

Abstract
Avian influenza virus subtype H5N1 is a potential pandemic threat with human-adapted strains resistant to antiviral drugs. Although virtual screening (VS) against a crystal or relaxed receptor structure is an established method to identify potential inhibitors, the more dynamic changes within binding sites are neglected. To accommodate full receptor flexibility, we use AutoDock4 to screen the NCI diversity set against representative receptor ensembles extracted from explicitly solvated molecular dynamics simulations of the neuraminidase system. The top hits are redocked to the entire nonredundant receptor ensemble and rescored using the relaxed complex scheme (RCS). Of the 27 top hits reported, half ranked very poorly if only crystal structures are used. These compounds target the catalytic cavity as well as the newly identified 150- and 430-cavities, which exhibit dynamic properties in electrostatic surface and geometric shape. This ensemble-based VS and RCS approach may offer improvement over existing strategies for structure-based drug discovery.
Introduction
Avian influenza has received worldwide attention due to its rapid global spread via migratory birds and the growing number of human cases. The highly pathogenic avian influenza virus that the World Health Organization fears may cause a pandemic in humans comprises strains from the subtype H5N1 of influenza type A.(1) Subtypes of influenza virus are named based on the observed combinations of two viral surface membrane glycoproteins, hemagglutinin (HA)a and neuraminidase (NA), with 16 and 9 types known to date, respectively.(2) Three historical pandemic flu viruses were caused by H1N1 (1918), H2N2 (1957), and H3N2 (1968).
HA is involved in the attachment of viral particles to host cell surface glycoproteins, whereas NA plays a role in the release of newly synthesized viral particles (for a recent review, see ref (3)). NA, a membrane-bound homotetrameric enzyme, releases viral paricles by cleaving terminal sialic acid (SA) residues on the host cell surface proteoglycans, bound by HA.(4) As the NA active site is relatively well-conserved, it became a good target for structure-based antiviral drug development.(5) Commercial inhibitors such as zanamivir and oseltamivir are stockpiled by various countries in case of a pandemic outbreak. While both drugs are successful in treating several human cases,(6) drug-resistant strains have quickly emerged due to antigenic drift.7,8
In 2006, Russell et al. reported the first crystal structures of a group-1 NA (N1), which revealed the existence of a “150-cavity” that, under various crystallization conditions, closed upon ligand binding to the active site through movement of the 150-loop.(9) To further probe the structure and dynamics of N1, explicitly solvated molecular dynamics (MD) simulations on the tetrameric N1 systems with (holo) or without (apo) oseltamivir bound were performed.(10) The simulations indicated that the flexibility of the 150-loop may be even greater than previously anticipated and that the dynamics of the neighboring 430-loop also influenced the topology of the binding site.
More recently, representative conformations of the N1 binding site (including the flexible 150- and 430-loop regions), extracted from a clustering analysis of the N1 MD simulations, were used for computational solvent mapping (CS-Map), which assessed the binding affinity of small, solvent-sized probe molecules within these areas.(11) The mapping analyses revealed the presence of novel druggable hot spots in the 150- and 430-loop regions, providing further support for the feasibility of developing high-affinity inhibitors capable of binding these areas. Such inhibitors may be applicable to the group-1 NA’s including N1, N4, N5, and N8, which have nearly identical active site regions,(9) but not necessarily group-2 enzymes (N2, N3, N6, N7, and N9), which appear to lack such well defined cavities.
Structure-based drug discovery has made significant progress in the past 30 years,(12) benefiting from recent advances in high performance and distributed grid computing.(13) However, the drawbacks are well documented (reviewed in refs (14,17)), and only in the past decade have methods able to treat receptor flexibility been developed.18–25 A related challenge is the identification of drug leads out of large compound libraries through receptor-based virtual screening (VS). VS is a widely used method that has been shown to be successful in a variety of studies, although it also has many shortcomings (reviewed in refs (26) and (27)). The treatment of receptor flexibility within the scope of VS is still in its infancy and a very active area of research, as it is widely accepted that receptor flexibility plays an important role in molecular recognition. The use of multiple experimentally derived protein structures in VS has been shown to effectively improve enrichment factors28,29 and, most recently, aid in the discovery of novel compounds active against the p53 tumor repressor.(30)
The relaxed complex scheme (RCS), a hybrid computational method that combines the advantages of docking algorithms with dynamic structural information provided by MD simulations, has been successfully applied to a number of systems.22,23,31 Recently, raltegravir became the first FDA approved drug targeting HIV integrase,(32) and the treatment of receptor flexibility with MD simulations played a critical role in understanding the mechanism of action for this new class of inhibitors.(33) During MD simulations of the integrase enzyme in complex with the known inhibitor 5CITEP, a new pocket adjacent to the binding site was revealed, and docking of ligands into this new area indicated favorable binding.(34) The development of compounds capable of binding to both the original binding site and the new cavity, so-called “butterfly” compounds, formed the basis for this new class of integrase inhibitors.
In this work, we build upon these recent advances and present a novel, systematic, in silico lead identification strategy, with the goal of taking full receptor flexibility into account in the lead identification and scoring stages. VS experiments are carried out against the NCI diversity set (NCIDS) on a set of eight N1 representative receptor structures,(31) consisting of two crystal structures and the three most dominant cluster-representative receptor structures as extracted from the explicitly solvated apo and holo MD simulations.(10) The use of the most dominant MD-generated structures systematically incorporates a broader range of receptor configurations into the hit identification process, in contrast to screening the crystal structures alone. We anticipate that for very flexible receptors, such as N1, the incorporation of this variable structural information may identify new drug leads. Indeed, comparison of the cluster representative structures suggests that the remarkable flexibility of the 150- and 430-loops results in correspondingly different 150-, 430-, and binding site cavities.
This novel VS approach is further combined with the RCS, a lead validation and refinement technique in which the top N hits are redocked to the entire nonredundant receptor ensemble and rescored. Ultimately, we report 27 experimentally available compounds that are predicted to have a binding affinity equal to or better than four known N1 antivirals. Importantly, half of these compounds would have been neglected based on the crystal structure VS scores alone. Examination of the interactions of these top hits within the NA active site region reveals different classes of compounds with binding preference to the 150-, 430-, and catalytic cavities. Several compounds are predicted to interact with key residues across two cavities and may provide novel bridging compound scaffolds. We anticipate that this ensemble-based virtual screen with relaxed complex scheme (EVSRCS) approach may contribute to better structure-based drug design strategies for very flexible receptors.
Materials And Methods
Apo System Setup
Details of the simulation setup without any ligand bound are as previously described.(10)Briefly, the apo system was built with the avian H5N1 NA crystal structure 2HTY.(9) Protonation states for histidine residues were defined at an apparent pH 6.5 using the PDB2PQR web server.(35) All crystallographically resolved water oxygen positions were retained in the apo system, as well as Ca2+ ions, which are required for optimal NA function.(36) To mimic experimental inhibition assay conditions, a 150 mM NaCl salt bath was introduced. The simulated system contained 112311 atoms.
Holo System Setup
Details of the simulation setup with oseltamivir bound are as previously described.(10) Briefly, the 2HU0 structure used for the holo system initially had a single oseltamivir molecule bound in the active site of chain B.(9) To introduce oseltamivir within each of the other chains, chain B was aligned to chain A, C, and D by superimposition of the backbone Cα atoms, and the resulting transformation matrix was applied to the oseltamivir molecule. Amber9 was used to set up the oseltamivir-bound system using an identical protocol to the apo system, with the exception of the additional oseltamivir parameters. The composite tetrameric oseltamivir-bound system, comprising one oseltamivir molecule in each of the four active sites and calcium ions in 150 mM NaCl salt bath, contained 112457 atoms.
Molecular Dynamics Simulations
The apo and holo systems were energy minimized for 5 × 104 steps using NAMD version 2.6b1 (http://www.ks.uiuc.edu/Research/namd).(37) The MD methods have been previously described in refs 10,11. Free dynamics were subsequently performed using a 2 fs time step for a total of 40 ns for each system. Trajectories were generated on the San Diego Supercomputing Center’s DataStar machine (benchmark time of 0.3 days/ns using 112 parallel processors).
RMSD Clustering to Extract Representative MD Structures
To generate a reduced, representative set of N1 structures, root-mean-square difference (RMSD) conformational clustering was performed as previously described in ref (11) with the GROMOS++ analysis software,(38) part of the GROMOS05 software for biomolecular simulation (http://www.igc.ethz.ch/GROMOS). Briefly, for the apo and holo simulations, each chain of the tetramer was extracted at 10 ps intervals over the 40 ns simulation. The resulting 1.6 × 104 trajectory structures for each simulation were superimposed using all Cα atoms to remove overall rotation and translation. The RMSD-clustering was performed on the subset of 62 residues that line the entire binding-site area, which we define here as the binding-site residues: 117−119, 133−138, 146−152, 156, 179, 180, 196−200, 223−228, 243−247, 277, 278, 293, 295, 344−347, 368, 401, 402, and 426−441. These residues were clustered into batches of similar configurations using the atom-positional RMSD of all atoms (including side chains and hydrogens) as the similarity criterion. A cutoff of 1.3 Å was chosen after evaluation of the dependence of cluster populations against the total number of clusters found for each simulation using a cutoff in the range 1.0−1.5 Å. The central member cluster structure, the structure within the cluster having the smallest RMSD to all other structures within the cluster (i.e., the centroid), was chosen as the cluster representative structure. Fifty-one clusters were obtained for the apo system, and 27 clusters for the holo system, the three most dominant configurations represented about 21%, 20%, and 19% and 38%, 22%, and 14% of the apo and holo ensembles, respectively.(39) In Figure 2, we indicate the time points that correspond to each of the central member cluster structures. We do not mean to imply that all members of each cluster always form a continuous portion of the trajectory. In fact, although many structures near a similar time point can be found within any given cluster, multiple discontinuous time points are represented in the larger clusters. This indicates that areas of configurational space may be sampled multiple times within a single long trajectory.
