to that of the GH-type inverting phosphorylase, GH94. Our data strongly support the current classification of GLNBP homologues as one of the GH families, GH112.
Conformational Changes on Ligand Binding—The 18 subunit structures in five crystals that were determined can be classified into three distinctive conformational states (Table 2). Subunits in the “semiclosed” state always contain a GlcNAc or GalNAc molecule in the active site, whereas the “open” conformation has no ligand. The structural difference was not so large but was clear at important regions for sugar binding at subsite +1 (Fig. 4C). The formation of hydrophobic interactions induces the movement of Pro-161, Val-162, Tyr-165, Gln-217, and Trp-233 on sugar binding. The movements cause Cα atom shifts of about 1 Å around these residues, including the 160-170 helix (Fig. 4A, blue line). Notably, the Oδ2 atom of the catalytically important Asp-313 residue shifts about 1.6 Å to form a hydrogen bond with the O3 atom of GlcNAc/GalNAc. These structural changes can be interpreted as a small induced fit (see Movie S1).
Both of the two “closed” state subunits contain an anion
(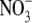 or
or
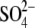 ) at their active sites
(Table 2, subunit A in
GlcNAc-NO3-EG and subunit D in GlcNAc-SO4). The mobile
domains in the closed state subunits (half-barrel unit and C-terminal domain)
are not involved in crystal packing, indicating that this feature is not a
crystal artifact. The closed state was characterized by a notable
conformational change at the half-barrel unit of the catalytic domain
(Fig. 4B). This
conformational change simultaneously creates a hydrogen bond network between
the anion and the three arginine residues in the half-barrel unit
(Fig. 4D). The
electrostatic interaction between these elements may drive the large
conformational change. Although the closed state subunits in both the
GlcNAc-NO3-EG and GlcNAc-SO4 complex forms exhibit
characteristic movement in the half-barrel unit, a large part of the latter
form was disordered (Fig. S2C). Therefore, anion binding seems to be
insufficient to complete the domain closure. The formation of hydrophobic
interactions between the Tyr-362 and Phe-364 side chains and the galactose
moiety of the substrate may be the key to ensure ligand holding and mask the
active site from solvent water.
) at their active sites
(Table 2, subunit A in
GlcNAc-NO3-EG and subunit D in GlcNAc-SO4). The mobile
domains in the closed state subunits (half-barrel unit and C-terminal domain)
are not involved in crystal packing, indicating that this feature is not a
crystal artifact. The closed state was characterized by a notable
conformational change at the half-barrel unit of the catalytic domain
(Fig. 4B). This
conformational change simultaneously creates a hydrogen bond network between
the anion and the three arginine residues in the half-barrel unit
(Fig. 4D). The
electrostatic interaction between these elements may drive the large
conformational change. Although the closed state subunits in both the
GlcNAc-NO3-EG and GlcNAc-SO4 complex forms exhibit
characteristic movement in the half-barrel unit, a large part of the latter
form was disordered (Fig. S2C). Therefore, anion binding seems to be
insufficient to complete the domain closure. The formation of hydrophobic
interactions between the Tyr-362 and Phe-364 side chains and the galactose
moiety of the substrate may be the key to ensure ligand holding and mask the
active site from solvent water.
In almost all chains, except for subunit A in the ligand-free form, residues 34-46 were disordered (Table 2). This region corresponds to the α3 helix of the TIM barrel architecture. Although secondary structure prediction suggests that this region tends to form a helix (data not shown), no secondary structure was formed even in subunit A of the ligand-free form (Fig. 2B). Therefore, the TIM barrel fold of GLNBP is incomplete, and the half-barrel unit is highly flexible at this region.
