

Abstract
Arsenic enhances skin tumor formation when combined with other carcinogens, including UV radiation (UVR). In this study we report that low micromolar concentrations of arsenite synergistically increases UVR-induced oxidative DNA damage in human keratinocytes as detected by 8-hydroxyl-2′-deoxyguanine (8-OHdG) formation. Poly(ADP-ribose) polymerase-1 (PARP-1) is involved in base excision repair, a process that repairs 8-OHdG lesions. Arsenite suppresses UVR-induced PARP-1 activation in a concentration-dependent manner. Inhibition of PARP-1 activity by 3-aminobenzamide or small interfering RNA silencing of PARP-1 expression significantly increases UVR-induced 8-OHdG formation, suggesting that inhibition of PARP-1 activity by arsenite contributes to oxidative DNA damage. PARP-1 is a zinc finger protein, and mass spectrometry analysis reveals that arsenite can occupy a synthetic apopeptide representing the first zinc finger of PARP-1 (PARPzf). When the PARPzf peptide is preincubated with Zn(II) followed by incubation with increasing concentrations of arsenite, the ZnPARPzf signal is decreased while the AsPARPzf signal intensity is increased as a function of arsenite dose, suggesting a competition between zinc and arsenite for the same binding site. Addition of Zn(II) abolished arsenite enhancement of UVR-stimulated 8-OHdG generation and restored PARP-1 activity. Our findings demonstrate that arsenite inhibits oxidative DNA damage repair and suggest that interaction of arsenite with the PARP-1 zinc finger domain contributes to the inhibition of PARP-1 activity by arsenite. Arsenite inhibition of poly(ADP-ribosyl)ation is one likely mechanism for the reported co-carcinogenic activities of arsenic in UVR-induced skin carcinogenesis.
Arsenic is a naturally occurring element that is present in food, soil, and water (1, 2). Environmental or occupational exposures to arsenic are associated with both acute and chronic toxic effects in humans, including increased incidence of skin, lung, liver, and urinary tract cancers (3). Although human epidemiological data link inorganic arsenic in drinking water with an elevated risk of non-melanoma skin cancer (4), arsenic as a sole agent is not an effective skin carcinogen in animal models (5). However, arsenite enhances tumor development in animals pretreated with other carcinogens (6), chronically stimulated by growth factors (7), or co-treated with UV radiation (UVR)2 (8). It has been reported that sodium arsenite concentration as low as 1.25 mg/liter (10 μm) in drinking water enhances UVR-induced tumorigenicity in mice (8), but the mechanisms underlying this observation are not fully understood.
Arsenite exposure generates reactive oxygen species (ROS), and we have
directly demonstrated that the production of
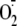 ,
H2O2, and ·OH in arsenite-exposed
keratinocytes is associated with DNA damage
(9,
10). Similarly UVR,
particularly UVA, generates ROS in the skin also leading to oxidative DNA
damage (11). Oxidative stress
plays a significant role in UVR-induced skin carcinogenesis
(12), and excessive ROS
generation causes a range of DNA damages, including DNA strand breaks
(13), DNA-protein cross-links
(14), deletion mutations
(15), and
8-hydroxyl-2′-deoxyguanine (8-OHdG)
(16). 8-OHdG is a biomarker of
oxidative stress and the major mutagenic form of oxidative DNA damage
(17). 8-OHdG lesions can lead
to G:C to T:A transversion mutations
(18,
19), and oxidative DNA lesions
are detected in many human tumors, including human squamous cell carcinoma
(reviewed in Ref. 18).
,
H2O2, and ·OH in arsenite-exposed
keratinocytes is associated with DNA damage
(9,
10). Similarly UVR,
particularly UVA, generates ROS in the skin also leading to oxidative DNA
damage (11). Oxidative stress
plays a significant role in UVR-induced skin carcinogenesis
(12), and excessive ROS
generation causes a range of DNA damages, including DNA strand breaks
(13), DNA-protein cross-links
(14), deletion mutations
(15), and
8-hydroxyl-2′-deoxyguanine (8-OHdG)
(16). 8-OHdG is a biomarker of
oxidative stress and the major mutagenic form of oxidative DNA damage
(17). 8-OHdG lesions can lead
to G:C to T:A transversion mutations
(18,
19), and oxidative DNA lesions
are detected in many human tumors, including human squamous cell carcinoma
(reviewed in Ref. 18).
The major pathway eliminating DNA base damage and helix distortions is excision repair, subdivided into nucleotide excision repair and base excision repair (20). Oxidized bases, apurinic/apyrimidinic sites, and DNA single strand breaks induced by ROS are repaired predominantly by the base excision repair pathway (17). There is evidence that diverse DNA repair systems, including base excision repair and nucleotide excision repair are inhibited by low, non-cytotoxic concentrations of carcinogenic metals such as Ni(II), Co(II), Cd(II), and As(III) (reviewed by Hartwig et al. (21)). Arsenite (AsIII) inhibits the activities of several DNA damage repair proteins, including poly(ADP-ribose) polymerase-1 (PARP-1), formamidopyrimidine-DNA glycosylase, and xeroderma pigmentosum group A protein (XPA), each containing a zinc finger DNA binding domain (22–24). Sub-micromolar concentrations of arsenite reduced PARP-1 activity in mammalian cells (24), and trivalent arsenicals released zinc from the zinc finger domain of human XPA protein (25). These findings suggest that DNA repair proteins with functional zinc finger motifs hold potential as targets for inhibition by arsenic.
