

Abstract
Microtubules are cytoskeletal components involved in multiple cell functions such as mitosis, motility, or intracellular traffic. In vivo, these polymers made of αβ-tubulin nucleate mostly from the centrosome to establish the interphasic microtubule network or, during mitosis, the mitotic spindle. Centrosomal P4.1-associated protein (CPAP; also named CENPJ) is a centrosomal protein involved in the assembly of centrioles and important for the centrosome function. This protein contains a microtubule-destabilizing region referred to as PN2-3. Here we decrypt the microtubule destabilization activity of PN2-3 at the molecular level and show that it results from the sequestration of tubulin by PN2-3 in a non-polymerizable 1:1 complex. We also map the tubulin/PN2-3 interaction both on the PN2-3 sequence and on the tubulin surface. NMR and CD data on free PN2-3 in solution show that this is an intrinsically unstructured protein that comprises a 23-amino acid residue α-helix. This helix is embedded in a 76-residue region that interacts strongly with tubulin. The interference of PN2-3 with well characterized tubulin properties, namely GTPase activity, nucleotide exchange, vinblastine-induced self-assembly, and stathmin family protein binding, highlights the β subunit surface located at the intermolecular longitudinal interface when tubulin is embedded in a microtubule as a tubulin/PN2-3 interaction area. These findings characterize the PN2-3 fragment of CPAP as a protein with an unprecedented tubulin sequestering mechanism distinct from that of stathmin family proteins.
Microtubules (MTs)6 are major components of the cytoskeleton shaped as tubes whose walls are composed of αβ-tubulin heterodimers (tubulin). During interphase, they form a dynamic network that participates in the organization of the cell architecture and serves as tracks for molecular motors that convey intracellular cargos and organelles throughout the cytoplasm. In dividing cells, during the M phase, this network is fully reorganized to form spindle MTs on which chromosome pairs align and segregate. To fulfill this wide range of functions, MTs alternate phases of growth and shrinkage in a process known as dynamic instability. The architecture of the MT array results from the action of endogenous proteins that affect MT nucleation and dynamics. MTs are nucleated by the γ-tubulin ring complex and the γ-tubulin small complex that, together with proteins that regulate their effects, are localized at the centrosome (1); in differentiated cells this is the primary MT organizing center. Endogenous proteins that regulate MT dynamics include MT-associated proteins that stabilize MTs and proteins that destabilize them like depolymerizing kinesins and tubulin-sequestering proteins. One of the best characterized families of tubulin-sequestering proteins is that of stathmin. Stathmin, the founding member of the family, was shown to regulate the amount of assembled MTs by binding tubulin and preventing its self-assembly in microtubules (2, 3). This group of proteins forms a tight ternary complex made of two tubulins and one stathmin family protein (4, 5).
Centrosomal P4.1-associated protein (CPAP; also named CENPJ) is a 1338-amino acid centrosomal protein identified as a partner of the head domain of the cytoskeletal protein 4.1R-135 (6). Homozygous mutations of the CPAP gene were found to be associated with an autosomal recessive form of primary microcephaly (7), suggesting that CPAP plays some key physiological role. Observations have been made that identify CPAP as an important factor of the centrosome cycle: it was observed to be associated with centrosomes along the cell cycle (6), and its depletion by RNA interference alters centrosome integrity and induces multipolar spindles (8). CPAP also plays a role in the centrosome function as anti-CPAP polyclonal antibodies inhibit microtubule nucleation by isolated centrosomes as observed with anti-γ-tubulin antibodies (6). Interestingly CPAP comprises a region (referred to as PN2-3; amino acid residues 311–422) that inhibits microtubule nucleation from the centrosome, impedes assembly of pure tubulin, and depolymerizes Taxol-stabilized MTs (9). When PN2-3 is transiently overexpressed, it localizes to the centrosome and in the cytosol and inhibits MT reassembly after cold depolymerization as does the full-length CPAP protein. Together these observations show that the PN2-3 region contains a novel microtubule-destabilizing domain (MDD) without homology with other known MT destabilizers (9).
We undertook a biochemical and structural analysis of the tubulin/PN2-3 interaction to help clarify its functioning at the molecular level. We show that the microtubule destabilization activity of PN2-3 results from its ability to sequester tubulin in a 1:1 complex that does not polymerize. Combining NMR and biochemical experiments, we identified the MDD of PN2-3 as a 76-amino acid region that targets a tubulin surface involving the longitudinal interdimer interface of the β subunit. Our data highlight the PN2-3 domain of CPAP as a unique tool to stabilize a single tubulin heterodimer that will be useful in particular for biochemical or structural purposes.
EXPERIMENTAL PROCEDURES
Proteins—Tubulin was purified from sheep brain crude extracts as described previously (10). Pure tubulin was stored at -80 °C in 50 mm MES-K, pH 6.8, 0.5 mm EGTA, 0.25 mm MgCl2, 0.05 mm EDTA, 33% glycerol, 0.1 mm GTP. Before use, an additional cycle of assembly/disassembly was performed to remove any non-functional protein. Tubulin concentration was determined by spectrophotometry using an extinction coefficient of 1.2 mg-1 × cm2 at 278 nm (11).