Figure 2.

(A) Binding energy spectra of two known antivirals and the natural sialic acid substrate (SA) from the RC redocking against the 27 holo ensemble cluster representatives. The bar graph shows the percent ensemble population represented by each cluster, ordered by their corresponding MD simulation time step. (B) Binding energy statistics of the five control ligands from the RC redocking. The standard deviation of each RC weighted arithmetic mean of the binding energies is represented by an error bar.
Virtual Screening with AutoDock
The three most dominant clusters from each of the apo and holo system and the two original crystal structures are used as receptors for virtual screening against the NCIDS library of compounds. AutoDockTools version 1.4.5(40) was used to add polar hydrogens and assign Gasteiger charges(41) to each of the eight NA structures. The PDB2PQR web server (http://nbcr.net/pdb2pqr) was used to simulate a biological assay pH of 6.5.(35) For each of the eight NA structures, AutoGrid version 4.0 was used to create affinity grids centered on the active site. Each grid enclosed an area of 64 Å by 72 Å by 66 Å with 0.375 Å spacing, and affinity grids were calculated for all of the following atom types: A (aromatic carbon), C, F, I, N, NA (hydrogen-bond-accepting N) Cl, O, OA (hydrogen-bond-accepting O), P, S, SA (hydrogen-bond-accepting S), Br, H, and e (electrostatic). All NA structures from simulations were aligned with the closed 150-loop crystal structure (2HU4, chain B) to maintain a consistent grid location.
The NCIDS is a collection of approximately 2000 compounds that are structurally representative of a wide range of pharmacophores.(42) AutoDockTools version 1.4.5 was used to merge nonpolar hydrogens, add Gasteiger charges to each ligand, and set up rotatable bonds through AutoTors. A number of ligands containing rare earth elements could not be processed and were excluded. A total of 1883 compounds from the NCIDS were included in the screen. Five additional compounds experimentally known to bind avian influenza N1 were also included in the screen: sialic acid (N-acetyl neuraminic acid, aka, NANA or Neu5Ac, SA), 2,3-didehydro-2-deoxy-N-acetyl neuraminic acid (DANA), oseltamivir, zanamivir, and peramivir (an experimental inhibitor in phase II clinical trials(43)).
AutoDock version 4.0.1 with the Lamarckian genetic algorithm was used to simulate ligand−receptor docking.(44) Docking parameters were optimized for the positive control docking of oseltamivir to the closed 150-loop conformation of NA (2HU4) and were as follows: trials of 100 dockings, population size of 200, random starting position and conformation, translation step ranges of 2.0 Å, rotation step ranges of 50°, elitism of 1, mutation rate of 0.02, crossover rate of 0.8, local search rate of 0.06, and 5 million energy evaluations. A total of 15104 docking jobs were run on the National Biomedical Computation Resource (NBCR, http://nbcr.net) Kryptonite cluster. Docked conformations were clustered using a tolerance of 2.0 Å RMSD using the external clustering script provided with AutoDockTools version 1.4.5.
Docking results were sorted by the lowest binding energy of the most populated cluster using AutoDockTools version 1.4.5, and the top 30 hits from each of the eight systems were chosen for further analysis. The top hits from each screen were initially filtered for druglikeness by their adherence to Lipinski’s “rule of fives”.(45) It is recommended that compounds should conform to two or more of these rules,(46) and we only considered compounds conforming all rules in this study, i.e., having no more than five hydrogen bond donors, no more than 10 hydrogen bond acceptors, a molecular weight under 500 g/mol, and a partition coefficient (log P) under 5.(45) Compounds were filtered after rather than prior to docking to prevent premature exclusion of compounds that do not meet this criterion. A brief analysis of top binders, however, revealed that most compounds that failed the Lipinski rules were unlikely drug candidates. Although some compounds that failed the Lipsinki rules also had good docking scores, this cutoff criterion was imposed to focus the scope of our search to the more promising compounds. The remaining hits were further filtered based on clustering criteria in AutoDock: we only consider a compound among the top hits if the most populated cluster included at least 25% of all docked conformations. Receptor−ligand interactions, including hydrogen-bonding interactions and molecular surfaces, were calculated in AutoDockTools version 1.4.5.47,48
A crucial metric for assessing the performance of high-throughput virtual screening is the extent to which a database of compounds could be enriched such that only a much smaller subset needs to be prepared and assayed to identify leads. The metric is often quantified by the enrichment factor (EF):(19)
where D is the total number of compound in the data set and S is a subset (e.g., < 2%) of the data set.
Possible Aggregator Identification
Top hits were compared to a list of 263 known false positives.(49) These so-called “promiscuous inhibitors” are irregular compounds that inhibit nonspecifically by forming aggregates onto which the protein will adsorb and thus block activity.49,50 A similarity search was performed to identify false-positive aggregators within the 27 top hits, and none were found using the Tanimoto index at 98% cutoff.
Relaxed Complex Scheme
To validate and refine the virtual screening results, redocking experiments were performed on the combined top 27 hits plus oseltamivir, zanamivir, peramivir, DANA, and SA. Each compound was redocked into the 27 central member cluster structures, which accounts for 100% of the ensemble variance from the holo simulation at 1.3 Å cutoffs. Following the same docking procedure and parameter set described in the previous section, 756 redocking calculations were distributed to the Pacific Rim Application and Grid Middleware Assembly (PRAGMA, http://www.pragma-grid.net) grid using the metascheduler Community Scheduler Framework (CSF4, http://sourceforge.net/projects/gcsf) command line client and the output transferred back to the NBCR kryptonite cluster. The binding free energies were extracted with AutoDockTools 1.4.5 scripts, and the ligand binding spectra were plotted for further analysis.
For the weight means and standard deviations calculations, the following formula was used.
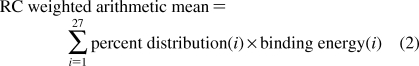 |
 |
i is the index number of each holo ensemble cluster, whose percent distribution sums up to 100%. The selection criteria are identical to that used in the earlier virtual screening steps. In cases where the AutoDock cluster size does not meet the 25% cluster size cutoff, the lowest binding energy of the most populated cluster is still used for calculation of the weighted mean of binding energies.
RC weighted harmonic mean is calculated by first converting the binding energies to inhibition constants Ki as follows:
where R is 1.98719 and T is 298.15. The weighted harmonic mean Ki value is then calculated from:
 |
At the end, Ki is converted back to the RC harmonic weighted mean binding energy.
Electrostatic Surface Calculation and Visualization
Electrostatic potentials were calculated using the Adaptive Poisson−Boltzmann Solver (APBS) and mapped to the reduced molecular surface by Python Molecular Viewer.48,51
Solvent Accessible Surface Area
To further quantify the changes in the binding site, the solvent accessible surface area was computed for each of the eight screened structures using VMD 1.8.6(52) with a probe radius of 1.4 Å. To maintain consistency in the comparisons to the crystal structures, the hydrogens were not included for the cluster representative structures.
Results
Structural Variability of the N1 Active Site
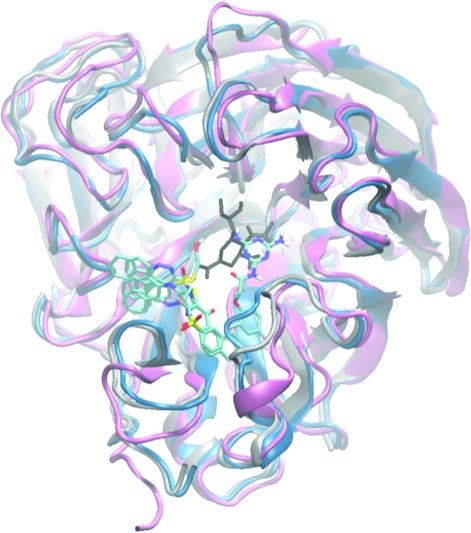
The two crystal structures of N1, 2HTY (apo crystal) with the “open 150-loop”, 2HU4 (holo crystal) with the “closed 150-loop”, and the corresponding three most dominant representative cluster members from the MD trajectories of the two structures are examined in parts A and B of Figure 1, respectively. The three most dominant clusters accounted for approximately 60% and 75% of the overall MD ensembles, respectively. Pairwise RMSD’s are presented in Table 1 for all structures over four α carbon subsets: (i) all N1 atoms, (ii) the 62 residues lining the binding site, (iii) the 150-loop residues 146−152, and (iv) the 430-loop residues 430−439. As shown in Table 1B and Figure 1, Apo-1 exhibits the biggest differences in the 430-loop compared to the other structures except for Apo-2. On the other hand, the apo crystal is significantly different from Apo-1, Apo-3, Holo-1, and Holo-3 but more similar to Apo-2 and Holo-1 in the 150-loop; the holo crystal exhibits 3 Å RMSD to all other structures in terms of the 150-loop. Overall, this analysis indicates the areas of greatest structural diversity and similarity among the different structures.
Figure 1.