In the current study, we report that environmentally relevant concentrations of arsenite interfere with repair of UVR-induced oxidative DNA damage. Arsenite concentrations that do not detectably increase 8-OHdG lesions enhance UVR-induced oxidative DNA damage. We find that (i) PARP-1 activation by UVR is diminished by 200 nm arsenite, (ii) UVR-induced 8-OHdG formation in DNA is enhanced in the presence of a PARP-1 inhibitor or PARP-1 siRNA, (iii) arsenite interacts with a synthetic peptide representing the first zinc finger of PARP-1, and (iv) inclusion of zinc ions (Zn(II)) counteracts arsenite-dependent inhibition of PARP-1 and enhancement of UVR-induced oxidative DNA damage. These data provide evidence that inhibition of DNA repair processes by arsenite may represent an underlying mechanism for the reported co-carcinogenic activities of arsenic and UVR in skin carcinogenesis and identify PARP-1 as a novel molecular target for arsenite-dependent disruption of DNA repair.
EXPERIMENTAL PROCEDURES
Cell Culture and Treatment—The human keratinocyte cell line (HaCaT) was generously provided by Dr. Mitch Denning (Loyola University Medical Center, Maywood, IL). HaCaT cells were maintained in Dulbecco's modified Eagle's medium/F-12 Ham's medium, supplemented with 10% newborn calf serum from Invitrogen, 4-fold concentration of minimal essential medium amino acids solution, 2 mm l-glutamine, and antibiotics (penicillin, 100 units/ml, and streptomycin, 50 μg/ml). The cells were cultured at 37 °C in 95% air/5% CO2 humidified incubators.
Stock solutions of sodium arsenite and zinc chloride at 10 mm were prepared in double-distilled water and sterilized using a 0.22-μm syringe filter. The working concentration was prepared by diluting the stock with Dulbecco's modified Eagle's medium/F-12 medium containing 0.1% (w/v) bovine serum albumin. For all experiments involving incubation with arsenite and/or zinc, HaCaT cells were rinsed with PBS and placed into Dulbecco's modified Eagle's medium/F-12 medium with 0.1% bovine serum albumin containing metal concentrations as indicated in the figure legends.
UVR Exposure—Cells were treated with arsenite and/or zinc as described in the figure legends. Cell culture medium was removed, cells were rinsed three times with PBS, and then cells were covered with a thin layer of PBS and placed on ice in the dark. Cells were maintained on ice during UVR exposure. Cells were exposed to 8 J/cm2 solar-simulated light using a 1000-watt Solar Ultraviolet Simulator (Oriel Corp., Stratford, CT). This solar simulator produces a high intensity UVR beam in both the UVA (320–400 nm) and UVB (280–320 nm) spectra. After UVR exposure, PBS was removed and replaced with serum-free medium. Cells were returned to incubators until collection for further experimental procedures.
HPLC-EC Detection of 8-OHdG—DNA extraction and hydrolysis procedures were performed according to Ding et al. (10). Briefly, cellular DNA was extracted using the DNA extraction kit from Wako Pure Chemical industry (Osaka, Japan). Following extraction, DNA samples were hydrolyzed and transferred to a 30,000-Da microfiltration tube, and centrifuged for 30 min at 10,000 × g at 4 °C prior to HPLC-EC analysis. HPLC-EC was performed with a 15-cm × 4.6-mm, 3-μm LC-18-DB column (Supelco, Bellefonte, PA) with a flow rate of 1.0 ml/min. The mobile phase consisted of methanol/50 mm KH2PO4 (5:95, v/v). The samples were analyzed by a separate UV detector ESA Model 520 (Chelmsford, MA) at 260 nm for dG and an ESA Model 5600 CoulArray linked in series for electrochemical (EC) detection of 8-OHdG. The level of 8-OHdG in sample DNA was expressed as the number of 8-OHdG per 106 dG.
Immunoperoxidase Staining for 8-OHdG—Immunoperoxidase staining for 8-OHdG was performed as described previously (26, 27). Briefly, cells were washed twice with PBS and fixed with methanol at -20 °C for 10 min followed by washing with PBS. Fixed cells were treated with RNase (100 μg/ml, Sigma) for 1 h at 37 °C and proteinase K (10 μg/ml, Sigma) for 10 min at room temperature. After rinsing with PBS, DNA was denatured by treatment with 4 n HCl for 10 min followed by pH adjustment with 50 mm Tris (pH 10) for 5 min at room temperature. Endogenous peroxidase was blocked by treating the cells with 3% H2O2 in methanol for 30 min at room temperature. After washing with PBS, the cells were treated with 10% normal horse serum at 37 °C for 1 h and then incubated with primary antibody 4E9 (Trevigen, Gaithersburg, MD, 1:50 dilution) at 4 °C overnight followed by goat antimouse IgG conjugated to biotin (Vector Laboratories, Burlingame, CA) at 37 °C for 30 min. ABC reagent, avidin conjugated to horseradish peroxidase (Vector Laboratories) was added, and the slides were incubated for 30 min at 37 °C followed by extensive washing. To localize peroxidase, cells were treated with diaminobenzidine (Sigma) for 10 min at room temperature. Finally, slides were washed with PBS, ethanol, and xylene and mounted using Permount. Images were obtained using an Olympus BH2-RFCA fluorescence microscope (Melville, NY) and Omegafire digital camera with MagnaFire 2.1 software (Optronix, Goleta, CA). The relative intensity of nuclear staining of 30–50 randomly selected cells was measured using the Image-Pro Plus (Media Cybernetics, Inc., Silver Spring, MD) software and expressed as arbitrary units.
Transfection of siRNA Duplexes—Transfection of siRNA was performed according to Liu et al. (28). Briefly, SMARTpool siRNA specific for human PARP-1 sequence (GenBank™ accession number: NM_001618) was obtained from Dharmacon Research, Inc. (Lafayette, CO). SiGLO RISK-Free siRNA (Dharmacon, Lafayette, CO), a fluorescently labeled siRNA, was used as a control siRNA. HaCaT cells were seeded at a density of 3 × 105 cells/well in 6-well plates the day before transfection in Dulbecco's modified Eagle's medium/F-12 containing 10% fetal bovine serum without antibiotics. Transfection of siRNAs was carried out using DharmaFECT transfection reagent (Dharmacon). DharmaFECT reagent was diluted 1:50 in serum-free Dulbecco's modified Eagle's medium/F-12 and incubated at room temperature for 5 min. In parallel, 2 μm siRNA in 1 × siRNA buffer (Dharmacon) was diluted 1:1 in serum-free Dulbecco's modified Eagle's medium/F-12. The two mixtures were combined and incubated for 20 min at room temperature prior to addition to cells with a final siRNA concentration of 100 nm. After 24 h, the medium was replaced with complete growth medium, and cells were collected at times as indicated in the figure legends. Specific silencing was confirmed by immunoblot analysis.