Standard recombinant DNA techniques were carried out as described previously (12). Restriction enzymes were from New England Biolabs (Surrey, British Columbia, Canada). The cDNA encoding the PN2-3 region of CPAP (amino acids 311–422 of CPAP) was amplified from a human cDNA library by PCR using the following primers: 5′-TATTATTCCATGGCACAAAAACATGATGATTCCTCAGAAG (forward) and 5′-TATTATAGGATCCTTACCGCTGGAGTTGCTGTCTATC (reverse) and the High Expand Fidelity Enzyme (Stratagene, La Jolla, CA). The PCR product was verified by sequencing (Genome Express, Meylan, France) after cloning in the pET-3d inducible expression vector (Novagen, Madison, WI). This cDNA contained an additional alanine codon located in the 5′ region immediately after the ATG for better Escherichia coli expression. PN2-3 amino acid residues were numbered with respect to the amplified sequence (Ala1, Gln2, Lys3, His4, Asp5,..., Arg113). PN2-3 was expressed in the BL21(DE3) E. coli strain. Bacteria were grown in LB medium at 37 °C, and expression was induced with 0.4 mm isopropyl β-d-thiogalactoside when A600 reached ∼0.5. Bacteria were pelleted 3 h later (4000 × g for 15 min at 4 °C) and resuspended in 20 mm sodium phosphate, pH 6.8, 1 mm EGTA containing the antiprotease Complete mixture (Roche Applied Science). After 3 × 1-min sonication on ice, the lysate was centrifuged again (4000 × g for 15 min at 4 °C), and the supernatant (S1) was boiled for 10 min before another centrifugation step (100,000 × g for 1 h at 4 °C) to yield the boiled S2 supernatant. The S2 supernatant was then purified by cation-exchange chromatography on an SP-Sepharose Fast Flow column (Amersham Biosciences) equilibrated with 20 mm sodium phosphate, 1 mm EGTA, pH 6.8. Proteins were eluted with a 0–1 m NaCl linear gradient in the same buffer. The eluted fractions were analyzed by SDS-PAGE with Coomassie Blue staining. PN2-3-positive fractions were pooled and concentrated by ultrafiltration using a Centriprep 3000 device (Millipore, Bedford, MA). Finally protein extracts were loaded on a Superdex 75 16/60 size exclusion chromatography column (Amersham Biosciences) equilibrated with 20 mm sodium phosphate, 200 mm NaCl, 1 mm EGTA, pH 6.8. Pure PN2-3 fractions were detected by SDS-PAGE and concentrated as above. Concentration of purified PN2-3 was determined by amino acid analysis. For NMR analyses, 15N-labeled PN2-3 or 15N, 13C-labeled PN2-3 were produced using standard protocols in M9 medium supplemented with 15NH4Cl or 15NH4Cl/d-[13C]glucose. The purification procedure and protein concentration determination were identical to those used for unlabeled PN2-3.
The recombinant stathmin-like domain of RB3 (RB3SLD) and stathmin were produced and purified as described previously (4). The stathmin N-cap (also named I19L) is a peptide in the N-terminal part of the stathmin protein capping the α-tubulin longitudinal interface (13). Its sequence (IQVKELEKRASGQAFELIL) corresponds to human stathmin residues 6–24. It was obtained by chemical synthesis from Epytop (Nîmes, France).
In Vitro Tubulin Polymerization Assay—MT assembly was monitored turbidimetrically at 350 nm in an Ultrospec 3000 spectrophotometer (GE Healthcare) thermostated at 37 °C (1-cm light path). Experiments were carried out in 50 mm MES-K, pH 6.8, 30% glycerol, 0.5 mm EGTA, 6 mm MgCl2, 0.5 mm GTP. Tubulin polymerization was evaluated in each sample by the difference between the 37 °C steady state absorbance and the residual absorbance after subsequent cold-induced depolymerization, which ensures that the absorbance at 350 nm taken into account results from MT assembly and not from cold stable aggregates (3). Differences between the two values were used to construct critical concentration plots representing the amount of polymerized tubulin observed at steady state versus the total concentration of tubulin measured in the absence or presence of PN2-3 or stathmin as described previously (3).
NMR Spectroscopy—All 1H NMR experiments were carried out at 293 K on a Bruker Avance 600-MHz NMR spectrometer equipped with a cryoprobe, and data were processed using XWINNMR (Bruker) software. Sodium [3-trimethylsilyl-2,2′,3,3′-2H4]propionate (TSP-d4) was used as an internal reference for proton chemical shifts.
Free and Tubulin-bound PN2-3—Spectra of PN2-3 free and bound to tubulin were collected with 0.05 mm 15N-labeled samples in 500-μl volumes. The tubulin/PN2-3 interaction was studied with a molar ratio of 1:1. Buffer conditions were 50 mm sodium phosphate, pH 6.8, 0.02% NaN3, 10% D2O supplemented with 150 mm NaCl for free PN2-3. 1H-15N HSQC experiments were carried out with 2048 data points × 1024 increments × 32 scans and a spectral width of 1500 and 1520 Hz for 1H and 15N dimensions, respectively. The data were zero-filled to give a 4096 × 2048 data matrix prior to Fourier transformation. Structures of PN2-3 were calculated using 50 torsion angle constraints derived from TALOS predictions (14). A total of 200 structures were generated with a standard simulated annealing protocol using Crystallography and NMR System (CNS) 1.1 (15). Of these calculated structures, the quality of the 15 structures with the lowest total energy was analyzed, and the seven best structures without torsion angle violations were selected and visualized with MOLMOL 2.6 (16).
Simultaneous Binding of PN2-3 and Stathmin N-cap to Tubulin— 60-μl samples containing 1 mm peptide and 20 μm tubulin (50:1 peptide:protein ratio) in 50 mm sodium phosphate buffer, pH 6.8 and 0.02% NaN3 in 10% D2O were placed in a 1.7-mm-diameter capillary tube. One-dimensional proton spectra were acquired with 64 scans and 16,000 data points. One-dimensional saturation transfer difference NMR spectroscopy (STD-NMR) experiments (17) of stathmin N-cap in interaction with tubulin preincubated or not with 50 μm PN2-3 or stathmin were recorded with 1024 scans. The protein resonances were saturated at 0 or 10 ppm (40 ppm for reference spectra) with a cascade of 40 selective Gaussian-shaped pulses of 50-ms duration with a 1-ms delay between each pulse, resulting in a total saturation time of 2.04 s. Subtraction of saturated spectra from reference spectra was performed by phase cycling.