Three cluster representative structures for the three most dominant clusters from the (A) apo and (B) holo simulations. The structures from the most dominant clusters are colored in pink (Apo-1, Holo-1), the second most dominant in green (Apo-2, Holo-2), and the third most dominant in orange (Apo-3, Holo-3). The apo crystal structure (open 150-loop) is shown in silver, and the holo crystal structure (closed 150-loop) is shown in blue. The 150- and 430-loops are indicated.
Table 1. Distance Matricesa.
 |
Distance matrices based on pairwise RMSD’s calculated using (A) all N1 atoms (lower triangle) and the 62 residues lining the binding site (upper triangle); (B) the 150-loop residues (lower triangle) and the 430-loop residues (upper triangle). In (B), the RMSD values of the Holo-1 cluster representative to the other structures are shown in grey, with the arrows illustrating the originating holo1 label for the 150-loop (solid arrow) and 430-loop (broken arrow). Bold numbers indicate the smallest RMSD value for a structure and italics indicate the largest value for a structure in comparison to all the other seven structures.
Positive Controls
The docking conditions for VS have been optimized using the oseltamivir-bound holo crystal structure (). Oseltamivir docks to the apo and holo crystal structures in a manner consistent with its crystallized conformation. The overall RMSD (all-atoms) of the predicted binding pose compared to the crystal structure pose is 1.8 Å for the Holo crystal structure and 2.1 Å for the apo crystal structure (). In the redocking of oseltamivir to the closed N1 structure, the differences between docked and crystal poses lie primarily in the orientation of the pentyl ether group, which interacts with the protein through variable hydrophobic interactions. The negatively charged carboxylate group is anchored through electrostatic interactions to the arginine trio (R118, R292, and R371) and its position does not differ significantly between the docked and crystal poses. The successful redocking of oseltamivir confirms that AutoDock 4.0.1 is able to emulate experimental binding with an excellent degree of accuracy and precision, serving as a positive control to validate our virtual screening hits.
To examine whether the screening conditions offer satisfactory discrimination of known NA inhibitors and to provide positive controls for our larger scale VS analysis, we docked the set of five known inhibitors to the holo crystal (with oseltamivir in the crystal structure removed) as well as the 27 clusters obtained from the MD trajectory clustering analysis (Figure 2). Both weighed harmonic mean and weighted arithmetic mean were used to calculate the binding affinity estimates, and both yield qualitatively similar results for the five control compounds (Figure 2B). In this work, we chose the weighed harmonic mean to rank the top hits because it tends to favor the lower binding energies, which should be given more weight in the evaluation of the overall binding affinity, and its better performance is supported by previous studies.53,54
For the control compounds, the binding energy difference fluctuates from 2.5 to 5 kcal/mol for DANA and zanamivir, respectively. For zanamivir, the experimental IC50 is from 0.5−2.5 µM,(55) the range we observed is from 0.05 µM (−10 kcal/mol) to 215 µM (−5 kcal/mol). In agreement with what is known experimentally of the sialic-acid analogs, oseltamivir is predicted to be the best overall binder, followed by zanamivir and DANA. However, peramivir is much more potent than DANA, oseltamivir, and zanamivir, but its average binding profile is closer to that of DANA (Figure 2). As the cluster size represented by each structure used in the redocking varies, as shown in the lower half of the Figure 2B, the weighted harmonic and arithmetic mean binding energies for each compound is calculated and the ranking is consistent with the visual examination described above.
Ensemble-Based Virtual Screens Define Top 27 Hits
The VS results performed with the two crystal structures and 6 most dominant MD ensemble representative structures are summarized in Only hits that are in the top 30 and have at least 25% of the total population in their most populated cluster are selected from each set of VS results and indicated in bold. As is evident, the number of structure variants captured different compounds with binding energies ranging from −11.6 to −9.25 kcal/mol. Note that at this cutoff of −9.25 kcal/mol, peramivir and DANA are actually not selected, which will be discussed later. As some hits are ranked among the top 30 in more than one structure or cluster representative, a total of 27 top hits are reported in this work (see Table 3for a preview). Given the size of the screening library employed here (1883 compounds), an enrichment factor of 20 is achieved for the top 2% ranked list and 40 for the top 1% ranked list.
Table 3. Final Set of Recommended Compoundsa.
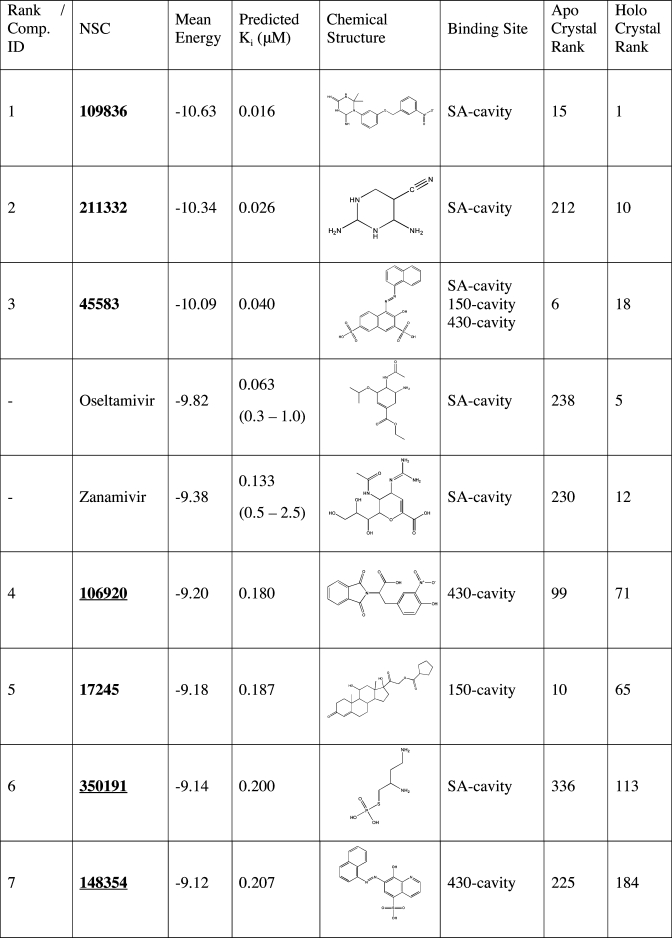 |
Table 3. Continued.
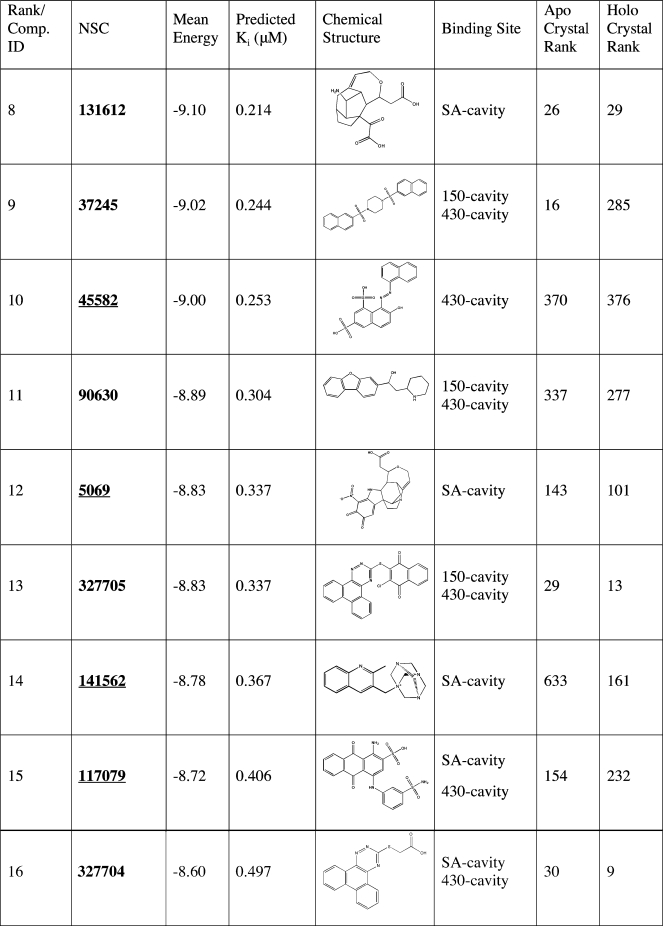 |
Table 3. Continued.
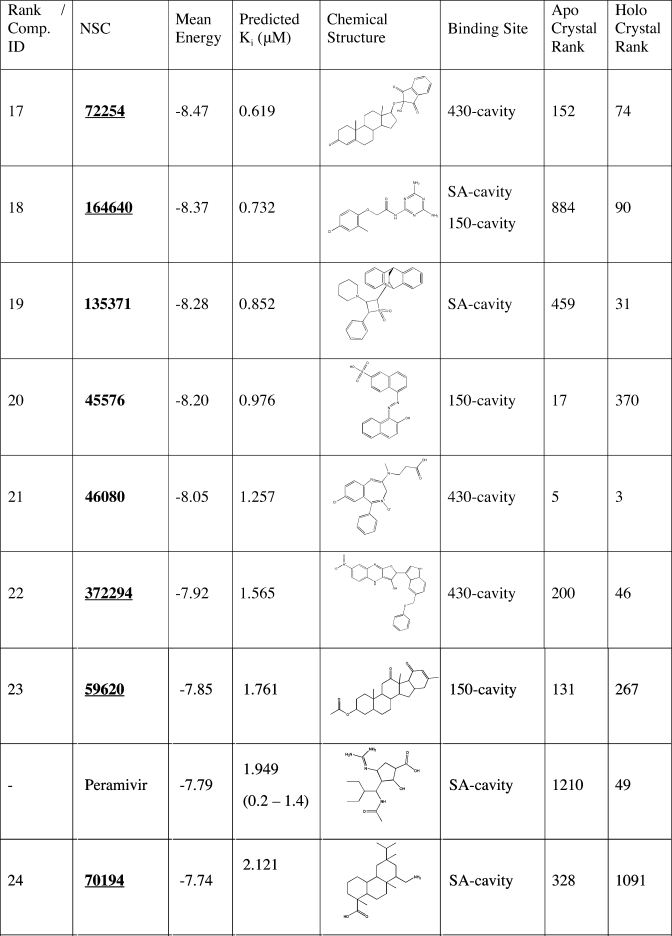 |
Table 3. Continued.