Measurement of PARP-1 Activity—PARP-1 activation forms protein-conjugated poly(ADP-ribose) (29), so steady-state levels of poly(ADP-ribosyl)ation in intact cells can be detected by in situ immunofluorescence detection of poly(ADP-ribose) using a mouse monoclonal antibody (10H) raised against poly(ADP-ribose) (30). Cells were grown as monolayers on coverslips, treated as indicated in the figure legends, rinsed with ice-cold PBS, and fixed in ice-cold 10% trichloroacetic acid for 20 min followed by successive 10-min washing in 75%, 90%, and absolute ethanol (-20 °C). Coverslips were air-dried, rehydrated in PBS, and incubated in blocking reagent (1% bovine serum albumin in PBS) at 37 °C for 1 h. The coverslips were then incubated with mouse monoclonal antibody (10H) raised against poly(ADP-ribose) (Axxora, LLC, San Diego, CA, diluted to 10 μg/ml) in blocking reagent in a humid chamber at 37 °C for 1 h, followed by repeated PBS washes. The secondary, fluorescein isothiocyanate-conjugated anti-mouse antibody (Chemicon, Temecula, CA, diluted 1:200 in blocking reagent) was applied, and samples were incubated in a humid chamber at 37°C for 1 h in the dark. Cover slips were mounted on microslides in Vectashield mounting medium containing 2 μg/ml 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA) to visualize nuclei. Images were obtained using an Olympus BH2-RFCA fluorescence microscope (Melville, NY) and Omegafire digital camera with MagnaFire 2.1 software (Optronix). The National Institutes of Health ImageJ program (rsb.info.nih.gov/ij/) was used for the quantitative analysis of PARP-1 immunofluorescence intensity after adjusting threshold levels to eliminate background staining. Values shown represent the average of all cells in each of three independent experiments. As a separate measure of PARP activity, protein extracts were assayed for PARP activity using the HT Colorimetric PARP/Apoptosis Assay kit (Trevigen, Inc., Gaithersburg, MD) according to the manufacturer's instructions.
Mass Spectrometry Analysis—The peptide (grasckkcsesipkdkvphwyhfscfwkv) derived from the first zinc finger of human PARP-1 (apoPARPzf) was commercially synthesized by Genemed Synthesis Inc., (San Antonio TX). Lyophilized peptide was suspended at a concentration of 1 mm in 20 mm Tris buffer, pH 6.8, containing 1 mm dithiothreitol to allow the Cys residues to remain in the reduced state. Stock solutions of metal ions were prepared at a concentration of 1 m in 20 mm Tris buffer, pH 6.8. For experimental analysis both metal ions and peptide were diluted to working concentrations in 20 mm Tris, pH 6.8. Aliquots of 200 μm apoPARPzf were incubated 20 mm arsenite (as AsNaO2) alone or for competition studies with 200 mm Zn(II) (as ZnCl2) for 15 min at 25 °C followed by addition of arsenite at 4 mm, 10 mm, and 20 mm and further incubation for 15 min at 25 °C. After the total incubation time, the samples were diluted 200 times in 10 mg/ml α-cyano-4-hydroxycinnamic acid (Sigma-Aldrich) in a 1:1 (v/v) acetonitrile/water solution, and 2 μl of each sample was deposited in duplicate on the MALDI plate, allowed to dry at room temperature and analyzed by mass spectrometry. MALDI time-of flight (TOF) analyses were performed on a Applied Biosystems 4700 Proteomics Analyzer (TOF/TOF, Applied Biosystems/MDX Sciex, Foster City, CA) operating in MS reflector-positive ion mode. The total acceleration voltage was 20 kV. Desorption was performed using a Nd:YAG laser (355 nm, 3-ns pulse width, and 200-Hz repetition rate). Mass spectra were acquired with a total of 1000 laser pulses over a mass range of m/z range from 1000 to 4000 Da using a focus mass of 3400 Da. Final mass spectra were the summation of eight sub-spectra, each acquired with 125 laser pulses.
Statistical Analysis—Data were analyzed using the Student's t test or analysis of variance (ANOVA). Differences between means were regarded as significant if p < 0.05, and significant differences are indicated by an asterisk.
RESULTS
Low Concentrations of Arsenite Enhance UVR-induced Oxidative DNA Damage in Human Keratinocytes—UVB (290–320 nm) and UVA (320–400 nm) both act as complete carcinogens through DNA damage and regulation of tumor-promoting signal transduction cascades (31). UVA comprises >90% of the solar UVR at the Earth's surface, and ROS induced by UVA exposure leads to oxidative modifications such as hydroxylation of dG, protein-DNA cross-linking, base loss, and strand breaks (32), whereas UVB primarily causes direct DNA damage (11). Arsenite exposure also leads to ROS generation and oxidative DNA damage (10), so we investigated whether low concentrations of arsenite could amplify UVR-induced oxidative DNA damage as measured by 8-OHdG formation in a human keratinocyte cell line (HaCaT). Treatment of HaCaT cells with low concentrations of arsenite (1–3 μm) for 24 h did not increase 8-OHdG levels above that detected in untreated control cells (Fig. 1). Exposure of cells to 8 J/cm2 UVR increased 8-OHdG formation 4- to 5-fold as measured by HPLC-EC detection (Fig. 1, A and C) and immunoperoxidase staining (Fig. 1B). Pretreatment with ≥ 2 μm arsenite for 24 h significantly increased 8-OHdG formation beyond that detected in cells exposed to UVR alone (Fig. 1, A and B). This increase due to arsenite pretreatment was evident 30 min after UVR exposure and persisted for at least 240 min (Fig. 1C). These findings demonstrate that pretreatment of cells with low concentrations of arsenite enhances UVR-induced oxidative DNA damage.