Circular Dichroism—Far-UV circular dichroism (CD) spectra and thermal unfolding profiles were recorded on a Jasco J-810 spectropolarimeter equipped with a Peltier temperature-controlled single cell holder. Measurements were done in a 0.1-cm-path length quartz cell with 10 μm PN2-3 in a 20 mm sodium phosphate, pH 6.5, 1 mm EGTA, 1.65 mm NaCl buffer. Spectra are the average of five accumulations from 260 to 185 nm, and the thermal unfolding profile was recorded at 222 nm from 4 to 60 °C with a 1 °C min-1 ramping rate. The CD signal (millidegrees) was converted to mean residual ellipticity ([θ], degrees·cm2·dmol-1) defined as [θ] = [θ]obs(10cln)-1 where [θ]obs (millidegrees) is the experimental ellipticity, c (mol·dm-3) is the protein concentration, l (cm) is the cell path length, and n is the number of residues in the protein. The percentage of helix was calculated from the following equation: f = ([θ]222 - [θ]R)/([θ]Hn - [θ]R) where f is the α-helical fraction of protein residues, [θ]222 is the mean residue ellipticity value at 222 nm, [θ]R (1580) is the mean residue ellipticity for pure random coil, and [θ]Hn is the mean residue ellipticity for a helix of length n. This last factor is derived from the equation [θ]Hn = [θ]H∞ (1 - k/n) where [θ]H∞ (-39,500) is the mean residue ellipticity for pure helix of infinite length and k is a wavelength-dependent constant (2.57 for λ = 222 nm) (18).
Secondary Structure Prediction—The secondary structure of PN2-3 was predicted using PROF (19), NNPREDICT (20), PSIPRED (21), and PSA (22).
Tubulin·Colchicine GTPase Activity—Measurements of the colchicine-enhanced GTPase activity of tubulin were performed at a 20 μm tubulin concentration in 80 mm PIPES-K, pH 6.8, 1 mm MgCl2, 0.5 mm EGTA, 150 μm [γ-32P]GTP (GE healthcare). The tubulin·colchicine complex (Tc; prepared as described previously (23)) was incubated with increasing amounts of PN2-3 during 15 min at 4 °C. Measurements were started after prewarming the corresponding samples for 6 min at 37 °C. Free inorganic phosphate was quantified by extracting it as a phosphomolybdate complex as described previously (24). Briefly 50-μl aliquots were taken off in the time course and mixed immediately with 1.5 ml of an ice-cold 10 mm ammonium molybdate solution in 1 m HCl and 4% HClO4. H3PO4 (0.2 mm) was added as a carrier. After extraction by 3 ml of cyclohexane:isobutyl alcohol:acetone (5:5:1) saturated with water, 1 ml of the organic phase was mixed with 10 ml of scintillation solution for radioactivity counting. The radioactivity of 150 μm [γ-32P]GTP was taken as a reference to quantify GTP hydrolysis. During the 24-min time course in which the reaction was monitored, the rate of GTP hydrolysis remained constant. The total GTPase activity of each sample was deduced from the slope of the plot of the concentration of inorganic phosphate as a function of time, and the specific activity of tubulin was derived by dividing the background-subtracted total activity by the tubulin concentration.
Vinblastine-induced Tubulin Assembly—10 μm tubulin or Tc with or without PN2-3 was incubated with 10–100 μm vinblastine for 20 min at room temperature in 15 mm PIPES-K, pH 6.8, 300 μm MgCl2, 200 μm EGTA, 100 μm GDP. After high speed centrifugation (300,000 × g for 15 min at 20 °C), the supernatant and pellet were analyzed by SDS-PAGE on Coomassie Blue-stained gels.
Size Exclusion Chromatography—Samples were analyzed by gel filtration at a 0.5 ml/min flow rate on a Superose 12 HR 10/30 column (GE Healthcare) previously equilibrated with 80 mm PIPES-K, pH 6.5, 1 mm EGTA, 5 mm MgCl2. The column was calibrated with ribonuclease A (Stokes radius (RS) = 16.4 Å), chymotrypsinogen A (RS = 20.9 Å), ovalbumin (RS = 30.5 Å), bovine serum albumin (RS = 35.5 Å), aldolase (RS = 48.1 Å), catalase (RS = 52.2 Å), ferritin (RS = 61 Å), and thyroglobulin (RS = 85Å) (gel filtration calibration kit, GE Healthcare). 100-μl samples at a 10 μm tubulin concentration either alone or with RB3SLD and/or PN2-3 were analyzed. The elution profiles were recorded at 280 nm where we essentially monitor tubulin because RB3SLD and PN2-3 do not absorb light significantly at this wavelength. The Stokes radius was determined graphically on a plot of (-log Kav)½ as a function of the Stokes radius (25) constructed with the above mentioned standard proteins. Kav is calculated as (Ve - V0)/(Vt - V0) where Ve represents the elution volume at the top of the peak, V0 is the void volume of the column (determined with blue dextran), and Vt is the total volume of the gel bed (determined as the elution volume of acetone).
Nucleotide Exchange—The exchange of GDP for GTP at the nucleotide exchangeable site (on β-tubulin) was estimated by measuring the displacement of “cold” GDP by [α-32P]GTP following a procedure adapted from Bai et al. (26). GDP-tubulin (10 μm) was incubated for 5 min on ice with varying amounts of PN2-3 in 80 mm PIPES-K, pH 6.9, 0.5 mm MgCl2.50 μm [α-32P]GTP was added, and the reaction mixture was incubated for an additional 15 min. Tubulin was separated from unbound nucleotide by rapid gel filtration on a Micro Bio-Spin P6 column (Bio-Rad) previously equilibrated with the same buffer (27). There is no release of tubulin-bound nucleotide during centrifugation (28). We also quantified the free nucleotide that was eluted from the column. The radioactivity of the tubulin-containing eluted fraction was counted, corrected for eluted free nucleotide, and compared with the radioactivity of an [α-32P]GTP sample of known concentration.
Surface Plasmon Resonance (SPR)—The BIAcore 2000 system, sensor chips SA (i.e. streptavidin-coated, thus allowing coupling with biotin), and HBS buffer (10 mm HEPES, pH 7.4, 150 mm NaCl, 3.4 mm EDTA, 0.005% (v/v) surfactant P20) were from BIAcore AB (Uppsala, Sweden). One flow cell was used as a reference. Another flow cell was coupled with 300 resonance units of RB3SLD that was specifically biotinylated on an N-terminal tag as described previously (29). To check whether PN2-3 binds to the tubulin·RB3SLD complex (T2R), the coupled sensor chip was first loaded with 20 μm tubulin in 80 mm PIPES-K, pH 6.8, 1 mm EGTA, 5 mm MgCl2 at a 10 μl/min flow rate at 20 °C. PN2-3 at concentrations ranging from 16 to 820 nm was then flowed over the sensor chip. As a control, stathmin was injected at 1 μm concentration. For analysis, the reference flow cell sensorgram was subtracted from the corresponding sensorgrams to take into account base-line drift, bulk, and nonspecific interaction contributions. The sensor chip was regenerated using 30 μl of 50 mm HEPES, pH 7.4, 500 mm NaCl, 3 mm EDTA, 0.005% surfactant P20.