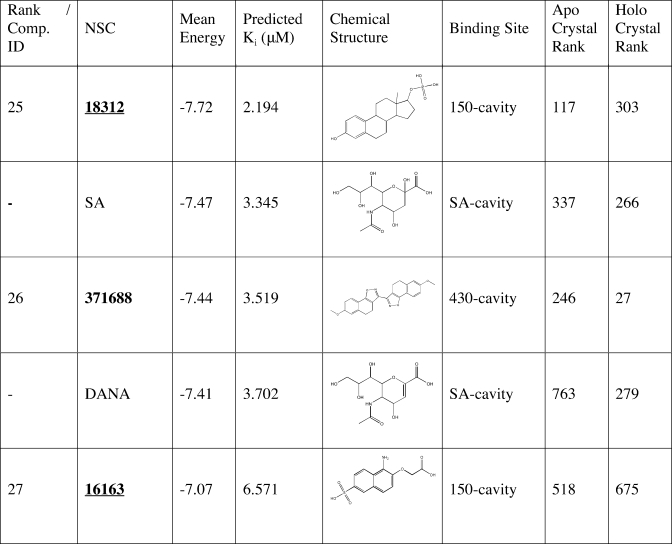 |
The top 27 recommended compounds ranked by their weighted harmonic mean energy (kcal/mol) from the RCS. The first column lists the final compound rank, which is also each compounds’ identification(42) (Comp ID). Also listed are the predicted Ki (µM) as calculated from the weighted harmonic mean binding energy, the chemical structure, and the predicted binding sites. Experimental IC50 values (nM/L) for oseltamivir, zanamivir, and peramivir are denoted with parenthesis.(55) For comparison, the last two columns indicate the compounds’ relative rank (among all 1883 NCIDS compounds and the five control compounds screened) in the crystal structure screenings. Underlined hits are those identified from the ensemble screens but ranked very poorly in the crystal structure screens.
Rescoring Using the Relaxed Complex Scheme
The selected top hits are redocked into the 27 nonredundant structures representing the entire holo MD ensemble. For each compound, the mean and minimum binding energies of a compound docked to the three most dominant clusters (ensemble mean), all 27 holo-ensemble cluster representative structures (RC mean), as well as the weighted harmonic mean of the binding energies to the full ensemble (RC weighted harmonic mean), are plotted in Figure 3. For most of the compounds, the RC mean and the ensemble mean differ by less than 0.5 kcal/mol. Only three compounds, 3, 4, and 15, had binding energies differing by more than 1 kcal/mol. In addition, when the RC weighted harmonic mean method is used with compounds that may not have met the 25% cluster size cutoff, DANA and peramivir are ranked higher than several of the top 27 hits.
Figure 3.

Binding energy statistics of the 27 top hits and five control compounds. Ensemble mean and ensemble minimum are the arithmetic mean and minimum of the binding energies obtained from screening against the three most dominant holo-ensemble structures. RC weighted harmonic nean, RC mean and RC minimum are the weighted harmonic mean, arithmetic mean, and minimum of the binding energies obtained from the RC redocking against the 27 holo ensemble cluster representatives. The percentages of the represented ensemble populations are shown in the bar graph scaled by the right axis.
Ligand Binding Cavities in N1
The active site of N1 may be best described in three cavities for convenience: (i) the sialic acid (SA) cavity and (ii) the 150-cavity, which have been previously named,4,9 and (iii) the 430-cavity, the presence and size of which is controlled by movement of the 430-loop. An examination of the top 27 hits and their interactions with the crystal structures and the cluster representatives clearly indicates that three cavities are areas of favorable binding (Figures 4 and 5, and Table 3). The residues lining the three binding site cavities, which interact with the identified top hits and known inhibitors, are summarized in Table 2 and are illustrated in for selected compounds. The specific cavities each ligand interacts with are summarized in Table 3.
Figure 4.

Selected hits clustered in the (A) apo crystal, (B) Apo-1, (C) Apo-2, and (D) Apo-3 structures. The electrostatic surface maps were generated using APBS and PMV using 85% opacity for the surface (scale shown in panel D).
Figure 5.

Selected hits clustered in the (A) holo crystal, (B) Holo-1, (C) Holo-2, and (D) Holo-3 structures. The electrostatics surface map was generated using APBS and PMV using 85% opacity for the surface (scale shown in panel D).
Table 2. Binding Cavity Residuesa.
| sialic acid cavity | R118, E119, L134, V149*, K150*, D151, R152, S153,P154, R156, W178, S179, S195, G196, I222, R224, E227, S246, E276, E277, R292, N294, Y347,R371, Y406 |
| 150-cavity | V116, I117, R118, L134, T135, Q136, S145, G147,T148, V149*, K150*, D151,R156, R430, P431,I437, W438, T439 |
| 430-cavity | N325, P326, Y347, N369, S370, R371, W403, S404,Y406, I427, R428, G429, R430, P431, K432,E433, I437, W438, T439 |
Italicized residues are those required for catalysis. Bold face identify those highlighted by the CS-Map analysis.(39) Underlined residues participate in interactions in more than one cavity across “cavity boundaries”. An asterisk indicates the residues that are found in the 150-cavity, but line the SA cavity when the 150-loop assumes a closed conformation.
The electrostatic surface maps for these ensemble structures are included to provide an additional perspective of the dynamic properties of these cavities. As shown in Figures 4 and 5, the SA cavity is largely negatively charged, whereas the 430-cavity is relatively positively charged and the 150-cavity remains relatively neutral in our docking conditions. The variations in the electrostatic surfaces are due to the changing surface topology (e.g., exposed residues) between the different structures.
The solvent accessible surface areas (SASA) of the cavities also vary greatly. To compare the changes in the binding site, the SASAs were computed for the 62 residues lining the binding site and are as follows in decreasing order (in Å2): Apo-1, 6125; Holo-3, 5844; Apo-2, 5812; Apo-3, 5793; Holo-2, 5731; Holo-1, 5658; 2HTY, 5467; 2HU4, 5197.
The apo ensemble representative structures illustrate the large changes in the SASAs during the apo simulation. In Apo-1, the 430-cavity is wide open (Figure 2b); the 430-loop Cα RMSD is almost 4 Å from either of the crystal structures (Table 1), and this is reflected in the largest exhibited SASA. Apo-2, however, with the 150-loop Cα RMSD at 1.14 Å from the Apo crystal structure, has a much larger 3.88 Å RMSD from the holo crystal structure. This shift has an effect of reducing the size of the 150-cavity. Apo-3 exhibits significant outward movement of both the 430-loop and the 150-loop, with the 150 loop Cα RMSD 1.17 Å from the Apo crystal and 3.93 Å from the holo crystal.
Overall, the holo ensemble generated structures exhibit a tighter range of values for their respective SASAs as compared to the Apo ensemble. Holo-1, which represents 40% of the MD snapshots, is most similar to Apo-2 in the 150-loop Cα RMSD, and with a similar 150-cavity empty with no ligand bound. Holo-2 shows significant movement of the 150-loop, with the 150 cavity smaller, yet allowing a bridging compound to interact in the cavity. Holo-3, which presents the largest SASA of the holo ensemble representative structures and shows significant changes in its electrostatic surface properties, is also capable of binding bridging compounds.
Characteristics of Top Hits
The two top ranked hits, 1 and 2, both contain a nitrogen-heavy six-membered ring. Compound 2 has a pyrimidinecarbonitrile scaffold and is the parental compound for several existing compounds found in the patent database. It binds consistently in the SA cavity regardless of 150- or 430-loop motion (). In its lowest energy pose docked to Apo-3, it forms hydrogen bonds to framework residues E119 and E227,(56) non-hydrogen bonding interactions with catalytic residues D151, R152, and Y406, and with framework residue W178.(56) Compound 1 has a triazinane scaffold, derivatives of which are also found in the patent database with biological effects (Figure 6a). In the lowest energy conformation docked to the holo crystal structure, its diimino-triazinane functional group forms hydrogen bonds to framework residues E119 and S179 and the hydoxy(oxido)amino functional group forms hydrogen bonds to the catalytic arginine trio (R118, R292, and R371) in the same manner as the carboxylate functional group of oseltamivir.(56) Compound 1 also forms non-hydrogen-bonding interactions with known catalytic residues D151, R224, E276, and Y406 and framework residues R156, W178, E227, and E277.(56) Compound 1 was ranked high when the VS was conducted with the holo crystal alone. However, Compound 2 dropped to no. 212 when the apo crystal is used.
Figure 6.

