

Abstract
S-Adenosylmethionine decarboxylase (AdoMetDC) is a critical enzyme in the polyamine biosynthetic pathway and depends on a pyruvoyl group for the decarboxylation process. The crystal structures of the enzyme with various inhibitors at the active site have shown that the adenine base of the ligands adopts an unusual syn conformation when bound to the enzyme. To determine whether compounds that favor the syn conformation in solution would be more potent AdoMetDC inhibitors, several series of AdoMet substrate analogues with a variety of substituents at the 8-position of adenine were synthesized and analyzed for their ability to inhibit hAdoMetDC. The biochemical analysis indicated that an 8-methyl substituent resulted in more potent inhibitors, yet most other 8-substitutions provided no benefit over the parent compound. To understand these results, we used computational modeling and X-ray crystallography to study C8-substituted adenine analogues bound in the active site.
Introduction
S-Adenosylmethionine decarboxylase (AdoMetDCa ) is a pyruvoyl dependent decarboxylase and a critical enzyme in the polyamine biosynthetic pathway, which is found in mammals, Protista, and many other species.1−4 The polyamines putrescine, spermidine, and spermine are essential for cell growth and play important roles in cell proliferation and differentiation.5−7 Polyamines have been found to be elevated in various types of cancer including non-small-cell lung cancer, prostate cancer, melanoma, and pancreatic cancer.8,9 Polyamine levels in cells depend on the polyamine biosynthetic and catabolic pathways as well as on import and export of polyamines across the cellular membrane. Altering regulation of the key enzymes in the polyamine pathway is a therapeutic strategy for treatment of various types of cancers. AdoMetDC catalyzes the conversion of S-adenosylmethionine (AdoMet) to decarboxylated S-adenosylmethionine (dcAdoMet), which then donates the aminopropyl group to putrescine or spermidine to form spermidine and spermine, respectively. AdoMetDC is at a key branch point in the pathway and its action commits AdoMet to polyamine biosynthesis and removes it from the pool available for methyl transfer to a variety of substrates.
Attempts to regulate polyamine levels have resulted in the development of inhibitors that target the biosynthetic enzymes ornithine decarboxylase (ODC),(10) AdoMetDC, and the catabolic enzyme spermidine/spermine N1-acetyltransferase (SSAT).(11) The best-known inhibitor of ODC is α-difluoromethylornithine (DFMO), which irreversibly inactivates the enzyme. The success of DFMO in cancer therapy has been limited as the cells compensate for the decreased synthesis of polyamines through increased cellular uptake of polyamines.(12) DFMO is currently being investigated as a chemopreventive agent against carcinogenesis.13−17 The development of drugs to inhibit AdoMetDC (Figure 1A) started with the competitive inhibitor methylglyoxal bis(guanylhydrazone) 1 (MGBG), which is similar to spermidine in structure.(18) Use of 1 caused extreme toxicity in humans, and many analogues of 1 were developed in attempts to decrease the toxicity. One such AdoMetDC inhibitor that resulted was 4-amidinoindan-1-one-2′-amidinohydrazone 2 (CGP48664A(19)), which progressed into clinical trials as a cancer chemotherapeutic agent.(19) Alternatively, inhibitors such as 5′-deoxy-5′-[(3-hydrazinopropyl)methylamino]adenosine 3(MHZPA), 5′-deoxy-5′-[(3-hydrazinoethyl)methylamino)adenosine 4 (MHZEA), and 5′-[(2-aminooxyethyl)methylamino]-5′-deoxyadenosine 5 (MAOEA), which are structural analogues of the natural substrate, were developed (Figure 1B). These compounds inactivate AdoMetDC by forming a Schiff base to the active site pyruvoyl group.(20) Another known nucleoside inhibitor of AdoMetDC is 5′-[[(Z)-4-amino-2-butenyl]methylamino]-5′-deoxyadenosine. This butenyl analogue was designed as an enzyme-activated irreversible inhibitor,(21) but subsequent experiments showed that it acted via transamination of the pyruvate prosthetic group.(20)
Figure 1.

Previously described inhibitors of hAdoMetDC.
The crystal structure of AdoMetDC and its S68A and H243A mutants were solved to aid understanding of the mechanisms of decarboxylation and autoprocessing.22−24 The crystal structures of AdoMetDC with inhibitors like 5, 3, and the methyl ester of S-adenosylmethionine (MeAdoMet) have been solved previously.(25) These structures show that the adenine base of the inhibitors assumes an unusual syn conformation within the active site. The preference for the unusual conformation has led us to develop new structural analogues of AdoMet with modifications on the adenine base and to investigate, through biochemical analysis, computational modeling and analysis of crystal structures, whether these compounds would be more potent inhibitors of AdoMetDC than the unsubstituted parent compounds. Substitution at the 8-position of adenine is expected to result in ligands that favor the syn conformation in solution, and it was hoped that this would increase their ability to inhibit AdoMetDC. We now describe the synthesis of several series of structural analogues of AdoMet with 8-substituted adenine and present AdoMetDC inhibition data. We report the crystal structures of the AdoMetDC F223A mutant complexed with MeAdoMet and the wild-type protein complexed with several 8-substituted inhibitors.