RESULTS
PN2-3 Sequesters Tubulin in a 1:1 Complex—The effect of PN2-3 on tubulin polymerization at steady state was quantified using critical concentration plots derived from turbidimetric measurements (Fig. 1). In the buffer used for this experiment, the critical concentration of tubulin for microtubule assembly was 2 μm. In the presence of PN2-3, the critical concentration plots were shifted while remaining parallel to the control straight line observed with tubulin alone. This indicates that PN2-3 prevents polymerization of the same amount of MTs at all tubulin concentrations by sequestering tubulin in a non-polymerizable state. The amount of unassembled tubulin in complex with PN2-3 can be derived from the shift of the plot. In the presence of 4 or 8 μm PN2-3, the apparent critical concentration increased to 5.5 and 10 μm, respectively, in good agreement with a stoichiometry of one tubulin sequestered per PN2-3. As a control, we checked that the addition of 2 μm stathmin (known to form a 2:1 tubulin·stathmin complex (T2S)) results in a shift similar to that observed with 4 μm PN2-3. These data show that PN2-3 acts as a tubulin-sequestering protein forming with tubulin a binary 1:1 complex that does not participate in microtubule assembly.
FIGURE 1.

PN2-3 inhibits MT assembly by sequestering tubulin in a 1:1 complex. Critical concentration plots for microtubule assembly in the absence (•) and in the presence of 4 (▪) or 8 μm (♦) PN2-3 or of 2 μm stathmin (×) are displayed. Abs, absorbance.
PN2-3 Folds into a Short Helical and Unstable Structure in Solution—We used far-UV CD spectroscopy to characterize the PN2-3 structure in solution. The spectrum recorded at 20 °C has one minimum centered at 222 nm characteristic of proteins with α-helical content (Fig. 2A). The mean residue ellipticity value at 222 nm translates into about 20% helical content in PN2-3, corresponding to 23 α-helical residues. The spectrum also displays strong negative values at 195–200 nm, a feature characteristic of unfolded proteins (30). The largely unfolded nature of PN2-3 was further confirmed by recording its thermal unfolding profile from 4 to 60 °C. It shows a reversible (data not shown) non-cooperative transition (Fig. 2B), which means that PN2-3 unfolds non-cooperatively between 4 and 40 °C, a known behavior of intrinsically unstructured proteins.
FIGURE 2.

CD analysis of PN2-3. A, far-UV CD spectrum of 10 μm PN2-3 recorded at 20 °C. B, thermal unfolding profile of 10 μm PN2-3 recorded at 222 nm. deg, degrees.
We also investigated the PN2-3 structure by NMR spectroscopy. The two-dimensional 1H-15N HSQC of free PN2-3 revealed a weak dispersion of correlation peaks, another indication that this subdomain does not have a well defined tertiary structure when isolated in solution (Ref. 31 and Fig. 3A). The recent and mostly complete 1H-15N and 13C free PN2-3 NMR assignments allowed us to probe the presence of PN2-3 regions containing regular secondary structure. Using the chemical shift deviations from random coil values of Cα atoms (32), we delimitated a 25-amino acid region (residues Phe29 to Glu53; PN2-3 numbering) containing significant α-helical structure (31). Taking also the chemical shift deviation values of Hα and carbonyl atoms into account (supplemental Fig. 1), we then determined the α-helical region more accurately and restricted it to residues Phe29 to Glu51 in agreement with the helical content determined by CD spectroscopy. An ensemble of PN2-3 structures calculated with TALOS (14) torsion angle constraints recapitulating these data is depicted in supplemental Fig. 1B.
FIGURE 3.

NMR analysis of the tubulin/PN2-3 interaction. A, 1H-15N HSQC spectra of free PN2-3 (black; the resonances of interacting residues are labeled) and bound to tubulin (red). B, sequence of PN2-3. The tubulin-binding residues (Val10 to Lys85) delimiting the PN2-3 MDD are in black.
The secondary structure predictions of PN2-3 using several algorithms (19–22) point out additional stretches that may fold into α-helices (supplemental Fig. 1A). Overall these programs predict an N-terminal helix longer than the one experimentally observed, from residues Glu9 to Ala52, and two additional shorter ones in the C-terminal part of the sequence (Leu73–Gln84 and Lys105–Leu111). To summarize, the experimental CD spectroscopy and NMR data indicate that PN2-3 has a limited secondary structure content, whereas predictions suggest more extensive α-helical folding.
A 76-amino Acid Region of PN2-3 Binds Strongly to Tubulin— We investigated the interaction of PN2-3 with tubulin by chemical shift perturbation mapping (33), i.e. by analyzing the modifications of the two-dimensional 1H-15N HSQC NMR spectrum (disappearances, attenuations, or shifts of cross-peaks) of PN2-3 upon tubulin binding using 15N-labeled PN2-3 and unlabeled tubulin. Because of the large size of the tubulin·PN2-3 complex (∼110 kDa), the PN2-3 1H-15N cross-peaks of residues that interact strongly with tubulin are expected to become invisible. The comparison of the HSQC spectrum of tubulin·PN2-3 at a 1:1 ratio with that of free PN2-3 (Fig. 3A) indicates that more than half of 1H-15N cross-peaks disappear. A peak by peak analysis using our assignment of free PN2-3 (31) allowed us to determine that the resonances significantly affected in the presence of tubulin correspond to residues Val10 to Lys85 (Fig. 3B); they define the tubulin-binding region of PN2-3. This 76-amino acid domain that interacts strongly with tubulin encompasses the α-helix region (residues 29–51) observed in free PN2-3. Interestingly it is predicted to fold into two helices (Val10–Ala52 and Leu73–Gln84) separated by a proline-rich region (Pro55, Pro57, Pro62, and Pro65); it is possible that these helical regions fold as predicted upon tubulin binding.