Unusual Deformations Found in TIM Barrel Scaffold—The TIM barrel is the most common protein fold and is adopted by about 10% of proteins with known three-dimensional structures (64). The functional versatility of TIM barrel proteins is generally based on the stability of the barrel architecture as well as the variability of the βα-loops at the C-terminal ends of the β-strands (“catalytic face”). An unusual example of major conformational changes of a TIM barrel scaffold on substrate-binding is reported for methylmalonyl-CoA mutase from Propionibacterium shermanii (65). In the absence of substrate, its barrel is widely open, and the β1 and β2 strands are completely split apart. When a substrate binds, the barrel closes to form a complete barrel with the substrate inside it. The barrel domain can be divided into two rigid bodies. The first rigid body comprises the β6, β7, β8, and β1 strands, and the second comprises the β2, β3, β4, and β5 strands. They relatively rotate 18° along the axis parallel to the barrel strands. Both cases of the barrel deformations of GLNBP and methylmalonyl-CoA mutase occur in about the half-unit of the barrel that contains four β-strands. Based on several structural observations and experiments, it is presumed that the TIM barrel proteins have evolved by duplication and fusion of ancestral half-barrel proteins (64, 66-68). The structural change of GLNBP is distinct from methylmalonyl-CoA mutase, since the rotation axis of its half-barrel unit is perpendicular to the barrel strands. Despite the large deformation, the barrel β-strands of GLNBP retain their hydrogen bonding networks that make the circular β-sheet even in the closed state. To the best of our knowledge, this type of large conformational change of a TIM barrel is an unprecedented case. Because the source organisms of GH112 homologues are basically limited to enteric bacteria or human-related microbes, they may be relatively “newly evolved” enzymes in a relationship with animals, which abundantly display glycoconjugates containing LNB or GNB on their cell surface. Such an intricate adaptation to the substrate by deformation of the TIM barrel scaffold, instead of varying the C-terminal loops, is a unique example of molecular evolution.
Supplementary Material
Acknowledgments
We thank the staff of the Photon Factory for the x-ray data collection.
The atomic coordinates and structure factors (codes 2ZUS, 2ZUT, 2ZUU, 2ZUV, and 2ZUW) have been deposited in the Protein Data Bank, Research Collaboratory for Structural Bioinformatics, Rutgers University, New Brunswick, NJ (http://www.rcsb.org/).
This work was supported by the Program for Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN) and Japan Society for the Promotion of Science for Young Scientists Research Fellowship 17-00182 (to M. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2, Figs. S1-S4, and Movie S1.
Footnotes
The abbreviations used are: GNB, galacto-N-biose; LNB, lacto-N-biose I; Gal-HexNAcP, β-1,3-galactosyl-N-acetylhexosamine phosphorylase; GLNBP, galacto-N-biose/lacto-N-biose I phosphorylase; GH, glycoside hydrolase; Gal1P, α-d-galactose 1-phosphate; GNBP, galacto-N-biose phosphorylase; LNBP, lacto-N-biose I phosphorylase; GT, glycosyltransferase; ASU, asymmetric unit; EG, ethylene glycol; r.m.s., root mean square; A4-β-galactosidase, β-galactosidase from T. thermophilus A4.