(A) The apo crystal structure (2HTY) is shown above in silver with top hits 1 and 21 bound. These two compounds dock similarly in both crystal structures and their overlapping positions in both are pictured above. (B) A large number of hits docked to the wide-open 150-cavity in Apo-1, shown above in pink with compound 27 bound.
Potential 150- and 430-Cavity Binders
A number of the compounds targeting the 150-cavity would not have been discovered or ranked extremely low (Table 3) based on the crystal structure screens alone. Some of the highest affinity hits to the open and wide-open 150-cavity are steroids such as 5, 23, and 25. These compounds interact with the 150-cavity primarily through hydrophobic interactions, specifically to hydrophobic residues V116, I117, L134, T135, S145, T148, V149, P431, I437, W438, and T439. Several other naphthalene-based compounds also bind with high affinity in the 150-cavity, interacting through hydrophobic interactions to the hydrophobic naphthalene group and through hydrogen-bonding interactions to polar functional groups on the naphthalene base (Table 3). These compounds include compounds 3, 9, 13, and 20.
Similar to the wide-open 150-cavity, the open 430-cavity is seen only in MD simulations and not observed in the crystal structures. Thirteen hits to the 430-cavity were identified among the top 27 hits (Table 3). The 430-cavity also seems to favor naphthalene-based compounds, namely compounds 9, 13, 7, and 10. These compounds interact through hydrophobic interactions to hydrophobic residues P326, Y347, S370, W403, S404, Y406, I427, G429, P431, I437, W438, and T439 in the 430-cavity. Compound 7 is one example of a naphthalene-based compound that binds with high affinity to the open 430-cavity.
Other compounds with hydrophobic benzene rings, similar to naphthalene, also bind in the open 430-cavity with high affinity, for example, compounds 4, 11, 16, 21, 22, and 26. Compound 21 is an interesting benzodiazepine that is highly ranked in the crystal structure screens but poorly ranked in the apo- and holo-ensemble screens and the RCS redocking (Table 3).
Potential Bridging Compounds
Of the top 27 hits, seven hits dock simultaneously to more than one cavity (Table 3). Four hits dock in the SA cavity and either the 150-cavity or the 430-cavity (see for examples of the apo ensemble). These hits are of particular interest because they may be linked to known inhibitors to form a novel, bridged inhibitor that binds with higher affinity.
Three hits, compounds 3, 15, and 16, dock primarily with hydrophobic interactions in the 430-cavity and extend a polar functional group into the SA cavity. Compound 18 appears to be a particularly promising cavity-bridging compound. The compound contains a diamino-triazine group that binds in the SA cavity and forms hydrogen bonds to catalytic residue R118 and to framework residues S179 and E277.(56) Of these three residues, S179 and E277 are also predicted to mediate hydrogen-bonding by the CS-Map analysis.(39) It also interacts through nonbonded interactions to catalytic residues D151 and Y406 and framework residues E119, R156, W178, and E227.(56)
Compound 18 binds simultaneously to the open 150-cavity through hydrophobic contacts to residues I117, T135, Q136 and T439, which are mediated by a methyl-chlorobenzene group on the ligand. Unlike the other three hits that bind in the SA cavity, compound 18 need not be linked to SA. It is predicted to be a compound that will naturally bridge the catalytic residues in the SA cavity to the high-affinity open 150-cavity.
Discussion
Ensemble-Based Virtual Screening and Relaxed Complex Scheme
The development of computational tools for computer-aided drug design depends on the critical compromise between accuracy and computational costs. Ideally, the most reliable prediction of molecular affinity can be obtained through rigorous free energy calculations of the ligand-binding process (ref (57) and references within). Although the theory and methods are well established for calculating free energies, in practice, however, they are still prohibitively expensive to be employed in virtual screening of drug-like databases.
Sampling Space for Receptor Flexibility
To account for receptor flexibility in ligand binding, we have included representative snapshots from the MD clustering analysis as part of the receptor ensemble, in addition to the crystal structures. In total, the four concatenated monomer trajectories (extracted from the tetramer simulation) are equivalent to 160 ns of MD simulation. By choosing the three most dominant clusters from each simulation, the coverage of 60% of the apo simulation and 75% of the holo simulation accounts for a generous sampling of configurational space for N1. Still, the question remains whether the reduced set of cluster representative structures are biologically relevant. We have used the set of known inhibitors to investigate the behavior of the system. As shown in Figure 2, some configurations, e.g., 3.41, 29.68 ns, seem unfavorable to all the inhibitors, with as much as 5 kcal/mol difference observed for zanamivir. It is also evident that the three most dominant clusters, which correspond to 2.65, 5.76, and 22.02 ns, do not always have the lowest binding energy, which is often found in clusters with less than 0.01% of the total population of MD snapshots. However, considering that the dominant configurations are most likely to be encountered by a ligand, we conjecture that the dominant clusters will be good candidates to represent the diversity of configurational space, within the sampling limitations of the MD simulations.
Rescoring the Top Hits
The top compounds were redocked into the entire, nonredundant holo-ensemble. There are several reasons to use the holo-enzyme. First, the oseltamivir-bound holo-ensemble accounts for ligand-induced effects on the binding site. Second, any inhibitors may have to compete for the binding site with a favored natural substrate already bound. Third, the holo ensemble contains oseltamivir, an analogue of the natural substrate SA. In fact, from this study, as shown in Figure 2B, the Apo-1 cluster representative has oseltamivir bound at the 150-cavity, albeit with a higher binding energy (−7.22 kcal/mol) This further suggests that the holo ensemble is more appropriate to use for rescoring the hits. Last, the choice to use a holo-ensemble has also been shown to perform well in other works on the RC methodology(31) and the discovery of inhibitors against an RNA editing ligase in Trypanosoma brucei, the parasite responsible for African sleeping sickness.(58) The latter study used the QR-factorization method to extract a nonredundant ensemble of MD snapshots and the RCS for rescoring of hits from a VS against a single crystal structure.
A preliminary study using the known set of inhibitors indicated that the weighted harmonic mean method is able to rank the controls relative to one another for three out of the four inhibitors (Figure 3). The natural substrate SA ranks closely to DANA, with oseltamivir and zanamivir correctly ranked with higher affinity. The exception is peramivir, which is ranked lower than what is expected based on its experimental IC50 values. Closer examination of the N8 with peramivir bound (2HTU)(9) and the docking pose of peramivir in 2HTY revealed no differences in the position and orientation of the ligand (data not shown). It is possible that the current docking condition does not account for stabilizing interactions conferred by the solvent, water molecules, or other potential ligand induced conformational changes. On the other hand, the standard error (−2 kcal/mol) of AutoDock4 may be the limiting factor and the rescoring and reranking would be able to discriminate the true differences between some hits. This is consistent with the general observation that docking programs are good at separating the binders from nonbinders, but poor at ordering the hits based on their affinity.(59) However, by taking into account the different distributions of the cluster size, giving more weight to the clusters with more substantial populations in the calculation of the mean binding energy, the weighted harmonic mean method may be able to improve the rankings, and may offer an interesting area for future methodological development.
For example, for each compound, we have examined the ensemble minimum binding energy, the lowest value from one of the three most dominant clusters, and the RC minimum binding energy, from the cluster with the lowest binding energy of all the 27 clusters (Figure 3). Eleven out of the 27 compounds had the ensemble minimum in the most abundant cluster, but only one of which, compound 10, had a RC minimum also in an abundant cluster. Overall, six out of 27 compounds had the ensemble minimum and RC minimum occurring in abundant clusters. The co-occurrence of ensemble minimum (lowest binding energy of one of the top three clusters) and the RC minimum (lowest possible observed energy) may indicate that a highly abundant configurational space (i.e., ensemble of substates) is also favored by the ligand for most stable interaction. In fact, four out of the six co-occurrences occurred for compounds ranked within the top 10 hits, and all six ranked within the top 20 hits. In terms of binding energy, four out the six had identical binding energy between the ensemble minimum and RC minimum; the other two had binding energy differing less than 1 kcal/mol. Although the most populated structures may be most relevant to the initiation of binding events, it seems unlikely that they would be truly precatalytic states.
Some compounds that score poorly in the crystal-structure-only screen score well when MD structures are used, demonstrating the advantages of using MD structures to expand the description of the receptor’s structure. Conversely, other compounds that score well in the crystal-structure-only screen move down the list when the MD structures are included (e.g., compound 21) (Figure 6). This reordering happens for two reasons: first, because other compounds move up the list by producing better binding energies, and second, because some compounds produce a significantly worse binding energy when the MD structures are included. For example, compound 21 appears to move down the list due to a combination of both reasons mentioned above. Other compounds rank better in the MD structures and thus move up the list. Closer examination of compound 21 reveals that its position in the 430-cavity is compromised when the 430-loop opens during the MD simulation; as the loop opens, compound 21 loses contact with several stabilizing residues and thus the affinity is reduced. Therefore, loop motion and the subsequent interactions gained and/or lost is the cause of many compounds’ movements up and/or down the reranked list.
Rescoring a Larger List of Hits
In , we noted that the cutoff of −9.25 kcal/mol meant that peramivir and DANA would not have been selected. On the other hand, when these compounds were rescored together with the other top hits, taking into account all the lowest binding energies regardless of the size of the most populated cluster, peramivir and DANA actually ranked better than some of the top hits. These data suggest that, given the current standard error of 2 kcal/mol for AutoDock4, and the somewhat arbitrary nature of the 25% cutoff, the weighted harmonic mean or some improved knowledge-based heuristics may be applied to rescore an expanded list of hits and potentially recover some lost hits (i.e., false negatives).