FIGURE 1.

Effect of arsenite on UVR-induced oxidative DNA damage. A, HaCaT cells were incubated with the indicated concentrations of arsenite in serum-free medium for 24 h and then exposed to 8 J/cm2 UV radiation on ice. 8-OHdG was measured by HPLC-EC 120 min after UVR exposure. The experiment was repeated at least three times. Bars represent the mean ±S.D.; *, p < 0.05 compared with UVR alone. B, HaCaT cells were incubated with 2 μm arsenite in serum-free medium for 24 h and then exposed to 8 J/cm2 UV radiation on ice. 8-OHdG was detected by immunoperoxidase staining (upper panel) 120 min after UV exposure and quantified using image analysis software (lower panel). The arrows illustrate examples of 8-OHdG-positive staining. C, HaCaT cells were incubated with 2 μm arsenite (As) in serum-free medium for 24 h and then exposed to 8 J/cm2 UV radiation on ice. After UVR exposure, cells were rinsed and incubated in serum-free medium for the indicated times with “0” representing cell collection on ice immediately after UVR exposure. Each experiment was repeated at least three times. Bars represent the mean ±S.D.; *, p < 0.05 compared with UVR alone.
Arsenite Inhibits UVR-induced PARP-1 Activity—Inhibition of DNA repair is one proposed mechanism to account for arsenic-induced carcinogenicity (21). PARP-1 is a DNA binding zinc finger protein involved in base excision repair, a process responsible for repair of 8-OHdG lesions in DNA (33). PARP-1 activity was detected in intact cells using in situ immunological detection of poly(ADP-ribose) (30). HaCaT cells were exposed to 8 J/cm2 UVR, and PARP-1 activity was measured over time. The product of PARP activity, poly(ADP-ribose), was detected 10 min after UVR exposure, maximal at 20 min and waned by 2 h (Fig. 2A). To test the impact of arsenite on PARP-1 activation, HaCaT cells were pretreated with different concentrations of arsenite (0.2–2.0 μm) for 24 h, exposed to 8 J/cm2 UVR, and poly(ADP-ribose) was measured 20 min after UVR exposure. UVR-induced PARP-1 activation was suppressed in cells incubated with arsenite (Fig. 2, B and C). A concentration-dependent inhibition of PARP-1 activity was observed with significant decrease in PARP-1 activity evident at arsenite concentrations as low as 0.2 μm (Fig. 2B). Cell treatment with 2 μm arsenite nearly abolished UVR-induced PARP-1 activation as detected by in situ analysis (Fig. 2B) or measurement of PARP-1 activity in cell extracts (Fig. 2C). These findings illustrate that low concentrations of arsenite effectively inhibit PARP-1 activity.
FIGURE 2.

Arsenite inhibition of UVR-induced PARP-1 activation. A, HaCaT cells were exposed to 8 J/cm2 UV radiation on ice, and then rinsed and incubated with serum-free medium for the indicated times. Subsequently, PARP-1 activity was detected by in situ immunochemical detection of poly(ADP-ribose). Results are representative of at least three experiments. B, HaCaT cells were incubated with the indicated concentrations of arsenite in serum-free medium for 24 h, then exposed to 8 J/cm2 UV radiation and placed on ice. After UVR exposure, cells were rinsed and incubated with serum-free medium for 20 min, and PARP-1 activity was detected as in A. Images were obtained using an Olympus BH2-RFCA fluorescence microscope and Omegafire digital camera with MagnaFire 2.1 software and quantified as described under “Experimental Procedures.” *, p < 0.05 compared to UVR alone. C, HaCaT cells were incubated with the indicated concentrations of arsenite in serum-free medium for 24 h, and then exposed to UVR as described above. Cells were incubated for 20 min following UVR exposure, and cell protein extracts were assayed for PARP activity assessed using the HT Colorimetric PARP/Apoptosis Assay kit (Trevigen, Inc., Gaithersburg, MD) according to the manufacturer's instructions. NT = untreated.
Effect of PARP-1 Inhibition on UVR-induced 8-OHdG Generation—To directly test the importance of PARP-1 activity in the repair of 8-OHdG, we conducted experiments using the PARP-1 inhibitor 3-aminobenzamide (3-AB) (35) and PARP-1 siRNA knockdown approaches. UVR-induced PARP-1 activity was significantly suppressed in cells treated with 3 mm 3-AB (Fig. 3A). The level of PARP-1 inhibition by 3-AB was comparable to that observed in cells treated with 2 μm arsenite (Fig. 3A, lower panels). Similarly, UVR-induced 8-OHdG formation was enhanced in cells treated with 3-AB as detected by immunoperoxidase staining (Fig. 3B) or HPLC-EC detection (Fig. 3C). This finding implicates PARP-1 activity in the repair of oxidative DNA damage in response to UVR.
FIGURE 3.

Inhibition of PARP-1 increases UVR-induced oxidative DNA damage. A, HaCaT cells were incubated with 2 μm arsenite or 3 mm 3-AB in serum-free medium for 24 h and then exposed to 8 J/cm2 UV radiation on ice. After UVR exposure, cells were rinsed and incubated with serum-free medium for 20 min, and PARP-1 activity was detected as described in the legend to Fig. 2A. Results are representative of at least three experiments. B, HaCaT cells were incubated with 2 mm 3-AB in serum-free medium for 24 h and then exposed to 8 J/cm2 UV radiation on ice. 8-OHdG was detected by immunoperoxidase staining (upper panel) 120 min after UVR exposure and quantified using image analysis software (lower panel). The experiment was repeated at least three times. *, p < 0.05 compared with UVR alone. C, same as B, but 8-OHdG was measured by HPLC-EC. Bars represent the mean of three independent experiments ±S.D.; *, p < 0.05 compared with UVR alone.