References
- 1.Kitaoka, M., Tian, J., and Nishimoto, M. (2005) Appl. Environ. Microbiol. 71 3158-3162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nishimoto, M., and Kitaoka, M. (2007) Biosci. Biotechnol. Biochem. 71 1587-1591 [DOI] [PubMed] [Google Scholar]
- 3.Derensy-Dron, D., Krzewinski, F., Brassart, C., and Bouquelet, S. (1999) Biotechnol. Appl. Biochem. 29 3-10 [PubMed] [Google Scholar]
- 4.Nakajima, M., and Kitaoka, M. (2008) Appl. Environ. Microbiol. 74 6333-6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantarel, B. L., Coutinho, P. M., Rancurel, C., Bernard, T., Lombard, V., and Henrissat, B. (2008) Nucleic Acids Res. 37 D233-D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakajima, M., Nihira, T., Nishimoto, M., and Kitaoka, M. (2008) Appl. Microbiol. Biotechnol. 78 465. [DOI] [PubMed] [Google Scholar]
- 7.McDonel, J. L. (1980) Pharmacol. Ther. 10 617-655 [DOI] [PubMed] [Google Scholar]
- 8.Petit, L., Gibert, M., and Popoff, M. R. (1999) Trends Microbiol. 7 104-110 [DOI] [PubMed] [Google Scholar]
- 9.Motes, M. L., DePaola, A., Cook, D. W., Veazey, J. E., Hunsucker, J. C., Garthright, W. E., Blodgett, R. J., and Chirtel, S. J. (1998) Appl. Environ. Microbiol. 64 1459-1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hlady, W. G., and Klontz, K. C. (1996) J. Infect. Dis. 173 1176-1183 [DOI] [PubMed] [Google Scholar]
- 11.van den Steen, P., Rudd, P. M., Dwek, R. A., and Opdenakker, G. (1998) Crit. Rev. Biochem. Mol. Biol. 33 151-208 [DOI] [PubMed] [Google Scholar]
- 12.Mocchetti, I. (2005) Cell Mol. Life Sci. 62 2283-2294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hakomori, S. I. (2008) Biochim. Biophys. Acta. 1780 325-346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Erney, R. M., Malone, W. T., Skelding, M. B., Marcon, A. A., Kleman-Leyer, K. M., O'Ryan, M. L., Ruiz-Palacios, G., Hilty, M. D., Pickering, L. K., and Prieto, P. A. (2000) J. Pediatr. Gastroenterol. Nutr. 30 181-192 [DOI] [PubMed] [Google Scholar]
- 15.Daniels, G. (2002) Human Blood Groups, 2nd Ed., Blackwell Science, Oxford
- 16.Asakuma, S., Urashima, T., Akahori, M., Obayashi, H., Nakamura, T., Kimura, K., Watanabe, Y., Arai, I., and Sanai, Y. (2008) Eur. J. Clin. Nutr. 62 488-494 [DOI] [PubMed] [Google Scholar]
- 17.Magnani, J. L., Steplewski, Z., Koprowski, H., and Ginsburg, V. (1983) Cancer Res. 43 5489-5492 [PubMed] [Google Scholar]
- 18.Springer, G. F. (1984) Science 224 1198-1206 [DOI] [PubMed] [Google Scholar]
- 19.Springer, G. F., Desai, P. R., Wise, W., Carlstedt, S. C., Tegtmeyer, H., Stein, R., and Scanlon, E. F. (1990) Immunol. Ser. 53 587-612 [PubMed] [Google Scholar]
- 20.Saavedra, J. M., Bauman, N. A., Oung, I., Perman, J. A., and Yolken, R. H. (1994) Lancet 344 1046-1049 [DOI] [PubMed] [Google Scholar]
- 21.Gibson, G. R., and Roberfroid, M. B. (1995) J. Nutr. 125 1401-1412 [DOI] [PubMed] [Google Scholar]
- 22.van den Broek, L. A. M., Hinz, S. W. A., Beldman, G., Vincken, J. P., and Voragen, A. G. J. (2008) Mol. Nutr. Food Res. 52 146-163 [DOI] [PubMed] [Google Scholar]
- 23.Yasui, H., Shida, K., Matsuzaki, T., and Yokokura, T. (1999) Antonie Van Leeuwenhoek 76 383-389 [PubMed] [Google Scholar]