Dynamic Nature of N1 Active Site
The MD simulations offer representative conformations of the protein beyond what is provided by the static crystal structures alone, and they offer a possible description of the energy landscape where ligand−receptor interactions take place. The visual examination of the electrostatic surface maps for the different cavities, the various 150- and 430-loop configurations, and the SASA analyses indicate that at the monomer level, the N1 active site is highly dynamic.
Electrostatic Gradient and Substrate Movement
At the monomeric level, the dynamic opening and closing of the 150- and 430-cavities, with the corresponding change in the electrostatic field, may help target and orient the appropriate substrate to the catalytic site. In Figure 2B, oseltamivir interacts in the 150-cavity of the Apo-1 cluster representative structure, which represents a population size of about 20%. In Figure 3, oseltamivir is very stably found in the SA cavity of the holo clusters. It is possible that the open 150-cavity provides an initial binding site, helping to guide the substrate into contacts with the catalytic residues through further closing motion of the 150-loop. Both crystallographic evidence and the MD simulations indicate that this closed 150-loop is stabilized by oseltamivir in the active site,9,10 primarily through interactions with D151 and R152. Collectively, these observations may suggest that the closed substate is preferred when ligands are bound in the active site.
Interestingly, on the other side of the 430-cavity is an area known to be a second SA binding site in N9(60) and N2,(61) with hemadsorption activity.62,63 Notably, sialic acid (in the boat, chair, or twist conformation) shows a preference to bind near this alternate site or the 430-cavity in 12 out of 18 docking experiments, albeit with lower energy and, often, lower clustering percentage than the SA cavity (data not shown). Moreover, the electrostatic surface potential maps indicate that this alternate SA binding site may exhibit a weak negative electrostatic potential. Therefore it is possible that this alternate site acts to attract SA or to extend its interactions with the N1 binding site. The comparatively stronger electrostatic potential in the SA cavity may provide an electrostatic gradient to steer SA into the requisite catalytic location. The effect of electrostatics is well-known to play an influential role in controlling the diffusion of substrates into charged active site areas, as shown in the acetylcholinesterase system.64,65 More extensive electrostatic calculations are required to test this hypothesis in the N1 system.
Hot Spots for Ligand−Protein Interactions
Previously, a CS-Map analysis on the N1 cluster representative structures assessed the druggability of the newly defined areas identified in the MD simulations.(11) Using a complementary computational physics-based approach, CS-Map identified suitable sites for potential ligand interactions, which were presented as probe clusters near the 150- and 430-loop regions. The consensus approach adopted in the CS-Map analysis indicates that despite protein motion, the existence of hot spots over the binding site may persist, and that these areas may be relevant for drug discovery and design, as has also been shown through a variety of other methods.18,25,66–70 The work we present here provides support for the CS-Map analysis through the docking of the NCIDS compound library to these novel hot spots (Table 2). Many of the ligands we present make contacts to the residues that were found to mediate a consistent probe contact, such as R152, S153, etc., in the SA cavity, Q136 and S145, etc., in the 150-cavity, and N369, S370 in the 430-cavities, as illustrated in Figure 6 and . The CS-Map analysis also predicted that Q136 and T439 are important mediators of nonbonded interactions and our docking studies support that claim.(39) We anticipate that ligands that make contact to these hot spots are more likely to be biologically relevant.
Hit Identification and Hit to Lead Optimizations
The computationally derived potential hits as listed in Table 3 are ranked using the RC weighted harmonic means, and their predicted site(s) of interactions are indicated. The names of these compounds are shown in , along with the mean binding energy values shown in Figure 3.
Fragment-Based Approach
The differential preference of binding cavities by the top hits for the apo ensemble of receptors is illustrated in . The individual hits to each cavity may offer good starting points for hit to lead optimization, especially for those interested in using fragment-based approaches. Compounds that are predicted to bridge two cavities simultaneously are especially interesting, particularly those that bridge either the 430- or 150-cavity to the SA cavity. In theory, these compounds should interfere with the binding of SA to the catalytic residues, represented in italics in Table 2, while providing additional selectivity for N1 through binding to the 150- or 430-cavities. Clearly, these compounds will require further optimizations because their current binding energies and predicted Ki’s are not necessarily higher than those compounds that only interact with a single cavity. However, they provide an initial set of experimentally testable predictions regarding the capability of any compounds to bind to these newly identified, druggable areas. Additional fragment-based approaches (either computational or experimental) to bridge known SA cavity binders with 150- and/or 430-cavity binding compounds can easily be envisioned.
Drug Resistant Mutations
The influenza virus undergoes constant antigenic drift with a very high mutation rate yielding many different strains except for the essential catalytic residues that are conserved across species. Therefore, it is important to consider known mutations and avoid compounds that interact with experimentally verified mutational hot spots. One set of the published mutations found in vivo or in vitro for H5N1 was H274Y and N294S.(71)H274Y results in resistance to oseltamivir, but not to zanamivir, whereas N294S results in low resistance to oseltamivir. None of the 27 top hits are predicted to make contacts to H274. Compound 1 makes a nonbonded contact to N294 in Holo-1; no other hits do. In addition, the E276D mutation has been shown to confer resistance without sacrificing the viability or genetic stability of the virus.(72) E276 is located directly in the sialic acid cavity and illustrates that even a subtle reduction in side chain length can be enough to confer resistance, further underscoring the need to develop new antivirals that target alternate areas such as the 150- and 430-cavities. Other mutations have been identified in group-2 neuraminidases (N2, N9) and influenza type-B viruses, but it is often observed that mutant residues in one group are unrelated to those in another.(73) Furthermore, when mutations from N2 are introduced into H5N1, they result in significantly reduced viral viability.(74)
Another intriguing consideration is that the inhibitor does not necessarily need to make contact with the mutated residues in order for it to confer resistance. For example, oseltamivir does not make immediate contacts with H274, but the mutation to tyrosine very likely causes tertiary changes in the protein that affect oseltamivir binding. Additionally, it has been shown that changes in the HA glycosylation patterns can confer antiviral resistance in an NA-independent manner.75,76 Although our understanding of the complex interplay of interactions that confer resistance is incomplete, these factors can still be considered in the selection and design of N1 antivirals.
Conclusions
Through eight different virtual screens of the NCI diversity set, which employed structures taken from crystallographic studies as well as representative structures from apo and holo MD simulations, 27 drug-like ligands have been identified, which are predicted to bind with high affinity to avian influenza N1. Fourteen of these compounds are unique hits that would not have been found based on the crystal structure screens alone, and they include compounds that bind to flexible loop regions unseen in the crystal structures. Importantly, the docking of thousands of compounds to the N1 binding site elucidated specific regions of high ligand-binding affinity in the flexible 150- and 430- cavities. These cavities and the residue contacts observed among the top hits strongly agree with the location of hot spots and residues predicted to mediate receptor−ligand interactions by a recent CS-Map analysis. The top hits are validated and ranked through redocking experiments into a set of structures representing the holo ensemble within the RCS framework. This novel strategy for identifying NA inhibitors may facilitate the design of better drugs in the fight against global pandemic influenza and be applicable in structure-based drug discovery initiatives.
Acknowledgments
L.S.C, D.X., W.W.L., and P.W.A. are supported by NIH P41 RR08605 (to P.W.A.) and TATRC W81XWH-07-2-0014 (to W.W.L. and P.W.A.). L.S.C. is also supported by NSF INT 0407508 to the PRIME program. R.E.A. is funded in part by NIH F32-GM077729 and MRAC CHE060073N. The work is also funded by NIH GM31749, NSF MCB-0506593, and MCA93S013 (to J.A.M.). Additional support from the Howard Hughes Medical Institute, San Diego Supercomputing Center, Accelrys, Inc., the W.M. Keck Foundation, the National Biomedical Computation Resource, and the Center for Theoretical Biological Physics is gratefully acknowledged.
Supporting Information Available
Additional tables and figures, including receptor-specific binding energies and detailed docking poses for top hits. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: VS, virtual screening; RCS, relaxed complex scheme; RC, relaxed complex; MD, molecular dynamics; HA, hemagglutinin; NA, neuraminidase; CS-Map, computational solvent mapping; NCIDS, National Cancer Institute diversity set; EVSRCS, ensemble-based virtual screen with relaxed complex scheme; DANA, 2,3-didehydro-2-deoxy-N-acetyl neuraminic acid; SA, sialic acid; RMSD, root-mean-square deviation; NBCR, National Biomedical Computational Resource; PRAGMA, Pacific Rim Application and Grid Middleware Assembly; CSF4, Community Scheduler Framework; APBS, Adaptive Poisson−Boltzmann Solver; NSC, National Service Center; VMD, visual molecular dynamics; NAMD, not another molecular dynamics; SASA, solvent accessible surface area.
Supplementary Material
References
- Abdel-Ghafar A. N.; Chotpitayasunondh T.; Gao Z.; Hayden F. G.; Nguyen D. H.; de Jong M. D.; Naghdaliyev A.; Peiris J. S.; Shindo N.; Soeroso S.; Uyeki T. M. Update on avian influenza A (H5N1) virus infection in humans. N. Engl. J. Med. 2008, 358 (3), 261–273. [DOI] [PubMed] [Google Scholar]
- The Influenza (Flu) Viruses; Centers for Disease Control: Atlanta, GA, 2007; http://www.cdc.gov/flu/about/fluviruses.htm(accessed November 19, 2007).