PN2-3 Interferes with Tubulin Longitudinal Associations— We next mapped the PN2-3 footprint on tubulin through its interference with several well characterized tubulin properties. We first evaluated its effect on the tubulin GTPase activity in solution, a process that requires tubulin-tubulin longitudinal associations (23). Whereas this activity is very low, it is enhanced to a measurable amount when tubulin is in complex with colchicine (Tc) (34). We found the GTPase activity of Tc to be 5.5 ± 0.7 10-3 min-1 in good agreement with previous results (23). This activity is inhibited by PN2-3, a stoichiometric amount of which is sufficient for complete inhibition (Fig. 4A). We checked that GTP hydrolysis did not differ whether GTP was added to tubulin before or after PN2-3; as PN2-3 inhibits nucleotide exchange (see below), this ensures that it also inhibits GTP hydrolysis.
FIGURE 4.

PN2-3 prevents tubulin longitudinal associations. A, dose-dependent effect of PN2-3 on the GTPase activity of 20 μm tubulin·colchicine. Error bars were calculated from the deviations from a straight line of the variations of inorganic phosphate released as a function of time as described previously (47). Below 20 μm PN2-3 the data were fitted linearly and correspond to the stoichiometric inhibition of the Tc GTPase activity by PN2-3. Above 20 μm PN2-3, there is no residual activity within experimental error. B, inhibition of the vinblastine-induced tubulin helical assembly by PN2-3. The vinblastine-dependent tubulin/tubulin association was assayed by analyzing the tubulin content in the supernatant (S) and pellet (P) after centrifugation of mixtures of the relevant proteins with vinblastine at the indicated concentrations as described under “Experimental Procedures.” The vinblastine-induced assembly (top) is inhibited by PN2-3 (bottom). C, PN2-3 interferes with the binding of RB3SLD to tubulin. The gel filtration chromatography elution profiles of 10 μm tubulin either alone (continuous line), with 5 μm RB3SLD (long dashes), with 10 μm PN2-3 (alternating short and long dashes), or with 5 μm RB3SLD and 10 μm PN2-3 (short dashes) are displayed. Abs, absorbance; mAU, milliabsorbance units.
We also studied the effect of PN2-3 on vinblastine-induced tubulin self-assembly. Vinblastine induces the formation of spirals (35) by promoting tubulin-tubulin longitudinal associations into curved assemblies (36). As this leads to large oligomers, this process is conveniently monitored in spin-down assays. Using a 10 μm tubulin concentration, we found that about half of the tubulin is pelleted in the presence of 10 μm vinblastine and that this proportion increases at higher vinblastine concentrations (Fig. 4B). In the presence of 10 μm PN2-3, tubulin remains soluble even at a 100 μm vinblastine concentration (Fig. 4B). Identical results were obtained with Tc (data not shown).
We draw three conclusions about the tubulin/PN2-3 interaction from these results. (i) They indicate that, in addition to tubulin, PN2-3 also interacts with Tc as also confirmed by gel filtration chromatography (data not shown). Therefore PN2-3 also interacts with tubulin maintained in a bent conformation (37). (ii) The “titration-like” shape of the inhibition curve of the GTPase activity of 20 μm Tc (Fig. 4A) implies that PN2-3 binds to Tc with an affinity constant (Ka) higher than 106 m-1. Incidentally the stoichiometric inhibition fully confirms the 1:1 tubulin:PN2-3 stoichiometry derived from MT assembly inhibition. (iii) Finally because the Tc GTPase activity and the vinblastine-induced assembly depend on tubulin-tubulin longitudinal associations (23, 36) our results indicate that PN2-3 prevents these associations.
One implication of these results is that any property that depends on tubulin-tubulin longitudinal associations may be affected by PN2-3. This is in particular the case of the formation of tubulin·stathmin-like domain (SLD) complexes. SLDs form with tubulin a protofilament-like 2:1 tubulin·SLD complex (T2SLD) in which longitudinal interactions are established between the two tubulin molecules (38, 39). We evaluated the interference of PN2-3 with the formation of T2SLD by recording in a gel filtration chromatography assay the partition of tubulin among T2SLD and tubulin·PN2-3 in the presence of variable amounts of PN2-3. In this experiment we used the complex of tubulin with RB3SLD (T2R) as it does not dissociate appreciably on a gel filtration column (4). When a sample comprising 10 μm tubulin, 10 μm PN2-3, and 5 μm RB3SLD was analyzed, the gel filtration chromatogram displayed two peaks (Fig. 4C). The first one elutes at the same volume as T2R, and the second one elutes slightly before the tubulin peak and corresponds to tubulin·PN2-3. We also checked that the more PN2-3 we added the more the tubulin amount eluting in the second peak increased. These results indicate that PN2-3 binds tightly to tubulin in a way that prevents the formation of the T2R complex. The observation that the tubulin·PN2-3 peak is only slightly shifted from that of tubulin (the Stokes radius changes from 44 to 48 Å) argues in favor of the formation of a complex that comprises one tubulin molecule.
PN2-3 Targets the β-Tubulin Longitudinal Interface—To determine which of the α or the β subunit longitudinal interfaces is targeted by PN2-3, we studied the effects of PN2-3 binding on two tubulin properties depending specifically on residues located at these interfaces. First we recorded the interference of PN2-3 with the tubulin nucleotide exchange. Whereas the α-tubulin-bound GTP is non-exchangeable, the guanine nucleotide bound to the β subunit is freely exchangeable in solution (40). Moreover and as opposed to GTP hydrolysis, this is a unimolecular process as it occurs at the solvent-exposed nucleotide site in T2R, a complex that does not undergo tubulin-like longitudinal associations (36). All the solvent-exposed residues that constitute the exchangeable nucleotide binding site are located at the β-tubulin longitudinal intermolecular interface where the opening of the site is found (41); it is therefore expected that occluding this opening or preventing movements of the loops surrounding this site, which implies contacting this β subunit interface, inhibits nucleotide exchange. Indeed the only compounds known to inhibit nucleotide exchange completely and whose binding site on tubulin has been defined bind to that interface (42). No effect of PN2-3 on nucleotide exchange of 10 μm tubulin was detected up to the addition of a nearly stoichiometric amount of PN2-3, whereas we recorded a dose-dependent inhibition at higher PN2-3 concentration (Fig. 5A). This means that, at substoichiometric PN2-3 concentration and during the 15-min incubation of the samples, each tubulin molecule is uncomplexed for a sufficient time for nucleotide exchange to happen. This is consistent with the known nucleotide dissociation kinetic constant (koff = 0.1 s-1 (43)) and shows that PN2-3 dissociation from tubulin is not rate-limiting in our experimental conditions. Inhibition of nucleotide exchange by PN2-3 is evidenced by observations at higher PN2-3 concentrations with completeness being reached at 20 μm PN2-3. This strongly suggests that PN2-3 contacts the β-tubulin longitudinal interface.