- 24.Gagnon, M., Kheadr, E. E., Le Blay, G., and Fliss, I. (2004) Int. J. Food Microbiol. 92 69-78 [DOI] [PubMed] [Google Scholar]
- 25.Moro, G., Arslanoglu, S., Stahl, B., Jelinek, J., Wahn, U., and Boehm, G. (2006) Arch. Dis. Child. 91 814-819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Iwabuchi, N., Takahashi, N., Xiao, J. Z., Miyaji, K., and Iwatsuki, K. (2007) Microbiol. Immunol. 51 649-660 [DOI] [PubMed] [Google Scholar]
- 27.Suzuki, R., Wada, J., Katayama, T., Fushinobu, S., Wakagi, T., Shoun, H., Sugimoto, H., Tanaka, A., Kumagai, H., Ashida, H., Kitaoka, M., and Yamamoto, K. (2008) J. Biol. Chem. 283 13165-13173 [DOI] [PubMed] [Google Scholar]
- 28.Nishimoto, M., and Kitaoka, M. (2007) Appl. Environ. Microbiol. 73 6444-6449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujita, K., Oura, F., Nagamine, N., Katayama, T., Hiratake, J., Sakata, K., Kumagai, H., and Yamamoto, K. (2005) J. Biol. Chem. 280 37415-37422 [DOI] [PubMed] [Google Scholar]
- 30.Katayama, T., Sakuma, A., Kimura, T., Makimura, Y., Hiratake, J., Sakata, K., Yamanoi, T., Kumagai, H., and Yamamoto, K. (2004) J. Bacteriol. 186 4885-4893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wada, J., Ando, T., Kiyohara, M., Ashida, H., Kitaoka, M., Yamaguchi, M., Kumagai, H., Katayama, T., and Yamamoto, K. (2008) Appl. Environ. Microbiol. 74 3996-4004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Penders, J., Thijs, C., Vink, C., Stelma, F. F., Snijders, B., Kummeling, I., van den Brandt, P. A., and Stobberingh, E. E. (2006) Pediatrics 118 511-521 [DOI] [PubMed] [Google Scholar]
- 33.Bezirtzoglou, E., Maipa, V., Chotoura, N., Apazidou, E., Tsiotsias, A., Voidarou, C., Kostakis, D., and Alexopoulos, A. (2006) Comp. Immunol. Microbiol. Infect. Dis. 29 345. [DOI] [PubMed] [Google Scholar]
- 34.Haarman, M., and Knol, J. (2005) Appl. Environ. Microbiol. 71 2318-2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Harmsen, H. J. M., Wildeboer-Veloo, A. C. M., Raangs, G. C., Wagendorp, A. A., Klijn, N., Bindels, J. G., and Welling, G. W. (2000) J. Pediatr. Gastroenterol. Nutr. 30 61-67 [DOI] [PubMed] [Google Scholar]
- 36.Parracho, H., McCartney, A. L., and Gibson, G. R. (2007) Proc. Nutr. Soc. 66 405-411 [DOI] [PubMed] [Google Scholar]
- 37.Ward, R. E., Ninonuevo, M., Mills, D. A., Lebrilla, C. B., and German, J. B. (2006) Appl. Environ. Microbiol. 72 4497-4499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knol, J., Scholtens, P., Kafka, C., Steenbakkers, J., Gross, S., Helm, K., Klarczyk, M., Schopfer, H., Bockler, H. M., and Wells, J. (2005) J. Pediatr. Gastroenterol. Nutr. 40 36-42 [DOI] [PubMed] [Google Scholar]
- 39.Withers, S. G., Wakarchuk, W. W., and Strynadka, N. C. (2002) Chem. Biol. 9 1270-1273 [DOI] [PubMed] [Google Scholar]
- 40.Fushinobu, S., Hidaka, M., Miyanaga, A., and Imamura, H. (2007) J. Appl. Glycosci. 54 95-102 [Google Scholar]
- 41.Kitaoka, M., and Hayashi, K. (2002) Trends Glycosci. Glycotechnol. 14 35-50 [Google Scholar]
- 42.Nishimoto, M., and Kitaoka, M. (2007) Biosci. Biotechnol. Biochem. 71 2101-2104 [DOI] [PubMed] [Google Scholar]
- 43.Otwinowski, Z., and Minor, W. (1997) Methods Enzymol. 276 307-326 [DOI] [PubMed] [Google Scholar]