Results
Chemical Synthesis
Our synthetic efforts relating to AdoMetDC date back many years, when we prepared an early series of related compounds that included 3 and 5.(26) In our current research, we have prepared a series of compounds with various 8-substituents on an adenosine template having a chain extension at C-5′. These compounds fall into four broad categories with respect to the various substituents at C-5′, and synthetic schemes will be organized based upon these categories. For comparison purposes, we have included available compounds with an 8-H within the four categories. End groups of the C-5′ substituent such as an aminooxyalkyl will bind covalently and to a large extent irreversibly to the pyruvoyl group within the active site of the enzyme, while groups ending in an amide will not even bind reversibly covalently to the pyruvoyl group. Amino end groups will bind covalently, but entirely reversibly, while a hydrazide group binds with some reversibility. In addition to these compounds, we have prepared several compounds without a chain extension at C-5′, i.e., compounds that do not reach the vicinity of the pyruvate group within the binding site.
The syntheses of some of the 8-unsubstituted compounds date back to our earlier work,(26) and these compounds dictated our initial synthetic approaches. We began by assuming that we needed to block the 2′- and 3′-hydroxyl groups, which we did with an isopropylidene group. Later, we discovered that it was possible to conduct the chemistry without blocking these two hydroxyl groups and that the new schemes were superior to those that utilized a blocking−deblocking sequence. In situations where we had already prepared a target compound utilizing a blocked precursor, we did not go back and resynthesize the compound without using a blocking group, and the schemes below reflect that fact. Scheme 1 presents the precursor nucleoside series 8 and 9 that we have used along with their syntheses.
Scheme 1.

(a) (CH3)4Sn or (CH3CH2)4Sn, HMDS/dioxane, NMP, (Ph3P)4Pd, 110 °C; (b) CH3NH2, MeOH, 110 °C; (c) C6H5B(OH)2, K2CO3, (Ph3P)4Pd, 1,2-DME-H2O (2:1), 90 °C; (d) SOCl2, CH3CN/pyridine, 0 °C−RT, NH4OH, RT; (e) MsCl, pyridine, 0 °C; (f) 33% CH3NH2, EtOH, RT (9e,f,g) or 90 °C (9a−d); (g) NaOMe/MeOH, RT.
Target compounds with an aminooxyalkylamino side chain at C-5′ were prepared using two different routes, as shown in Scheme 2. In our original sequence, which utilized a 2′,3′-O-isopropylidene group for protection, we generated the hydroxyalkylamino precursor 15 by displacement of a tosyl group with the requisite amine. Using N-hydroxyphthalimide, triphenylphosphine, and DEAD,(27) the aminooxy precursor 16 was produced and then converted to the desired target 5 under acidic conditions. Later, we found that it was more effective to first generate the aminooxy precursors ethyl N-(2-bromoethoxy)ethanimidate(28) and ethyl N-(N-4-bromobutoxy)ethanimidate(29) (Scheme 2), which could be appended to C-5′ by halide displacement with a 5′-methylamino-5′-deoxynucleoside to produce product series 11 and 13. Initially, we carried out this displacement with an isopropylidene protecting group on the nucleoside but subsequently determined that the reaction works as well or better without the protecting group. By the above means targets 12a−c and 14a−f were prepared.
Scheme 2.

(a) CH3(OEt)C=NO(CH2)2Br (ref (28)), DMF, DIEA, 50 °C; (b) 1 N H2SO4, RT; (c) CH3(OEt)C=NO(CH2)4Br (ref (29)), DMF, DIEA, 50 °C; (d) 2-(methylaminoethanol), RT; (e) N-hydroxyphthalimide, PPh3, DEAD, THF (ref (40)), RT; (f) 1 N H2SO4, 60 °C.
All of the amides and hydrazides were made by similar procedures, as shown in Scheme 3. The 5′-methylamino-5′-deoxynucleosides were treated with the appropriate ω-chloroester, followed by treatment with either ammonia or hydrazine. If an isopropylidene group was involved, then it was removed with an acidic deprotection step. In this manner targets 17d−f, j−m, with two different linker lengths and various 8-substituents, were prepared.
Scheme 3.

(a) Cl(CH2)nCO2Et (n = 1 or 2), DMF, DIEA, 60 °C; (b) NH3/MeOH, RT; (c) 1 N H2SO4, RT; (d) NH2NH2, H2O, EtOH, reflux.