To further investigate the specificity of this observation for PARP-1, we conducted a parallel experiment using PARP-1 siRNA to silence PARP-1 gene expression. PARP-1 protein and mRNA expression was decreased by >80% in HaCaT cells transfected with PARP-1 siRNA (data not shown). This level of silencing effectively disrupted PARP-1 activation by UVR as detected in intact cells using immunological detection of poly(ADP-ribose) (Fig. 4A, upper panel) and PARP-1 activity in cell extracts (Fig. 4A, lower panel). PARP-1 silencing increased UVR-induced 8-OHdG formation in DNA (Fig. 4B). The enhancement of 8-OHdG formation was equivalent in cells treated with 2 μm arsenite or transfected with PARP-1 siRNA (Fig. 4B). No significant additional increase in 8-OHdG generation by UVR was observed in PARP-1-silenced cells pretreated with arsenite (Fig. 4B). These findings suggest that PARP-1 plays an important role in the repair of 8-OHdG and that arsenite augments UVR-induced 8-OHdG formation predominantly through inhibition of PARP-1 activity.
FIGURE 4.

PARP-1 gene silencing increases UVR-induced oxidative DNA damage. A, cells were transfected with control or PARP-1 siRNA as described under “Experimental Procedures.” Upper panel: PARP-1 activity was detected by in situ immunochemical detection of poly (ADP-ribose). Lower panel: PARP-1 activity was measured in cell extracts as described in the legend to Fig. 2C. Values shown represent the mean of three independent experiments ±S.D.; *, p < 0.05 compared with UVR alone. B, cells were transfected with control or PARP-1 siRNA as described under “Experimental Procedures.” Cells were incubated without or with 2 μm arsenite in serum-free medium for 24 h, then exposed to 8 J/cm2 UV radiation and placed on ice. 8-OHdG was measured by immunoperoxidase staining (upper panel) and quantified using image analysis software (lower panel) 120 min after UVR exposure. Each experiment was repeated at least three times. Bars represent the mean ±S.D.; *, p < 0.05 compared with UVR alone. No significant differences were detected between the UVR plus As(III) and UVR plus PARP-1 siRNA groups.
Evidence for the Zinc Finger Domain of PARP-1 as a Site of Molecular Interaction with Arsenite—Zinc finger proteins are defined by a domain containing zinc complexed through four invariant cysteine and/or histidine residues (36). Zinc finger proteins have been identified as potential targets for arsenite toxicity (21, 37) through direct (metal exchange, mixed metal complex formation) and indirect (oxidative assault of adventitious metal species) mechanisms (38). Therefore, we investigated arsenite interactions with a synthetic peptide representing the first zinc finger of PARP-1 (PARPzf) using mass spectrometry. Incubation of arsenite with the PARPzf apopeptide (3453.8 Da) leads to the formation of a new molecule, AsPARPzf, at 3525.7 Da. The 72-Da shift corresponds to the covalent binding of arsenite to PARPzf after the release of 3 hydrogens from the 3 cysteine residues of this C3H1 zinc finger peptide (Fig. 5). This finding demonstrates that arsenite can interact with a zinc finger domain of PARP-1. Incubation of Zn(II) with the PARPzf apopeptide produces ZnPARPzf with a 63-Da increment (peak 3518.2), suggesting that the apopeptide must release 2 hydrogens before binding to Zn(II) (Fig. 6, A and B). When the PARPzf peptide is preincubated with Zn(II) followed by incubation with increasing concentrations of arsenite, the ZnPARPzf signal is decreased in a dose-dependent manner while the signal intensity of AsPARPzf is increased (Fig. 6, C–E). This result suggests a competition between Zn(II) and arsenite for the same binding site on the PARPzf peptide and provides evidence that disruption of zinc finger function may represent a mechanism for the observed inhibition of PARP-1 activity by arsenite.
FIGURE 5.

MALDI-TOF-MS analysis of As(III) binding to a PARPzf peptide. Arsenite was incubated with a PARPzf apopeptide (Apo-PARPzf) derived from the first zinc finger for 15 min at 25 °C as described under “Experimental Procedures.” After incubation the samples were diluted 200-fold in 10 mg/ml α-Cyano-4-hydroxycinnamic acid in a 1:1 (v/v) acetonitrile/water solution, and 2 μl of each sample was deposited in duplicate on the MALDI plate, allowed to dry at room temperature, and analyzed by MALDI-TOF performed on a Applied Biosystems 4700 Proteomics Analyzer (TOF/TOF) as described under “Experimental Procedures.” A 72-Da shift was observed between 3453.8- and 3525.7-Da peaks, which corresponds to one A(III) ion binding after the release of 3 hydrogens (As(III)-PARPzf).
FIGURE 6.

MALDI-TOF-MS analysis of As(III) displacement of Zn(II) from a PARPzf peptide. Zinc was incubated with a PARPzf apopeptide (Apo-PARPzf) for 15 min at 25 °C followed by addition of increasing concentrations of As(III) and further incubation for 15 min at 25 °C followed by MS analysis as described unde “Experimental Procedures” and the legend to Fig. 5. The indicated As(III) concentrations represent the final concentration after dilution for application to the MALDI plate. A, a prominent signal for the apo-PARPzf at m/z 3454.8 Da. B, Zn(II) binding to the apopeptide (Zn(II)PARPzf) and a 63-Da increment (peak 3518.2 Da) suggesting that the apopeptide must release 2 hydrogens before the Zn2+ binding. C–E indicate As(III) binding to the PARPzf as indicated by a 72-Da increase in the 3525.7-Da peak and a reduction of the 3518.4-Da peak (Zn(I-I)PARPzf). The decrease in the Zn(II)PARPzf peak as a function of increasing As(III) concentration suggests a competition between Zn(II) and As(III) for the same site of the PARPzf.