FIGURE 5.

PN2-3 targets the β-tubulin longitudinal interface. A, inhibition by PN2-3 of tubulin nucleotide exchange. The variation of the amount of exchanged nucleotide as a function of PN2-3 concentration is presented. Error bars are standard deviations from triplicate experiments. The experimental data points are joined by a smoothed curve. B, PN2-3 does not compete with the stathmin N-cap for tubulin binding. The one-dimensional (1D) 1H NMR spectrum of the stathmin N-cap in the presence of tubulin (top panel) and the one-dimensional STD-NMR spectra of the stathmin N-cap in the presence of tubulin (second panel), of tubulin·PN2-3 (third panel), or of T2S (fourth panel) are displayed.
In a second experiment, we studied the interference between PN2-3 and the stathmin N-terminal peptide (stathmin N-cap) for tubulin binding; this peptide has been shown to interact exclusively with the α-tubulin longitudinal interface (13, 37). This was investigated by STD-NMR, taking advantage of the folding of the stathmin N-cap upon tubulin binding (13). This technique allows characterization of the binding site of a ligand in fast exchange with its receptor by saturating the receptor protons (17). We previously used it to identify the protons of the stathmin N-cap in contact with tubulin (13). We found that the one-dimensional STD-NMR spectrum of the stathmin N-cap in the presence of tubulin was not altered by PN2-3, whereas it was dramatically flattened when stathmin was added as a positive control for competition (Fig. 5B). Therefore PN2-3 does not compete with the stathmin N-cap for tubulin binding. These results also suggest that PN2-3, the stathmin N-cap, and tubulin associate into a ternary complex. Taken together with the interference of PN2-3 with tubulin longitudinal associations, they reinforce the argument based on nucleotide exchange inhibition that PN2-3 targets the β-tubulin longitudinal intermolecular interface.
As one such interface is exposed in T2R (37, 42), this raises the possibility that T2R·PN2-3 complexes might form even though apparently mutually exclusive T2R and tubulin·PN2-3 complexes are detected by gel filtration (Fig. 4C). In an attempt to detect trace amounts of these putative complexes, we turned to an SPR experiment. T2R was immobilized through a biotinylated RB3SLD onto a streptavidin-functionalized sensor chip (29). When PN2-3 was injected, we did not observe any mass increase (Fig. 6). This indicates that the T2R·PN2-3 complex, if it forms, is not sufficiently abundant to be detected by this technique. Instead and as compared with controls with buffer alone or with stathmin, a dose-dependent decrease of the signal related to the dissociation of tubulin from RB3SLD coupled to the sensor chip was observed. This suggests that PN2-3 interferes with the T2R complex in a way that destabilizes it, a finding that remains to be confirmed in solution and will be investigated further.
FIGURE 6.

SPR investigation of the interaction of PN2-3 with T2R. Net sensorgrams after background subtraction are displayed. The sensor chip with immobilized RB3SLD was first loaded with 20 μm tubulin (between points A and B) and then washed with buffer (until point C). Between C and D, stathmin (1 μm) or PN2-3 (at concentrations ranging from 16 to 820 nm) were injected following which a final buffer wash was performed.
DISCUSSION
The PN2-3 fragment of CPAP contains an MT destabilizing motif that binds to αβ-tubulin (9). The effect of PN2-3 on the amount of polymerized tubulin provides a means to assess the interference of PN2-3 with microtubule assembly quantitatively. Critical concentration plots were derived from turbidimetric measurements (Fig. 1). A quantitative analysis demonstrates that PN2-3 sequesters tubulin in an unpolymerizable 1:1 complex and that its effect is satisfactorily described by the following scheme.
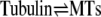 |
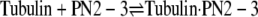 |
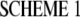 |
The critical concentration for microtubule assembly is the difference between the apparent critical concentration and the concentration of the tubulin·PN2-3 complex. It appears that it is not affected by PN2-3, suggesting that PN2-3 does not interact with microtubules appreciably in agreement with experimental results of the sedimentation of Taxol-stabilized MTs (9).
To define the mechanism of action of PN2-3 further, we characterized its structure in solution and mapped its interaction with tubulin both on its sequence and on the tubulin surface. NMR data show that PN2-3 does not fold into a well defined tertiary structure but that a 23-residue region (from residues 29 to 51) is predominantly α-helical in agreement with CD measurements. This helical content is very temperature-sensitive as PN2-3 unfolds reversibly upon heating in a broad and non-cooperative transition (Fig. 2B). Inspection of the amino acid composition (Fig. 3B) reveals that the PN2-3 sequence has a strong bias toward small and/or polar residues and a low content of hydrophobic amino acids: 73% of all residues are Gln, Asn, Ser, Pro, Glu, Lys, Gly, and Ala, whereas only 22% of all residues are Val, Leu, Ile, Met, Phe, Tyr, and Trp. Therefore both its sequence signature and its conformational behavior classify PN2-3 as an intrinsically disordered protein (44). Intrinsically disordered proteins have been found to fall in five broad categories, one of them being constituted of effectors that regulate large multiprotein complexes such as the ribosome or the cytoskeleton (45). The 1H-15N HSQC spectrum of a 1:1 mixture of PN2-3 with tubulin indeed shows that a 76-residue-long PN2-3 region (residues 10–85) binds tightly to tubulin (Fig. 3). Consistently we found that the PN2-3 10–85 fragment recapitulates all the functional properties of PN2-3 interaction with tubulin: it inhibits MT assembly and the tubulin GTPase activity, interferes with RB3SLD for tubulin binding, and inhibits vinblastine-mediated association as well as nucleotide exchange (data not shown); it defines the MDD of CPAP. This fragment is very similar to a 70-residue-long fragment of PN2-3 (residues 11–81 in our numbering) very recently shown to inhibit MT nucleation from centrosomes, and its limits are consistent with the observation that residues Lys68 and Arg69 are essential for tubulin binding (46). Interestingly the PN2-3 MDD comprises the isolated PN2-3 α-helix but is also predicted by several algorithms to contain an additional short helix (residues 73–84; supplemental Fig. 1) separated from the first one by a proline-rich region (Pro55, Pro57, Pro62, and Pro65). It is likely that it folds into a defined tertiary structure upon tubulin binding, and it is possible that this folded conformation has a higher helical content than isolated PN2-3, reminiscent of a number of unstructured proteins that fold only upon binding to their physiological partner (44). Finally we note that the NMR and the GTPase activity inhibition data demonstrate that the tubulin/PN2-3 association is tight (Ka > 106 m-1), suggesting that soluble αβ-tubulin is the physiological target of PN2-3. Because another CPAP domain binds autonomously to MTs (46) and because the whole protein has been shown to interact with γ-tubulin (6), CPAP emerges as a potential multifunctional MT regulator.