- De Clercq E.; Neyts J. Avian influenza A (H5N1) infection: targets and strategies for chemotherapeutic intervention. Trends Pharmacol. Sci. 2007, 28 (6), 280–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colman P., Anti-Influenza Drugs from Neuraminidase Inhibitors. In Structure-Based Drug Discovery: An Overview; Hubbard R. E., Ed.; Royal Society of Chemistry: Cambridge,, 2006. [Google Scholar]
- Kobasa D.; Kodihalli S.; Luo M.; Castrucci M. R.; Donatelli I.; Suzuki Y.; Suzuki T.; Kawaoka Y. Amino acid residues contributing to the substrate specificity of the influenza A virus neuraminidase. J. Virol. 1999, 73 (8), 6743–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew W.; Chen X.; Kim C. U. Discovery and development of GS 4104 (oseltamivir): an orally active influenza neuraminidase inhibitor. Curr. Med. Chem. 2000, 7 (6), 663–672. [DOI] [PubMed] [Google Scholar]
- Beigel J. H.; Farrar J.; Han A. M.; Hayden F. G.; Hyer R.; de Jong M. D.; Lochindarat S.; Nguyen T. K.; Nguyen T. H.; Tran T. H.; Nicoll A.; Touch S.; Yuen K. Y. Avian influenza A (H5N1) infection in humans. N. Engl. J. Med. 2005, 353 (13), 1374–1385. [DOI] [PubMed] [Google Scholar]
- de Jong M. D.; Tran T. T.; Truong H. K.; Vo M. H.; Smith G. J.; Nguyen V. C.; Bach V. C.; Phan T. Q.; Do Q. H.; Guan Y.; Peiris J. S.; Tran T. H.; Farrar J. Oseltamivir resistance during treatment of influenza A (H5N1) infection. N. Engl. J. Med. 2005, 353 (25), 2667–2672. [DOI] [PubMed] [Google Scholar]
- Russell R. J.; Haire L. F.; Stevens D. J.; Collins P. J.; Lin Y. P.; Blackburn G. M.; Hay A. J.; Gamblin S. J.; Skehel J. J. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 2006, 443 (7107), 45–49. [DOI] [PubMed] [Google Scholar]
- Amaro R. E.; Minh D. D.; Cheng L. S.; Lindstrom W. M. Jr.; Olson A. J.; Lin J. H.; Li W. W.; McCammon J. A. Remarkable loop flexibility in avian influenza N1 and its implications for antiviral drug design. J. Am. Chem. Soc. 2007, 129 (25), 7764–7765. [DOI] [PubMed] [Google Scholar]
- Landon M. R.; Amaro R. E.; Baron R.; Ngan C. H.; Ozonoff D.; Andrew McCammon J.; Vajda S. Novel Druggable Hot Spots in Avian Influenza Neuraminidase H5N1 Revealed by Computational Solvent Mapping of a Reduced and Representative Receptor Ensemble. Chem. Biol. Drug Des. 2008, 71 (2), 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard R. E.Structure-Based Drug Discovery: An Overview, 1st ed.; RSC Publishing: Cambridge, 2007; p 261. [Google Scholar]
- Foster I.; Kesselman C., The Grid 2: Blueprint for a New Computing Infrastructure, 2 ed.; Morgan Kaufmann Publishers, Inc.: San Francisco, 2004. [Google Scholar]
- Carlson H. A. Protein flexibility and drug design: how to hit a moving target. Curr. Opin. Chem. Biol. 2002, 6 (4), 447–452. [DOI] [PubMed] [Google Scholar]
- Teague S. J. Implications of protein flexibility for drug discovery. Nat. Rev. Drug Discovery 2003, 2 (7), 527–541. [DOI] [PubMed] [Google Scholar]
- Teodoro M. L.; Kavraki L. E. Conformational flexibility models for the receptor in structure based drug design. Curr. Pharm. Des. 2003, 9 (20), 1635–1648. [DOI] [PubMed] [Google Scholar]
- Wong C. F.; McCammon J. A. Protein flexibility and computer-aided drug design. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 31–45. [DOI] [PubMed] [Google Scholar]
- Carlson H. A.; Masukawa K. M.; Rubins K.; Bushman F. D.; Jorgensen W. L.; Lins R. D.; Briggs J. M.; McCammon J. A. Developing a dynamic pharmacophore model for HIV-1 integrase. J. Med. Chem. 2000, 43 (11), 2100–2114. [DOI] [PubMed] [Google Scholar]
- Cavasotto C. N.; Abagyan R. A. Protein flexibility in ligand docking and virtual screening to protein kinases. J. Mol. Biol. 2004, 337 (1), 209–225. [DOI] [PubMed] [Google Scholar]
- Cavasotto C. N.; Kovacs J. A.; Abagyan R. A. Representing receptor flexibility in ligand docking through relevant normal modes. J. Am. Chem. Soc. 2005, 127 (26), 9632–9640. [DOI] [PubMed] [Google Scholar]
- Kua J.; Zhang Y.; McCammon J. A. Studying enzyme binding specificity in acetylcholinesterase using a combined molecular dynamics and multiple docking approach. J. Am. Chem. Soc. 2002, 124 (28), 8260–8267. [DOI] [PubMed] [Google Scholar]
- Lin J. H.; Perryman A. L.; Schames J. R.; McCammon J. A. Computational drug design accommodating receptor flexibility: the relaxed complex scheme. J. Am. Chem. Soc. 2002, 124 (20), 5632–5633. [DOI] [PubMed] [Google Scholar]
- Lin J. H.; Perryman A. L.; Schames J. R.; McCammon J. A. The relaxed complex method: Accommodating receptor flexibility for drug design with an improved scoring scheme. Biopolymers 2003, 68 (1), 47–62. [DOI] [PubMed] [Google Scholar]
- Sherman W.; Day T.; Jacobson M. P.; Friesner R. A.; Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49 (2), 534–553. [DOI] [PubMed] [Google Scholar]
- Damm K. L.; Carlson H. A. Exploring experimental sources of multiple protein conformations in structure-based drug design. J. Am. Chem. Soc. 2007, 129 (26), 8225–8235. [DOI] [PubMed] [Google Scholar]
- Oprea T. I.; Matter H. Integrating virtual screening in lead discovery. Curr. Opin. Chem. Biol. 2004, 8 (4), 349–358. [DOI] [PubMed] [Google Scholar]
- Kitchen D. B.; Decornez H.; Furr J. R.; Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat. Rev. Drug Discovery 2004, 3 (11), 935–949. [DOI] [PubMed] [Google Scholar]
- Ferrari A. M.; Wei B. Q.; Costantino L.; Shoichet B. K. Soft docking and multiple receptor conformations in virtual screening. J. Med. Chem. 2004, 47 (21), 5076–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B. Q.; Weaver L. H.; Ferrari A. M.; Matthews B. W.; Shoichet B. K. Testing a flexible-receptor docking algorithm in a model binding site. J. Mol. Biol. 2004, 337 (5), 1161–1182. [DOI] [PubMed] [Google Scholar]
- Bowman A. L.; Nikolovska-Coleska Z.; Zhong H.; Wang S.; Carlson H. A. Small molecule inhibitors of the MDM2-p53 interaction discovered by ensemble-based receptor models. J. Am. Chem. Soc. 2007, 129 (42), 12809–12814. [DOI] [PubMed] [Google Scholar]
- Amaro R. E.; Baron R.; McCammon J. A.. An improved relaxed complex scheme for receptor flexibility in computer-aided drug design. J. Comput.-Aided Mol. Des. 2008, 10.1007/s10822-007-9159-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz M. Nguyen B.-Y. Gotuzzo F.; Mendo F.; Ratanasuwan W.; Kovacs C.; Zhao J.; Gilde L.; Isaacs R.; Teppler H.. Potent antiviral effect of MK-0518, novel HIV-1 integrase inhibitor, as part of combination ART in treatment-naive HIV-1 infected patients. In XVI International AIDS Conference; 16th International AIDS Conference, Toronto, Canada, August 13−18, 2006, 2006; no. ThLB0214.