Targets with an aminoalkylamino side chain at C-5′ were mainly prepared utilizing the displacement of a C-5′ leaving group with the unsymmetrical amine (Scheme 4). For example, treatment of 8a with 3-methylaminoethylamine produced a mixture of 18f and 19d, which were separated to afford pure 18f, our desired target. In the case where this procedure involved a starting material with an isopropylidene group, treatment with acid produced the desired final product. In early work, compounds 21c,d were prepared by treatment of a 5′-methylamino-5′-deoxynucleoside with 3-bromopropylphthalimide followed by two deprotection steps.
Scheme 4.

(a) CH3NH(CH2)nNH2 (n = 1 or 2), RT; (b) 1 N H2SO4, RT; (c) 3-bromopropylphthalimide, DMF, DIEA, 60 °C; (d) NH2NH2, H2O, reflux; (e) 1H-pyrazole-1-carboxamidine·HCl (ref (30)), DMF, DIEA, RT; (f) 3-(methylamino)propionitrile, RT (ref (31)); (g) NH2OH·HCl, MeOH, DMF, KOH, RT.
Building on the aminoalkylamino side chain, reaction of 18e with 1-carboxamidinopyrazole(30) produced the guanidine target 22a. In a related sequence, the target amidoxime 22c was prepared by treating 8j with 3-(methylamino)propionitrile to produce the nitrile 22b,(31) which was treated with hydroxylamine hydrochloride under basic conditions.
The 5′-dimethylamino and 5′-dimethylsulfonio compounds 23a,b and 25a−d were prepared by routine methods (Scheme 5). The dimethylamino group was introduced by displacement of a 5′-chlorine on 8a or 8g(32) with dimethylamine. The 5′-methylthio compounds 24a,b were treated with methyl bromide to produce 25a and 25c. Ion exchange was utilized to prepare the chloride salts 25b and 25d. 8-Methyl-5′-methylthio nucleoside 24a was prepared by displacement of the 5′-chlorine in 8a with sodium thiomethoxide.
Scheme 5.

(a) (CH3)2NH, 2 M solution in MeOH, 90 °C; (b) CH3SNa, DMF, RT; (c) CH3Br, Et2O, HCO2H, HOAc, RT; (d) IRA-400 (Cl−) ion exchange resin.
Modeling of MeAdoMet in the Active Site of AdoMetDC
The crystal structures of AdoMetDC complexed with MeAdoMet or the inhibitors 3 and 5 have shown that the ligand binds with the adenine base in the unusual syn conformation.(25) The active site residues of AdoMetDC with MeAdoMet bound are shown in Figure 2. However, NMR data, coupled with molecular modeling studies, suggest that in solution AdoMet assumes an anti conformation as an energy minimum.(33) A survey of crystal structures in which AdoMet is bound showed that the substrate assumes a range of glycosidic torsion angles but that the anti conformation is preferred.(33) To explain the conformational preferences and the related energetics of ligand binding to AdoMetDC, the modeling of MeAdoMet in the active site of AdoMetDC was done. Because MeAdoMet is tethered to the active site of AdoMetDC through covalent bonding to the pyruvoyl group, docking involving positional and orientational sampling was not performed. Instead, a conformational search to locate the populated low energy conformations of MeAdoMet in the AdoMetDC active site was performed using the mixed Monte Carlo/low mode conformational search method within the MacroModel program.34−36 The conformational search started with MeAdoMet in either the anti or syn conformation, and in each case the five lowest energy structures from the search exhibited a syn conformation for the adenine nucleoside. A superposition of the modeled structure with the crystal structure (Figure 2) indicates that the results of the conformational search match well with those observed crystallographically. Conformational searches were also done for AdoMet, 5′-deoxy-5′-(dimethylsulfonio)adenosine (MMTA), 3, and 5 binding to AdoMetDC, and each yielded a syn conformation for the glycosidic bond (data not shown). The ribose makes key hydrogen bonds to Glu247 and the adenine base stacks between Phe7 and Phe223 and also makes hydrogen bonds to the backbone amide and C-terminal carboxyl group of Glu67. These interactions together with π−π stacking of the adenine base with Phe223 and with Phe7 constrain the glycosidic bond to the syn conformation.
Figure 2.

Comparison in stereoview of the crystal structure of hAdoMetDC complexed with MeAdoMet to that of a structure derived from the modeling of the complex. The crystal structure has all atoms colored gray. For the model, the active site pyruvoyl group is shown in magenta and MeAdoMet carbon atoms are shown in green. MeAdoMet makes a Schiff base to the pyruvoyl group. The ribose makes two hydrogen bonds to Glu247 (shown as red dashed lines). The adenine base stacks between Phe223 and Phe7 in the unusual syn conformation. The hydrogen bonds between the adenine base and the backbone of Glu67 stabilize the syn conformation. The modeling result agrees well with the experimentally determined crystal structure.