Zinc Counteracts Arsenite Effects on UVR-induced PARP-1 Activity and DNA Damage—To determine whether arsenite interferes with zinc-dependent functions of PARP-1, the impact of supplemental zinc on arsenite inhibition of PARP-1 activity and increase in UVR-induced oxidative DNA damage was investigated. HaCaT cells were treated with 2 μm arsenite alone or in combination with increasing concentrations of Zn(II) for 24 h, then exposed to UVR. Arsenite inhibited PARP-1 activation, and inclusion of Zn(II) restored PARP-1 activity in a concentration-dependent manner (Fig. 7A). A significant increase in PARP-1 activity was evident at 0.5 μm Zn(II) with near complete restoration of PARP-1 activity apparent at 5 μm Zn(II). Similarly, Zn(II) reversed arsenite-dependent augmentation of UVR-induced 8-OHdG generation in a concentration-dependent manner (Fig. 7B). A significant decrease was evident at 1 μm Zn(II) with complete reversal of the arsenite response apparent at 5 μm Zn(II). These findings suggest that arsenite interferes with zinc-dependent functions of PARP-1, including activity and repair of oxidative DNA damage.
FIGURE 7.

Effect of zinc on arsenite-dependent inhibition of PARP-1 activity and enhancement of UVR-induced oxidative DNA damage. HaCaT cells were incubated with 2 μm arsenite alone or together with the indicated concentrations of zinc ions in serum-free medium for 24 h, then exposed to 8 J/cm2 UV radiation. A, zinc prevented arsenite inhibition of PARP-1 activity after UVR exposure. After UVR exposure, cells were rinsed and incubated with serum-free medium for 20 min, and PARP-1 activity was detected by in situ immunochemical detection of poly(ADP-ribose). Values shown represent the average fluorescent intensity of all cells in each of three independent experiments ±S.D. *, p < 0.05 compared with UVR plus 2 μm As(III). B, zinc-dependent reversal of arsenite-enhanced oxidative DNA damage. Cells were treated as in A. After UVR exposure, cells were rinsed and incubated with serum-free medium for 120 min and 8-OHdG was measured by HPLC-EC. Each experiment was repeated for at least three times. Bars represent the mean ±S.D. *, p < 0.05 compared with UVR plus 2 μm As(III).
DISCUSSION
Inorganic arsenite increases UVR-induced skin cancer in mice (8), and acts as a co-mutagen with benzo[a]pyrene in mouse skin by significantly increasing the average benzo[a]pyrene DNA adduct level (39, 40). The molecular mechanisms involved in these processes are not known, but inhibition of DNA repair by arsenite has been proposed. Our results demonstrate that low micromolar concentrations of arsenite do not cause significant 8-OHdG generation in DNA (Fig. 1); however, these low arsenite levels synergistically increase UVR-induced 8-OHdG levels (Fig. 1) and inhibit UVR-induced poly(ADP-ribosyl)ation (Fig. 2 B, and C). It is worth noting that the time dependence for arsenite enhancement of UVR-induced 8-OHdG formation (Fig. 1C) coincides with that observed for UVR-induced PARP-1 activation (Fig. 2A). PARP-1 plays multiple roles in cellular response to genotoxic insult, and our data show that pre-treating cells with a PARP-1 inhibitor or PARP-1 siRNA significantly increased UVR-induced 8-OHdG lesions (Figs. 3B, 3C, and 4B), thereby implicating PARP-1 in the repair of oxidative DNA damage. Based on reports that PARP-1 deficiency enhances cellular sensitivity to arsenite (41) and the exquisite sensitivity of PARP-1 for inhibition by arsenite (24, 42) (Fig. 2B), we further explored potential mechanisms to account for arsenite-dependent inhibition of PARP activity.
Our results and those of others suggest that DNA repair proteins, especially those with functional zinc finger motifs, hold potential as important targets for inhibition by arsenite. We find that arsenite exacerbates three distinct types of UV-induced DNA damage (oxidative/8-OHdG, strand break, and CPD (Fig. 1) (43, 44), and Zn(II) treatment reversed arsenite enhancement of UVR-induced 8-OHdG generation in DNA (Fig. 7B) and hydrogen peroxide-induced DNA strand break (45) in a concentration-dependent manner. These findings suggest that arsenite may compete with Zn(II) for positions within the zinc finger domains and thus interfere with protein function. In support of that hypothesis, data from Schwerdtle et al. (25) found that arsenite released zinc from the zinc finger domain of the nucleotide excision repair complex DNA repair protein xeroderma pigmentosum group a (XPA). Although arsenite caused a concentration-dependent zinc release from the XPA, the pentavalent methylated metabolites monomethylarsonic acid and dimethylarsinic acid did not effectively release zinc from the XPA zinc finger (25), and a recent electrospray ionization-MS study demonstrated that MMA(III) released Zn(II) from ZnXPAzf (46). Similarly, trivalent arsenicals, either methylated or not, were most effective at inhibiting poly(ADP-ribosyl)ation of isolated PARP-1 (42), and we find that pretreatment of HaCaT cells with pentavalent monomethylarsonic acid and dimethylarsinic acid before UVR exposure did not lead to the increased 8-OHdG generation in DNA (data not shown). Collectively, these findings suggest that there is preferential disruption of zinc finger function by trivalent forms of arsenic.