- 44.Bricogne, G., Vonrhein, C., Flensburg, C., Schiltz, M., and Paciorek, W. (2003) Acta Crystallogr. Sect. D Biol. Crystallogr. 59 2023-2030 [DOI] [PubMed] [Google Scholar]
- 45.de la Fortelle, E., and Bricogne, G. (1997) Methods Enzymol. 276 472-494 [DOI] [PubMed] [Google Scholar]
- 46.Terwilliger, T. C., and Berendzen, J. (1999) Acta Crystallogr. Sect. D Biol. Crystallogr. 55 849-861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vagin, A., and Teplyakov, A. (1997) J. Appl. Crystallogr. 30 1022-1025 [Google Scholar]
- 48.Emsley, P., and Cowtan, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2126-2132 [DOI] [PubMed] [Google Scholar]
- 49.Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. Sect. D Biol. Crystallogr. 53 240-255 [DOI] [PubMed] [Google Scholar]
- 50.DeLano, W. L. (2002) The PyMOL Molecular Graphics System, DeLano Scientific, San Carlos, CA
- 51.Kleywegt, G. J. (1996) Acta Crystallogr. Sect. D Biol. Crystallogr. 52 842-857 [DOI] [PubMed] [Google Scholar]
- 52.Morris, G. M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart, W. E., Belew, R. K., and Olson, A. J. (1998) J. Comput. Chem,. 19 1639-1662 [Google Scholar]
- 53.Hidaka, M., Honda, Y., Kitaoka, M., Nirasawa, S., Hayashi, K., Wakagi, T., Shoun, H., and Fushinobu, S. (2004) Structure 12 937-947 [DOI] [PubMed] [Google Scholar]
- 54.Hidaka, M., Kitaoka, M., Hayashi, K., Wakagi, T., Shoun, H., and Fushinobu, S. (2006) Biochem. J. 398 37-43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fushinobu, S., Mertz, B., Hill, A. D., Hidaka, M., Kitaoka, M., and Reilly, P. J. (2008) Carbohydr. Res. 343 1023-1033 [DOI] [PubMed] [Google Scholar]
- 56.Sinnott, M. L. (1990) Chem. Rev. 90 1171-1202 [Google Scholar]
- 57.Deslongchamps, P. (1993) Pure Appl. Chem. 65 1161-1178 [Google Scholar]
- 58.Davies, G. J., Ducros, V. M. A., Varrot, A., and Zechel, D. L. (2003) Biochem. Soc. Trans. 31 523-527 [DOI] [PubMed] [Google Scholar]
- 59.Espinosa, J. F., Montero, E., Vian, A., García, J. L., Dietrich, H., Schmidt, R. R., Martín-Lomas, M., Imberty, A., Cañada, F. J., and Jiménez-Barbero, J. (1998) J. Am. Chem. Soc. 120 1309-1318 [Google Scholar]
- 60.Turnbaugh, P. J., Ley, R. E., Hamady, M., Fraser-Liggett, C. M., Knight, R., and Gordon, J. I. (2007) Nature 449 804-810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Krissinel, E., and Henrick, K. (2004) Acta Crystallogr. Sect. D Biol. Crystallogr. 60 2256-2268 [DOI] [PubMed] [Google Scholar]
- 62.Hidaka, M., Fushinobu, S., Ohtsu, N., Motoshima, H., Matsuzawa, H., Shoun, H., and Wakagi, T. (2002) J. Mol. Biol. 322 79-91 [DOI] [PubMed] [Google Scholar]
- 63.Ring, M., and Huber, R. E. (1990) Arch. Biochem. Biophys. 283 342-350 [DOI] [PubMed] [Google Scholar]
- 64.Sterner, R., and Hocker, B. (2005) Chem. Rev. 105 4038-4055 [DOI] [PubMed] [Google Scholar]
- 65.Mancia, F., and Evans, P. R. (1998) Structure 6 711-720 [DOI] [PubMed] [Google Scholar]
- 66.Lang, D., Thoma, R., Henn-Sax, M., Sterner, R., and Wilmanns, M. (2000) Science 289 1546-1550 [DOI] [PubMed] [Google Scholar]
- 67.Hocker, B., Beismann-Driemeyer, S., Hettwer, S., Lustig, A., and Sterner, R. (2001) Nat. Struct. Biol. 8 32-36 [DOI] [PubMed] [Google Scholar]
- 68.Akanuma, S., and Yamagishi, A. (2008) J. Mol. Biol. 382 458-466 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.