Virtual Mutations in the Active Site of AdoMetDC
Virtual mutations were made to study the effect of various residues on the conformation of the bound nucleoside. Conformational searching with MacroModel employing the AdoMetDC F223A and F7A single amino acid mutants, with MeAdoMet in the active site, resulted in a mixture of syn and anti conformations in the low energy ensemble. With each of the mutations, the global minimum was an anti conformation of the adenine base, closely followed by a syn conformation with an energy difference of ∼2 kJ/mol. The global minimum energy conformation of the ligand bound in the anti conformation in the F223A mutant exhibits major changes compared to the second lowest energy conformer which adopts the syn conformation. In the F223A binding site, the ribose of the global minimum energy structure is displaced and makes hydrogen bonds to Glu247 and Cys226 instead of to Glu247 alone (Figure 3A). This change causes the ligand to twist back upon itself, the sulfonium stacks over the adenine base, and the adenine base makes three hydrogen bonds to Ser66. In the F7A binding site, the ligand assumes a similar conformation as with the F223A mutant. The Phe223 side chain undergoes a torsional change to accommodate the conformational change of the ligand and also stacks with the adenine base (Figure 3B). The presence of the anti conformation in low energy structures of the ligand in the enzyme active site where virtual mutations have been made suggests the importance of the phenyl groups in maintaining the syn conformation of the ligand within the wild-type enzyme binding site. However, since we observed a syn conformation of the nucleoside as the second lowest energy structure in our conformational search on the F223A mutant and since the relative energy of that structure compared to the global minimum (ΔE = 2.5 kJ/mol) is well within the error limit of our calculations, we were prompted to obtain the crystal structure of the F223A mutant complexed with MeAdoMet.
Figure 3.

Comparison in stereoview of modeling of hAdoMetDC F223A and hAdoMetDC F7A mutants, each complexed with MeAdoMet, with the crystal structure of the F223A mutant with MeAdoMet bound. Global minimum of modeling of MeAdoMet in the active site of the F223A mutant superposed with the crystal structure (A) and the F7A mutant (B) of hAdoMetDC (see for details). The crystal structure has all atoms colored gray. The pyruvoyl group is shown in magenta and the ligand carbon atoms are shown in green for the models. Hydrogen bonds are shown as dashed lines. The adenine base attains an anti conformation in the models. The ribose makes one hydrogen bond to Glu247 and the other to the backbone carbonyl of Cys226. The adenine base makes three hydrogen bonds to Ser66. In the F7A model (B), the Phe223 residue changes its conformation to stack with the adenine base of MeAdoMet in the anti conformation.
Structure of F223A Mutant Complexed with MeAdoMet
The structure of the F223A mutant is similar to that of the wild type protein.(22) The human AdoMetDC (hAdoMetDC) protomer has a four layer αββα fold in which two β-sheets are sandwiched between two layers of α-helices. The secondary structural elements are related by a pseudo 2-fold axis, suggesting that the protomer resulted from gene duplication. The proenzyme consists of 334 amino acid residues, and the enzyme undergoes autoprocessing to give the α and the β subunits.(22) The autoprocessing reaction yields the active enzyme with the pyruvoyl cofactor. The pyruvoyl group is located at the end of the N-terminal β-sheet and the active site involves residues from both of the β-sheets. The binding site of putrescine, which activates both the autoprocessing and decarboxylation reactions of hAdoMetDC, is located well away from the ligand binding site within the wild-type enzyme. Experimental conditions for the purification of the enzyme included putrescine at sufficient concentration to ensure high occupancy of the putrescine binding site. The loops between the residues 1−4, 21−27, 165−173, 288−299, and 329−334 are disordered in the crystal structures.
The crystal structure of hAdoMetDC F223A complexed with MeAdoMet was solved using molecular replacement. The difference Fo − Fc density shows that MeAdoMet is covalently bound to the enzyme, and the nucleoside adopts a clear syn conformation. As expected, the composite omit map density shows no density for the Phe223 side chain. The ribose makes two hydrogen bonds to Glu247, which anchor the ligand, and the base is held in syn conformation by stacking interactions with Phe7 and hydrogen bonds between the adenine and Glu67. One molecule of putrescine per monomer is present in the expected putrescine binding site. A superposition of the F223A/MeAdoMet structure and the wild type structure with MeAdoMet shows that there is no appreciable change in the position or conformation of the ligand (Figure 3A). The loops disordered in the mutant are also disordered in the wild type protein.