Although published reports suggest that arsenite disrupts zinc finger function of PARP-1 (25, 47), the mechanism has not been elucidated. Arsenite at low concentrations is highly selective in reacting with closely spaced (vicinal) dithiol groups in proteins, and PARP-1 contains such vicinal dithiol groups (34, 48). In this study we provide evidence that arsenite interacts with a PARP-1 zinc finger peptide (Fig. 5), and the interaction is at the expense of zinc binding (Fig. 6). This finding, coupled with the restoration of PARP-1 activity upon zinc supplementation (Fig. 7), supports a model whereby arsenite displacement of zinc from the PARP-1 zinc finger leads to decreased PARP-1 activity. Our results identify PARP-1 as a molecular target that links the impairment of DNA repair processes by very low concentrations of arsenite to enhanced UVR-induced oxidative DNA damage. Taken together, the present findings support the hypothesis that arsenite acts as a co-carcinogen for UVR-induced skin carcinogenesis, at least in part, through inhibition of DNA repair and suggest that disruption of the zinc-dependent functions of PARP-1 represents a likely underlying mechanism.
Acknowledgments
We thank Dr. Yan Ning for her assistance with the immunofluorescence studies and Dr. Charlotte Mobarak for assistance with the MS studies.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 ES012938 and R01 ES15826. This work was also supported by the University of New Mexico (UNM) Cancer Research and Treatment Center (Grant P30 CA118100), the UNM NIEHS Center (Grant P30 ES-012072), the UNM Proteomics/Mass Spectrometry facility (funded by Grant P30 CA118100), and the New Mexico Idea Networks for Research Excellence. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: UVR, UV radiation; As(III), sodium arsenite; 8-OHdG, 8-hydroxyl-2′-deoxyguanine; PARP-1, poly(ADP-ribose)polymerase-1; ROS, reactive oxygen species; XPA, xeroderma pigmentosum group A protein; PBS, phosphate-buffered saline; HPLC-EC, high-performance liquid chromatography-electrochemical detection; siRNA, small interfering RNA; MALDI-TOF, matrix-assisted laser desorption ionization time-of-flight; PARPzf, zinc finger of PARP-1.
References
- 1.Liao, X. Y., Chen, T. B., Xie, H., and Liu, Y. R. (2005) Environ. Int. 31 791-798 [DOI] [PubMed] [Google Scholar]
- 2.Patel, K. S., Shrivas, K., Brandt, R., Jakubowski, N., Corns, W., and Hoffmann, P. (2005) Environ. Geochem. Health 27 131-145 [DOI] [PubMed] [Google Scholar]
- 3.Smith, A. H., Hopenhayn-Rich, C., Bates, M. N., Goeden, H. M., Hertz-Picciotto, I., Duggan, H. M., Wood, R., Kosnett, M. J., and Smith, M. T. (1992) Environ. Health Perspect. 97 259-267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossman, T. G., Uddin, A. N., and Burns, F. J. (2004) Toxicol. Appl. Pharmacol. 198 394-404 [DOI] [PubMed] [Google Scholar]
- 5.Pershagen, G. (1981) Environ. Health Perspect. 40 93-100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamamoto, S., Konishi, Y., Matsuda, T., Murai, T., Shibata, M. A., Matsui-Yuasa, I., Otani, S., Kuroda, K., Endo, G., and Fukushima, S. (1995) Cancer Res. 55 1271-1276 [PubMed] [Google Scholar]
- 7.Germolec, D. R., Spalding, J., Yu, H. S., Chen, G. S., Simeonova, P. P., Humble, M. C., Bruccoleri, A., Boorman, G. A., Foley, J. F., Yoshida, T., and Luster, M. I. (1998) Am. J. Pathol. 153 1775-1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rossman, T. G., Uddin, A. N., Burns, F. J., and Bosland, M. C. (2001) Toxicol. Appl. Pharmacol. 176 64-71 [DOI] [PubMed] [Google Scholar]
- 9.Shi, H., Hudson, L. G., Ding, W., Wang, S., Cooper, K. L., Liu, S., Chen, Y., Shi, X., and Liu, K. J. (2004) Chem. Res. Toxicol. 17 871-878 [DOI] [PubMed] [Google Scholar]
- 10.Ding, W., Hudson, L. G., and Liu, K. J. (2005) Mol. Cell Biochem. 279 105-112 [DOI] [PubMed] [Google Scholar]
- 11.Halliday, G. M. (2005) Mutat. Res. 571 107-120 [DOI] [PubMed] [Google Scholar]
- 12.Agar, N. S., Halliday, G. M., Barnetson, R. S., Ananthaswamy, H. N., Wheeler, M., and Jones, A. M. (2004) Proc. Natl. Acad. Sci. U. S. A. 101 4954-4959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu, F., and Jan, K. Y. (2000) Free Radic. Biol. Med. 28 55-63 [DOI] [PubMed] [Google Scholar]
- 14.Ramirez, P., Del Razo, L. M., Gutierrez-Ruiz, M. C., and Gonsebatt, M. E. (2000) Carcinogenesis 21 701-706 [DOI] [PubMed] [Google Scholar]
- 15.Hei, T. K., Liu, S. X., and Waldren, C. (1998) Proc. Natl. Acad. Sci. U. S. A., 95 8103-8107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsui, M., Nishigori, C., Toyokuni, S., Takada, J., Akaboshi, M., Ishikawa, M., Imamura, S., and Miyachi, Y. (1999) J. Invest. Dermatol. 113 26-31 [DOI] [PubMed] [Google Scholar]