To get insight into the regions of tubulin that interact with PN2-3, we measured the PN2-3 interference with well characterized tubulin properties. We first showed that PN2-3 interferes with the longitudinal interactions of tubulin. The simplest explanation, involving only local effects, is that PN2-3 contacts tubulin residues in its longitudinal intermolecular interfaces either in the α or in the β subunit. To discriminate between these two possibilities, we checked the competition for tubulin binding of PN2-3 with the stathmin N-cap and evaluated the effect of PN2-3 on tubulin nucleotide exchange. Taken together, the results of these two experiments strongly suggest that PN2-3 contacts residues of the β-tubulin intermolecular longitudinal interface. Targeting that surface is an efficient mechanism for inhibition of MT growth and for interference with the formation of T2R as these two tubulin assemblies require proper longitudinal interactions to be established. We showed that PN2-3 sequesters tubulin away from these assemblies. The only other tubulin-sequestering proteins that have been extensively studied thus far belong to the stathmin family (4). They sequester tubulin by maintaining it in a curved assembly consisting of two heterodimers and by capping α-tubulin at one end of this assembly (37). The PN2-3 fragment of CPAP differs from them in two respects: it sequesters a single tubulin heterodimer and does so, at least in part, by capping its β subunit longitudinal interface.
In conclusion, this work validates PN2-3 and its 10–85 region as a single tubulin heterodimer-sequestering motif, a finding that, to the best of our knowledge, is not shared by other proteins. As such it is a valuable tool for the study and handling of unassembled tubulin, a protein well known to be fragile when kept in this state. In addition, as αβ-tubulin is abundant in the pericentriolar material throughout the cell cycle, its sequestration by PN2-3 is likely to be important for the function of CPAP in the centrosome.
Supplementary Material
Acknowledgments
The Région Ile de France, the Conseil Général de l'Essonne, Genopole®, Direction des Sciences du Vivant/Commissariatà l'Energie Atomique, and the Association Française contre les Myopathies are acknowledged for contributions to the NMR equipment.
This work was supported by INSERM, CNRS, Association pour la Recherche sur le Cancer (to P. A. C. and A. S.), and Association Française contre les Myopathies (to A. S.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. 1.
Footnotes
The abbreviations used are: MT, microtubule; CPAP, centrosomal P4.1-associated protein; MDD, microtubule-destabilizing domain; STD-NMR, saturation transfer difference NMR spectroscopy; SLD, stathmin-like domain; Tc, tubulin·colchicine complex; T2R, tubulin·RB3SLD complex; T2S, tubulin·stathmin complex; T2SLD, tubulin·SLD complex; MES, 4-morpholineethanesulfonic acid; HSQC, heteronuclear single quantum correlation; CD, circular dichroism; PIPES, 1,4-piperazinediethane-sulfonic acid; SPR, surface plasmon resonance.
References
- 1.Schiebel, E. (2000) Curr. Opin. Cell Biol. 12 113-118 [DOI] [PubMed] [Google Scholar]
- 2.Belmont, L. D., and Mitchison, T. J. (1996) Cell 84 623-631 [DOI] [PubMed] [Google Scholar]
- 3.Jourdain, L., Curmi, P., Sobel, A., Pantaloni, D., and Carlier, M. F. (1997) Biochemistry 36 10817-10821 [DOI] [PubMed] [Google Scholar]
- 4.Charbaut, E., Curmi, P. A., Ozon, S., Lachkar, S., Redeker, V., and Sobel, A. (2001) J. Biol. Chem. 276 16146-16154 [DOI] [PubMed] [Google Scholar]
- 5.Steinmetz, M. O. (2007) J. Struct. Biol. 158 137-147 [DOI] [PubMed] [Google Scholar]
- 6.Hung, L. Y., Tang, C. J., and Tang, T. K. (2000) Mol. Cell. Biol. 20 7813-7825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bond, J., Roberts, E., Springell, K., Lizarraga, S. B., Scott, S., Higgins, J., Hampshire, D. J., Morrison, E. E., Leal, G. F., Silva, E. O., Costa, S. M., Baralle, D., Raponi, M., Karbani, G., Rashid, Y., Jafri, H., Bennett, C., Corry, P., Walsh, C. A., and Woods, C. G. (2005) Nat. Genet. 37 353-355 [DOI] [PubMed] [Google Scholar]
- 8.Cho, J. H., Chang, C. J., Chen, C. Y., and Tang, T. K. (2006) Biochem. Biophys. Res. Commun. 339 742-747 [DOI] [PubMed] [Google Scholar]
- 9.Hung, L. Y., Chen, H. L., Chang, C. W., Li, B. R., and Tang, T. K. (2004) Mol. Biol. Cell 15 2697-2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Castoldi, M., and Popov, A. V. (2003) Protein Expr. Purif. 32 83-88 [DOI] [PubMed] [Google Scholar]
- 11.Detrich, H. W., III, and Williams, R. C. (1978) Biochemistry 17 3900-3907 [DOI] [PubMed] [Google Scholar]