Biochemical Analysis of Potential Inhibitors of hAdoMetDC
The demonstrated importance of the syn conformation of the adenine base of the AdoMet substrate for binding in the active site of the enzyme led us to explore whether this could be exploited in designing better hAdoMetDC inhibitors. It is known that 8-substitution on adenine rings causes the nucleotide to favor a syn conformation in solution.26,27,37,38 It was thought that structural analogues of AdoMet that preferred the syn conformation in solution would lead to improved hAdoMetDC inhibition. Modeling of the active site had indicated that there was sufficient room to accommodate even rather large substituents at C8 of adenine. Several series of AdoMet structural analogues were synthesized with substituents ranging from a methyl group to a phenyl group at the 8-position of adenine. Each of these compounds was then assayed for its ability to inhibit hAdoMetDC and IC50 values for the inhibition were determined (Table 1).
Table 1. Inhibition of hAdoMetDCa.
| compd | IC50 |
|---|---|
| 12a | 7 nM |
| 12b | 86 nM |
| 12c | <5% inhibition at 100 μM |
| 14a | 49 nM |
| 14b | <5% inhibition at 100 μM |
| 14c | 11 nM |
| 14d | 5 nM |
| 14e | 15 nM |
| 14f | 18 nM |
| 5 | 55 nM |
| 17d | 400 nM |
| 17e | 4 μM |
| 17f | <5% inhibition at 100 μM |
| 17j | 7 μM |
| 17k | 170 nM |
| 17l | 1.5 μM |
| 17m | 31 μM |
| 18a | 440 μM |
| 18b | <5% inhibition at 100 μM |
| 18d | 500 μM |
| 18e | <5% inhibition at 100 μM |
| 18f | 88 μM |
| 19a | <5% inhibition at 100 μM |
| 19b | <5% inhibition at 100 μM |
| 19c | <5% inhibition at 100 μM |
| 19d | <5% inhibition at 100 μM |
| 21c | 70 μM |
| 21d | 420 μM |
| 22a | <5% inhibition at 100 μM |
| 22c | 157 μM |
| 23a | 600 nM |
| 23b | 9 μM |
| 25b | 3 μM |
| 25d | 15 μM |
Each of the potential inhibitors was assayed for the ability to inhibit hAdoMetDC. At least five concentrations of each compound were used and IC50 values were calculated from curve fits to plots of inhibitor concentration versus % inhibition of hAdoMetDC.
The inhibitors tested fall into four groups as described in the “” section. One group (12a−c, 14a−f, 5) has an aminooxyalkyl side chain at C-5′, which can form a Schiff base with the pyruvate of AdoMetDC20,39−41 Compounds of this group were potent inhibitors, with a 4-aminooxybutyl group being slightly superior to a 2-aminooxyethyl addition. A second group of compounds (17d,e,f,j,k,l,m) had an amide or a hydrazide side chain at C-5′, and a third group of inhibitors (18a,b,d,e,f; 19a,b,c,d; 21c,d) had an aminoalkylamino side chain at C-5′. Also related to the third group by the synthetic method are 22a and 22c, which, respectively, have a guanidine and an amidoxime at the end of the C-5′ side chain. The compounds of groups 2 and 3 were less potent (particularly those with the aminoalkylamino, guanidine, or amidoxime side chain) but are more likely to be stable under in vivo conditions. The final group of compounds consisted of 5′-dimethylamino (23a,b) or 5′-dimethylsulfonio (25b,d) compounds. Compound 25d has been previously reported to be an AdoMetDC inhibitor with a Ki in the μM range.(32) As shown in Table 1, the replacement of sulfur by nitrogen slightly improves the AdoMetDC inhibition.
Within each of these groups, there was a consistent improvement of inhibitory activity when an 8-methyl substituent was added to the adenine ring. The reduction in the IC50 value varied from 3.4-fold for compound 14d to 15−17-fold for compounds 23a and 17d. There was an 8-fold increase in potency when an adenine 8-methyl substituent was added to compound 5, forming compound 12a. This is consistent with the concept that the 8-methyl substitution on adenine biases the corresponding nucleoside toward the syn conformation and that this is the form that is bound at the active site. An adenine 8-hydroxy substituent resulted in slightly increased potency compared to no substituent but was not as effective as the 8-methyl substituent (compare 14c to 14d and 14f). Larger 8-substitutions did not improve the effectiveness. An 8-phenyl addition to compounds 5, 14f, and 18d abolished the inhibitory activity. Smaller additions such as 8-ethyl (compare 14d and 14e, 17d and 17e, and 21c and 21d) or 8-methylamino (compare 12a and 12b and 21c and 18a) were tolerated but were worse than 8-methyl.