- 17.Barzilai, A., and Yamamoto, K. (2004) DNA Repair (Amst.) 3 1109-1115 [DOI] [PubMed] [Google Scholar]
- 18.Evans, M. D., Dizdaroglu, M., and Cooke, M. S. (2004) Mutat. Res. 567 1-61 [DOI] [PubMed] [Google Scholar]
- 19.Valko, M., Izakovic, M., Mazur, M., Rhodes, C. J., and Telser, J. (2004) Mol. Cell Biochem. 266 37-56 [DOI] [PubMed] [Google Scholar]
- 20.Begley, T. J., and Samson, L. D. (2004) DNA Repair (Amst.) 3 1123-1132 [DOI] [PubMed] [Google Scholar]
- 21.Hartwig, A., Asmuss, M., Ehleben, I., Herzer, U., Kostelac, D., Pelzer, A., Schwerdtle, T., and Burkle, A. (2002) Environ. Health Perspect. 110 Suppl. 5, 797-799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asmuss, M., Mullenders, L. H., Eker, A., and Hartwig, A. (2000) Carcinogenesis 21 2097-2104 [DOI] [PubMed] [Google Scholar]
- 23.Hartwig, A., Blessing, H., Schwerdtle, T., and Walter, I. (2003) Toxicology 193 161-169 [DOI] [PubMed] [Google Scholar]
- 24.Hartwig, A., Pelzer, A., Asmuss, M., and Burkle, A. (2003) Int. J. Cancer 104 1-6 [DOI] [PubMed] [Google Scholar]
- 25.Schwerdtle, T., Walter, I., and Hartwig, A. (2003) DNA Repair (Amst.) 2 1449-1463 [DOI] [PubMed] [Google Scholar]
- 26.Yarborough, A., Zhang, Y. J., Hsu, T. M., and Santella, R. M. (1996) Cancer Res. 56 683-688 [PubMed] [Google Scholar]
- 27.Kessel, M., Liu, S. X., Xu, A., Santella, R., and Hei, T. K. (2002) Mol. Cell Biochem. 234–235, 301-308 [PubMed] [Google Scholar]
- 28.Liu, W., Rosenberg, G. A., and Liu, K. J. (2006) J. Neurosci. Res. 84 360-369 [DOI] [PubMed] [Google Scholar]
- 29.Le Rhun, Y., Kirkland, J. B., and Shah, G. M. (1998) Biochem. Biophys. Res. Commun. 245 1-10 [DOI] [PubMed] [Google Scholar]
- 30.Kawamitsu, H., Hoshino, H., Okada, H., Miwa, M., Momoi, H., and Sugimura, T. (1984) Biochemistry 23 3771-3777 [DOI] [PubMed] [Google Scholar]
- 31.Bowden, G. T. (2004) Nat. Rev. Cancer 4 23-35 [DOI] [PubMed] [Google Scholar]
- 32.Ahmed, N. U., Ueda, M., Nikaido, O., Osawa, T., and Ichihashi, M. (1999) Br. J. Dermatol. 140 226-231 [DOI] [PubMed] [Google Scholar]
- 33.Dantzer, F., de La Rubia, G., Menissier-De Murcia, J., Hostomsky, Z., de Murcia, G., and Schreiber, V. (2000) Biochemistry 39 7559-7569 [DOI] [PubMed] [Google Scholar]
- 34.Yager, J. W., and Wiencke, J. K. (1997) Mutat. Res. 386 345-351 [DOI] [PubMed] [Google Scholar]
- 35.De Blasio, A., Musmeci, M. T., Giuliano, M., Lauricella, M., Emanuele, S., D'Anneo, A., Vassallo, B., Tesoriere, G., and Vento, R. (2003) Int. J. Oncol. 23 1521-1528 [PubMed] [Google Scholar]
- 36.Mackay, J. P., and Crossley, M. (1998) Trends Biochem. Sci. 23 1-4 [DOI] [PubMed] [Google Scholar]
- 37.Hartwig, A., and Schwerdtle, T. (2002) Toxicol. Lett. 127 47-54 [DOI] [PubMed] [Google Scholar]
- 38.Bal, W., and Kasprzak, K. S. (2002) Toxicol. Lett. 127 55-62 [DOI] [PubMed] [Google Scholar]
- 39.Evans, C. D., LaDow, K., Schumann, B. L., Savage, R. E., Jr., Caruso, J., Vonderheide, A., Succop, P., and Talaska, G. (2004) Carcinogenesis 25 493-497 [DOI] [PubMed] [Google Scholar]
- 40.Fischer, J. M., Robbins, S. B., Al-Zoughool, M., Kannamkumarath, S. S., Stringer, S. L., Larson, J. S., Caruso, J. A., Talaska, G., Stambrook, P. J., and Stringer, J. R. (2005) Mutat. Res. 588 35-46 [DOI] [PubMed] [Google Scholar]
- 41.Poonepalli, A., Balakrishnan, L., Khaw, A. K., Low, G. K., Jayapal, M., Bhattacharjee, R. N., Akira, S., Balajee, A. S., and Hande, M. P. (2005) Cancer Res. 65 10977-10983 [DOI] [PubMed] [Google Scholar]
- 42.Walter, I., Schwerdtle, T., Thuy, C., Parsons, J. L., Dianov, G. L., and Hartwig, A. (2007) DNA Repair (Amst.) 6 61-70 [DOI] [PubMed] [Google Scholar]
- 43.Qin, X. J., Hudson, L. G., Liu, W., Timmins, G. S., and Liu, K. J. (2008) Toxicol. Appl. Pharmacol. 232 41-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ding, W., Hudson, L. G., Sun, X., Feng, C., and Liu, K. J. (2008) Free Radic. Biol. Med. 45 1065-1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qin, X.-J., Hudson, L. G., Liu, W., Ding, W., Cooper, K. L., and Liu, K. J. (2008) Chem. Res. Toxicol. 9 1806-1813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Piatek, K., Schwerdtle, T., Hartwig, A., and Bal, W. (2008) Chem. Res. Toxicol. 21 600-606 [DOI] [PubMed] [Google Scholar]
- 47.Kitchin, K. T., and Wallace, K. (2005) Toxicol. Appl. Pharmacol. 206 66-72 [DOI] [PubMed] [Google Scholar]
- 48.Kitchin, K. T., and Wallace, K. (2008) J. Inorg. Biochem. 102 532-539 [DOI] [PubMed] [Google Scholar]