- Hazuda D. J.; Anthony N. J.; Gomez R. P.; Jolly S. M.; Wai J. S.; Zhuang L.; Fisher T. E.; Embrey M.; Guare J. P. Jr.; Egbertson M. S.; Vacca J. P.; Huff J. R.; Felock P. J.; Witmer M. V.; Stillmock K. A.; Danovich R.; Grobler J.; Miller M. D.; Espeseth A. S.; Jin L.; Chen I. W.; Lin J. H.; Kassahun K.; Ellis J. D.; Wong B. K.; Xu W.; Pearson P. G.; Schleif W. A.; Cortese R.; Emini E.; Summa V.; Holloway M. K.; Young S. D. A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase. Proc. Natl. Acad. Sci. U.S.A. 2004, 101 (31), 11233–11238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schames J. R.; Henchman R. H.; Siegel J. S.; Sotriffer C. A.; Ni H.; McCammon J. A. Discovery of a novel binding trench in HIV integrase. J. Med. Chem. 2004, 47 (8), 1879–1881. [DOI] [PubMed] [Google Scholar]
- Dolinsky T. J.; Czodrowski P.; Li H.; Nielsen J. E.; Jensen J. H.; Klebe G.; Baker N. A. PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35 (Web Server issue), W522-5–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong A. K.; Pegg M. S.; von Itzstein M. Influenza virus sialidase: effect of calcium on steady-state kinetic parameters. Biochim. Biophys. Acta 1991, 1077 (1), 65–71. [DOI] [PubMed] [Google Scholar]
- Phillips J. C.; Braun R.; Wang W.; Gumbart J.; Tajkhorshid E.; Villa E.; Chipot C.; Skeel R. D.; Kale L.; Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26 (16), 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen M.; Hunenberger P. H.; Bakowies D.; Baron R.; Burgi R.; Geerke D. P.; Heinz T. N.; Kastenholz M. A.; Krautler V.; Oostenbrink C.; Peter C.; Trzesniak D.; van Gunsteren W. F. The GROMOS software for biomolecular simulation: GROMOS05. J. Comput. Chem. 2005, 26 (16), 1719–1751. [DOI] [PubMed] [Google Scholar]
- Landon M. R.; Amaro R. E.; Baron R.; Ngan C. H.; Ozonoff D.; McCammon J. A.; Vajda S. Novel druggable hot spots in avian influenza neuraminidase H5N1 revealed by computational solvent mapping of a non-redundant receptor ensemble. Chem. Biol. Drug Des. 2008, 71, 106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanner M. F. A component-based software environment for visualizing large macromolecular assemblies. Structure 2005, 13 (3), 447–462. [DOI] [PubMed] [Google Scholar]
- Gasteiger J.; Marsili M. Iterative partial equalization of orbital electronegativity: a rapid access to atomic charges. Tetrahedron 1980, 36 (22), 3219–3228. [Google Scholar]
- NCIDS Diversity Set Information. http://dtp.nci.nih.gov/branches/dscb/diversity_explanation.html, (accessed November 19, 2007).
- Babu Y. S.; Chand P.; Bantia S.; Kotian P.; Dehghani A.; El-Kattan Y.; Lin T. H.; Hutchison T. L.; Elliott A. J.; Parker C. D.; Ananth S. L.; Horn L. L.; Laver G. W.; Montgomery J. A. BCX-1812 (RWJ-270201): discovery of a novel, highly potent, orally active, and selective influenza neuraminidase inhibitor through structure-based drug design. J. Med. Chem. 2000, 43 (19), 3482–3486. [DOI] [PubMed] [Google Scholar]
- Morris G. M. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19 (14), 1639–1662. [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46 (1−3), 3–26. [DOI] [PubMed] [Google Scholar]
- Leach A. R.; Hann M. M.; Burrows J. N.; Griffen E. J., Fragment screening: an introduction. In Structure-Based Drug Discovery: An Overview; Hubbard R. E., Ed.; Royal Society of Chemistry: Cambridge, 2006; Vol. 2, pp 429−446 [Google Scholar]
- Sanner M. F. Python: a programming language for software integration and development. J. Mol. Graphics Modell. 1999, 17 (1), 57–61. [PubMed] [Google Scholar]
- Sanner M. F.; Olson A. J.; Spehner J. C. Reduced surface: an efficient way to compute molecular surfaces. Biopolymers 1996, 38 (3), 305–320. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Simeonov A.; Jadhav A.; Babaoglu K.; Inglese J.; Shoichet B. K.; Austin C. P. A high-throughput screen for aggregation-based inhibition in a large compound library. J. Med. Chem. 2007, 50 (10), 2385–2390. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Shelat A.; Doman T. N.; Guy R. K.; Shoichet B. K. High-throughput assays for promiscuous inhibitors. Nat. Chem. Biol. 2005, 1 (3), 146–148. [DOI] [PubMed] [Google Scholar]
- Baker N. A.; Sept D.; Joseph S.; Holst M. J.; McCammon J. A. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 2001, 98 (18), 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14 (1), 33–38. [DOI] [PubMed] [Google Scholar]
- Raffa R. B. Harmonic mean relationship between affinity for wild-type receptors and alanine-scan mutants. J. Theor. Biol. 2002, 218 (2), 207–214. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Y.; Thieme-Sefler A. M.; Maclean D.; McNamara D. J.; Dobrusin E. M.; Sawyer T. K.; Dixon J. E. Substrate specificity of the protein tyrosine phosphatases. Proc. Natl. Acad. Sci. U.S.A. 1993, 90 (10), 4446–4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubareva L. V.; Kaiser L.; Hayden F. G. Influenza virus neuraminidase inhibitors. Lancet 2000, 355 (9206), 827–835. [DOI] [PubMed] [Google Scholar]
- Colman P. M. Influenza virus neuraminidase: structure, antibodies, and inhibitors. Protein Sci. 1994, 3 (10), 1687–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipot C.; Pohorille A.. Free Energy Calculations. Springer: New York, 2007; Vol. 86. [Google Scholar]
- Amaro R. E.; Schnaufer A. Interthal H. Hol W. Stuart K. McCammon J. A.. Discovery of the first drug-like inhibitors of an essential RNA editing ligase in Trypanosoma brucei. 2008, in preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barril X.; Soliva R.. Molecular Modeling. In Structure-Based Drug Discovery, 1st ed.; Hubbard R. E., Ed.; RCS Publishing: Cambridge, 2007; pp 54−96. [Google Scholar]
- Varghese J. N.; Colman P. M.; van Donkelaar A.; Blick T. J.; Sahasrabudhe A.; McKimm-Breschkin J. L. Structural evidence for a second sialic acid binding site in avian influenza virus neuraminidases. Proc. Natl. Acad. Sci. U.S.A. 1997, 94 (22), 11808–11812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrosovich M. N.; Krauss S.; Webster R. G. H9N2 influenza A viruses from poultry in Asia have human virus-like receptor specificity. Virology 2001, 281 (2), 156–162. [DOI] [PubMed] [Google Scholar]
- Laver W. G.; Colman P. M.; Webster R. G.; Hinshaw V. S.; Air G. M. Influenza virus neuraminidase with hemagglutinin activity. Virology 1984, 137 (2), 314–323. [DOI] [PubMed] [Google Scholar]
- Kobasa D.; Rodgers M. E.; Wells K.; Kawaoka Y. Neuraminidase hemadsorption activity, conserved in avian influenza A viruses, does not influence viral replication in ducks. J. Virol. 1997, 71 (9), 6706–6713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilson M. K.; Straatsma T. P.; McCammon J. A.; Ripoll D. R.; Faerman C. H.; Axelsen P. H.; Silman I.; Sussman J. L. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science 1994, 263 (5151), 1276–1278. [DOI] [PubMed] [Google Scholar]
- Ripoll D. R.; Faerman C. H.; Axelsen P. H.; Silman I.; Sussman J. L. An electrostatic mechanism for substrate guidance down the aromatic gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. U.S.A. 1993, 90 (11), 5128–5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis S.; Kortvelyesi T.; Vajda S. Computational mapping identifies the binding sites of organic solvents on proteins. Proc. Natl. Acad. Sci. U.S.A. 2002, 99 (7), 4290–4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodford P. J. A computational procedure for determining energetically favorable binding sites on biologically important macromolecules. J. Med. Chem. 1985, 28 (7), 849–857. [DOI] [PubMed] [Google Scholar]
- Landon M. R.; Lancia D. R. Jr.; Yu J.; Thiel S. C.; Vajda S. Identification of hot spots within druggable binding regions by computational solvent mapping of proteins. J. Med. Chem. 2007, 50 (6), 1231–1240. [DOI] [PubMed] [Google Scholar]
- Mattos C.; Bellamacina C. R.; Peisach E.; Pereira A.; Vitkup D.; Petsko G. A.; Ringe D. Multiple solvent crystal structures: probing binding sites, plasticity and hydration. J. Mol. Biol. 2006, 357 (5), 1471–1482. [DOI] [PubMed] [Google Scholar]
- Mattos C.; Ringe D. Locating and characterizing binding sites on proteins. Nat. Biotechnol. 1996, 14 (5), 595–599. [DOI] [PubMed] [Google Scholar]
- Moscona A. Neuraminidase inhibitors for influenza. N. Engl. J. Med. 2005, 353 (13), 1363–1373. [DOI] [PubMed] [Google Scholar]
- Yen H. L.; Hoffmann E.; Taylor G.; Scholtissek C.; Monto A. S.; Webster R. G.; Govorkova E. A. Importance of neuraminidase active-site residues to the neuraminidase inhibitor resistance of influenza viruses. J. Virol. 2006, 80 (17), 8787–8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris O.; Lina B. Mutations of neuraminidase implicated in neuraminidase inhibitors resistance. J. Clin. Virol. 2007, 41, 13–19. [DOI] [PubMed] [Google Scholar]
- Yen H. L.; Ilyushina N. A.; Salomon R.; Hoffmann E.; Webster R. G.; Govorkova E. A. Neuraminidase inhibitor-resistant recombinant A/Vietnam/1203/04 (H5N1) influenza viruses retain their replication efficiency and pathogenicity in vitro and in vivo. J. Virol. 2007, 81 (22), 12418–12426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner R.; Matrosovich M.; Klenk H. D. Functional balance between haemagglutinin and neuraminidase in influenza virus infections. Rev. Med. Virol. 2002, 12 (3), 159–166. [DOI] [PubMed] [Google Scholar]
- Mishin V. P.; Novikov D.; Hayden F. G.; Gubareva L. V. Effect of hemagglutinin glycosylation on influenza virus susceptibility to neuraminidase inhibitors. J. Virol. 2005, 79 (19), 12416–12424. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.