Crystal Structures of hAdoMetDC Complexes
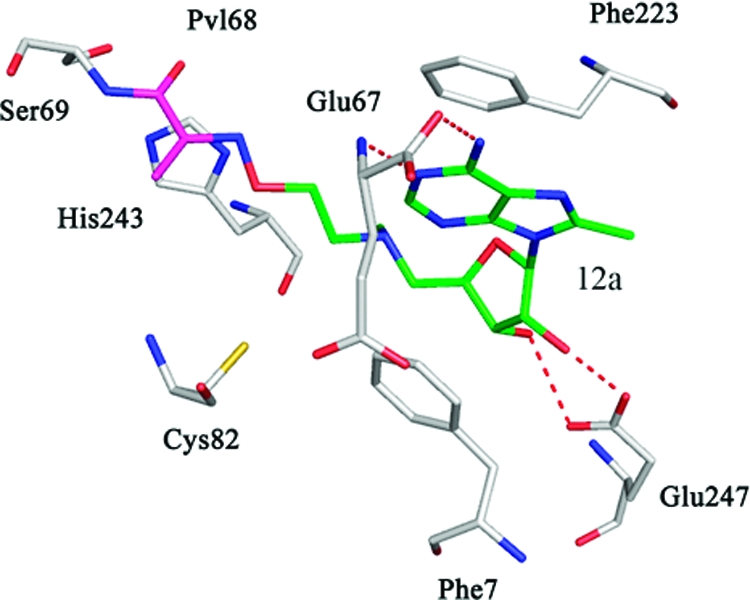
The crystal structure of native hAdoMetDC with 12a was solved using molecular replacement (Figure 4A). As noted above, 12a is structurally similar to the previously studied inhibitor 5 except that it has a methyl substitution at the 8-position on the adenine base. The electron density indicates that the amino terminus of 12a forms a Schiff base with the pyruvoyl group of the enzyme. The adenine base of 12a adopts a syn conformation in the crystal structure as expected. There is one molecule of putrescine bound in the putrescine binding site.
Figure 4.

Complexes of hAdoMetDC with inhibitors having 8-substitutions. The carbon atoms of the inhibitor are shown in green and the pyruvoyl group is shown in magenta. Water molecules are shown as red spheres and hydrogen bonds are shown as dashed lines. (A) Complex with 12a. The ligand makes a Schiff base linkage to the active site pyruvoyl group. (B) Complex with 14e. There is no evidence from the electron density for the formation of a Schiff base, and there is no density for the terminal three atoms of the ligand. The position of the terminal three atoms is determined by modeling. (C) Complex with 17d. The carboxamide terminus of the ligand makes hydrogen bonds to Leu65 and Ser229. (D) Complex with 17f. The carboxamide terminus of the ligand makes water mediated hydrogen bonds to Glu11 and Gly9. (E) Complex with 21c. The amino terminus of the ligand makes water mediated hydrogen bonds to Glu11 and Gly9. The inhibitors 17d, 17f, and 21c do not make a Schiff base to the enzyme and are hence competitive inhibitors. The adenine base of all the inhibitors attains a syn conformation.
The crystal structure of native hAdoMetDC with 14e was solved using molecular replacement (Figure 4B). Compound 14e is similar to 5 except for an ethyl substituent on the 8-position of the adenine base and two extra carbon atoms between the tertiary nitrogen (near ribose) and the terminal nitrogen. The presence of a three-carbon linker between the ribose and the amino terminus makes this ligand interesting to study. The electron density maps show no density for Schiff base formation between the pyruvoyl group and the amino terminus of the ligand. There is no density for the terminal three atoms of the ligand, but there is good density for the rest of the ligand. The position of the last three atoms was obtained by modeling them to an energetically favorable conformation using molecular modeling. The density around the pyruvoyl group fits it well and does not show any evidence of formation of a Schiff base. The ribose makes the critical hydrogen bonds to Glu247 and anchors the ligand. The nucleoside is held in the syn conformation and is stabilized by π−π stacking. The density of the ethyl substituent on the base is well defined indicating that the substituent is not disordered.
The crystal structures of hAdoMetDC with 17d (Figure 4C), 17f (Figure 4D), and 21c (Figure 4E) were also determined by molecular replacement. The three ligands have 8-methyl substituents; the first two have carboxamide end groups at the 5′-tail, while the third ligand has an amino group in this position. All three ligands showed clear electron density and all three ligands bound in the syn conformation.
Discussion
The active site of AdoMetDC contains a bound pyruvoyl cofactor. The interactions of various ligands at the active site were elucidated previously from the crystal structures obtained from complexes of the enzyme with the inhibitors 3 (PDB 1I79), 5 (PDB 1I72), MeAdoMet (PDB 1I7B), 1 (PDB 1I7C), and 2 (PDB 1I7M).(25) The crystal structure of MeAdoMet covalently bound to the enzyme most closely approximates the substrate AdoMet in the active site. The crystal structure shows key interactions of MeAdoMet with the enzyme including: (1) hydrogen bonding of the ribose oxygens with Glu247, (2) π−π stacking interactions of the adenine ring with Phe223 and Phe7, (3) hydrogen bonding of the 6-amino substituent of the adenine ring with Glu67, the C-terminal residue of the β-chain, and (4) hydrogen bonding of N-1 of the adenine ring with the backbone amide group of Glu67 (Figure 2A). Similar interactions are also present in the structures of 3 and 5 complexed with hAdoMetDC. The glycosidic angle for the adenine base in the structures ranges from 128° to 139°, which demonstrates a preference for the syn conformation of the nucleoside when bound to the active site. Crystal structures of 1 and 2 with the enzyme show that they stack between the two phenyl rings and make hydrogen bonds to Glu247.