- 12.Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY
- 13.Clement, M. J., Jourdain, I., Lachkar, S., Savarin, P., Gigant, B., Knossow, M., Toma, F., Sobel, A., and Curmi, P. A. (2005) Biochemistry 44 14616-14625 [DOI] [PubMed] [Google Scholar]
- 14.Cornilescu, G., Delaglio, F., and Bax, A. (1999) J. Biomol. NMR 13 289-302 [DOI] [PubMed] [Google Scholar]
- 15.Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T., and Warren, G. L. (1998) Acta Crystallogr. Sect. D Biol. Crystallogr. 54 905-921 [DOI] [PubMed] [Google Scholar]
- 16.Koradi, R., Billeter, M., and Wuthrich, K. (1996) J. Mol. Graph. 14 51-55 [DOI] [PubMed] [Google Scholar]
- 17.Mayer, M., and Meyer, B. (1999) Angew. Chem. Int. Ed. Engl. 38 1784-1788 [DOI] [PubMed] [Google Scholar]
- 18.Chen, Y. H., Yang, J. T., and Chau, K. H. (1974) Biochemistry 13 3350-3359 [DOI] [PubMed] [Google Scholar]
- 19.Ouali, M., and King, R. D. (2000) Protein Sci. 9 1162-1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kneller, D. G., Cohen, F. E., and Langridge, R. (1990) J. Mol. Biol. 214 171-182 [DOI] [PubMed] [Google Scholar]
- 21.McGuffin, L. J., Bryson, K., and Jones, D. T. (2000) Bioinformatics 16 404-405 [DOI] [PubMed] [Google Scholar]
- 22.Stultz, C. M., White, J. V., and Smith, T. F. (1993) Protein Sci. 2 305-314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang, C., Cormier, A., Gigant, B., and Knossow, M. (2007) Biochemistry 46 10595-10602 [DOI] [PubMed] [Google Scholar]
- 24.Carlier, M. F., Didry, D., and Pantaloni, D. (1987) Biochemistry 26 4428-4437 [DOI] [PubMed] [Google Scholar]
- 25.Siegel, L. M., and Monty, K. J. (1966) Biochim. Biophys. Acta 112 346-362 [DOI] [PubMed] [Google Scholar]
- 26.Bai, R. L., Pettit, G. R., and Hamel, E. (1990) J. Biol. Chem. 265 17141-17149 [PubMed] [Google Scholar]
- 27.Penefsky, H. S. (1977) J. Biol. Chem. 252 2891-2899 [PubMed] [Google Scholar]
- 28.Caplow, M., and Zeeberg, B. (1980) Arch. Biochem. Biophys. 203 404-411 [DOI] [PubMed] [Google Scholar]
- 29.Jourdain, I., Lachkar, S., Charbaut, E., Gigant, B., Knossow, M., Sobel, A., and Curmi, P. A. (2004) Biochem. J. 378 877-888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martin, S. R., and Schilstra, M. J. (2008) Methods Cell Biol. 84 263-293 [DOI] [PubMed] [Google Scholar]
- 31.Clement, M. J., Savarin, P., Coutant, J., Toma, F., and Curmi, P. (2008) Biomol. NMR Assign. 2 115-117 [DOI] [PubMed] [Google Scholar]
- 32.Wishart, D. S., Sykes, B. D., and Richards, F. M. (1992) Biochemistry 31 1647-1651 [DOI] [PubMed] [Google Scholar]
- 33.Zuiderweg, E. R. (2002) Biochemistry 41 1-7 [DOI] [PubMed] [Google Scholar]
- 34.David-Pfeuty, T., Simon, C., and Pantaloni, D. (1979) J. Biol. Chem. 254 11696-11702 [PubMed] [Google Scholar]
- 35.Weisenberg, R. C., and Timasheff, S. N. (1970) Biochemistry 9 4110-4116 [DOI] [PubMed] [Google Scholar]
- 36.Gigant, B., Wang, C., Ravelli, R. B., Roussi, F., Steinmetz, M. O., Curmi, P. A., Sobel, A., and Knossow, M. (2005) Nature 435 519-522 [DOI] [PubMed] [Google Scholar]
- 37.Ravelli, R. B., Gigant, B., Curmi, P. A., Jourdain, I., Lachkar, S., Sobel, A., and Knossow, M. (2004) Nature 428 198-202 [DOI] [PubMed] [Google Scholar]
- 38.Gigant, B., Curmi, P. A., Martin-Barbey, C., Charbaut, E., Lachkar, S., Lebeau, L., Siavoshian, S., Sobel, A., and Knossow, M. (2000) Cell 102 809-816 [DOI] [PubMed] [Google Scholar]
- 39.Steinmetz, M. O., Kammerer, R. A., Jahnke, W., Goldie, K. N., Lustig, A., and van Oostrum, J. (2000) EMBO J. 19 572-580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weisenberg, R. C., Borisy, G. G., and Taylor, E. W. (1968) Biochemistry 7 4466-4479 [DOI] [PubMed] [Google Scholar]
- 41.Lowe, J., Li, H., Downing, K. H., and Nogales, E. (2001) J. Mol. Biol. 313 1045-1057 [DOI] [PubMed] [Google Scholar]
- 42.Cormier, A., Marchand, M., Ravelli, R. B., Knossow, M., and Gigant, B. (2008) EMBO Rep. 9 1101-1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Amayed, P., Carlier, M. F., and Pantaloni, D. (2000) Biochemistry 39 12295-12302 [DOI] [PubMed] [Google Scholar]
- 44.Dyson, H. J., and Wright, P. E. (2005) Nat. Rev. Mol. Cell Biol. 6 197-208 [DOI] [PubMed] [Google Scholar]
- 45.Tompa, P. (2002) Trends Biochem. Sci. 27 527-533 [DOI] [PubMed] [Google Scholar]
- 46.Hsu, W. B., Hung, L. Y., Tang, C. J., Su, C. L., Chang, Y., and Tang, T. K. (2008) Exp. Cell Res. 314 2591-2602 [DOI] [PubMed] [Google Scholar]
- 47.Bevington, P. R., and Robinson, K. D. (1992) Data Reduction and Error Analysis for the Physical Sciences, 2nd Ed., McGraw-Hill, New York
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.