The molecular modeling of MeAdoMet in the active site of hAdoMetDC was performed by using mixed Monte Carlo/low mode conformational searching as described above. The glycosidic torsional angle was free to rotate during the conformational search, which would allow a wide range of rotamers that are compatible with the steric constraints of the active site before energy minimization. The low energy structures show that the adenine-derived nucleosides prefer the syn conformation in the active site of hAdoMetDC. Markham et al. have studied the conformational preferences of AdoMet in solution and in vacuo.(33) These studies based on 1H NMR and calculations based on NMR constraints have shown that AdoMet prefers an anti conformation in solution and a syn conformation in vacuo. In solution, the energy difference between the anti and the corresponding syn conformation, which includes steric, electrostatic, and the solvation contributions, is around −34 kJ/mol. However, these calculations were based on molecular mechanics without polarization effects and it is likely that the energy difference is much less negative. Our crystal structures and modeling results show that hAdoMetDC binds ligands in the syn conformation and that the energy difference is overcome by hydrogen bonding and π−π interactions with Phe7 and Phe233. Typical π−π interactions of parallel geometry account for a stabilization of 8−12 kJ/mol,(42) suggesting that other factors may be involved.
The roles of Phe223 and Phe7 in AdoMetDC were studied previously through crystal structures and kinetic experiments.(25) Kinetic data from reaction of hAdoMetDC F223A and F7A mutants with the substrate AdoMet have shown that there is a 45-fold reduction of efficiency (kcat/km) for the F7A mutant and a 1400-fold decrease with the F223A mutant. In addition, 1 and 2 show a significant increase in the IC50 values for both the F7A and F233A mutants when compared to the wild-type enzyme, with decrease in binding greater for F223A than for F7A. Therefore, we chose to investigate the structural and conformational properties of MeAdoMet in the active site of the F223A mutant.
Our conformational searches with virtual mutations were done to understand the roles of Phe223 and Phe7 in stabilizing the syn conformation. In contrast to calculations done with the wild type enzyme structure, in which only the syn conformation was observed for the ensemble of lowest energy structures, the global minimum from both the mutations has the base in an anti conformation and the next higher energy structure has the base in a syn conformation. The difference in the energy between these conformations is about 2 kJ/mol, which we estimate to be within the error limit of our molecular mechanics based calculations. The energy difference between the syn and anti conformation of both structures is low, and based on the X-ray structure of hAdoMetDC F223A with bound MeAdoMet, the enzyme binds the ligands in the syn conformation, suggesting that π−π interactions with Phe7 are sufficient to maintain the syn conformation. Thus, although the modeling studies were incapable of accurately predicting that the syn conformation of the nucleoside would be maintained in the F223A mutant, it was possible to infer from these studies that the binding affinity of the nucleoside for the enzyme would be diminished.
Our attempts to exploit the requirement by AdoMetDC for a ligand with a syn conformation were successful, as demonstrated by the 8- to 18-fold improvement in inhibition when a methyl group is attached to C-8. However, the larger substituents that we tested provided no benefit over the unsubstituted parent compounds. In fact, the 8-phenyl substituent rendered the compounds much less potent than the unsubstituted analogue. Modeling studies of the active site had indicated that there should be sufficient space to accommodate the larger groups with the adenine in the syn conformation. A more detailed look at the area occupied by adenine C-8 substituents has indicated that this area is near the solvent interface. On the basis of our biochemical results, although large 8-substituents were structurally compatible with the active site, the penalty of incompletely burying a large hydrophobic group within a hydrophobic cavity is apparently greater than the gain from favoring the syn conformation. We are now exploring the effect of more hydrophilic C-8 adenine substituents that should be more compatible with proximity to the solvent. Such substituents may be useful in maintaining the inhibitory potency associated with the syn structure while still allowing species specific binding.
Structures of many of the AdoMet analogues bound to AdoMetDC have shown that they inhibit the enzyme through Schiff base formation with the pyruvoyl group of the enzyme. The linker length between the tertiary ammonium/sulfur and the terminal nitrogen of those inhibitors is typically 3−4 atoms, which makes the formation of a Schiff base geometrically and sterically feasible. Compound 14e has a linker length of five atoms. The electron density map for the complex of 14e shows a break in the density after the pyruvoyl group, suggesting that there is no Schiff base formation. There is good density for the ligand except at the three terminal atoms, which are disordered and have no density. The positions of the last three atoms were fixed in an energetically favorable conformation using computer modeling. The five atoms of the linker region appear to cause a sterically unfavorable orientation for formation of the Schiff base. The ligand is still held rigidly in the active site by hydrogen bonds to Glu247 and the π−π stacking interactions with Phe7 and Phe223, and thus little movement is allowed to accommodate Schiff base formation for the longer linker region. Even though compound 14e is not covalently attached to the pyruvoyl group, its potency is better than 5 and nearly as good as compound 12a.
Conclusion
Previous structural studies showed that AdoMet binds to the active site of hAdoMetDC in the syn conformation, suggesting that adenosine analogues favoring the syn conformation in solution might be more potent inhibitors than corresponding analogues favoring the lower energy anti conformation. 8-Substituted nucleoside analogues favor the syn conformation because of unfavorable interactions in the anti conformation between a bulky 8-substituent and ribose. We used computer simulations to predict 8-substituted compounds that might bind to hAdoMetDC and synthesized and assayed the most promising candidates. We also determined crystal structures of several compounds bound to hAdoMetDC to validate the predictions; the structures confirmed that the 8-substituted analogues bound in the syn conformation and retained the previously identified features of AdoMet binding, namely, purine stacking between Phe7 and Phe223, and hydrogen bonding between the ribose hydroxyl groups and Glu247. A group of adenosine analogues was generated by varying the size and nature of the both the 8-substituent and the 5′-modification. In general, 8-substituted analogues bound with a potency of 8- to 18-fold higher compared to the corresponding compound with a hydrogen atom at the 8-position; however, 8-substituents larger than methyl often showed lower potency than the corresponding 8-H compound. The observation results from excessive solvent exposure for large 8-substituents. Computer modeling and X-ray crystallography were also used to understand the preference for the syn conformation. Modeling studies suggested an important role for the two active site phenylalanine residues in addition to Glu247; however, the crystal structure of the F223A mutant hAdoMetDC showed that AdoMet still binds in the syn conformation, suggesting that other factors that favor the syn conformation remain to be identified.
Experimental Section
Protein Production
For crystallography of wild type and F223A mutant hAdoMetDC, plasmids in the pQE30 vector in E. coli were produced as described previously.(25) This construct replaces the N-terminal methionine with MRGS(H)6GS− for purification by immobilized metal affinity chromatography. A different plasmid also based on the pQE30 vector was used for the production of protein for the hAdoMetDC enzyme assays. In this plasmid, the (H)6 tag was located at the carboxyl end replacing the terminal −QQQQQS. The position of the (H)6 tag did not alter the activity of the purified enzyme.
The wild type hAdoMetDC was purified based on the protocol described by Ekstrom et al.(22) The plasmid encoding the enzyme is in the pQE30 vector and was transformed into JM109 strain E. coli cells. The cells were grown as an overnight culture in LB media at 37 °C and then introduced into larger cell cultures with both of the cultures containing 100 mg/mL ampicillin. The cells were grown until they reached an OD600 of 0.6 and then were induced with 100 mg/L isopropyl β-d-thiogalactopyranoside (IPTG). The cells were allowed to grow overnight at 15 °C and were then harvested by centrifugation, washed using a wash buffer that contained 20 mM Na2HPO4, pH 7.0, 500 mM NaCl, 2.5 mM putrescine, 0.02% Brij-35 and 10 mM imidazole, and stored at −80 °C. The frozen cell pellet was thawed, suspended in the wash buffer, and lysed using a French press at 1500 psi. The cellular debris and the lysate were separated by centrifugation at 12000g. Talon metal affinity resin was equilibrated with the wash buffer, and then the lysate and the resin were gently spun together for 1.5 h. The resin was loaded onto a column and washed with a volume of wash buffer equivalent to 15−20 times the column volume. Next, the column was washed in the same manner with wash buffer containing 25 mM imidazole. The protein was then eluted with buffer containing 100−200 mM imidazole. The eluted protein solution was concentrated to around 10 mL and passed through a Sephadex G-75 column pre-equilibrated with 10 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid (HEPES), pH 7.5, 2.5 mM putrescine, 5 mM dithiothreitol (DTT), 0.1 mM ethylenediaminetetraacetic acid, 0.02% Brij-35, and 300 mM NaCl. The buffer was run through the column, and the fractions containing the protein were identified by UV absorbance at 280 nm. The protein was concentrated to ∼10 mg/mL and stored at −80 °C. The purification of the F223A mutant was similar to that of the native enzyme.
Structure Determination
The protein was thawed on ice and buffer exchanged to 10 mM HEPES, pH 7.5, 200 mM NaCl and 1 mM DTT using Bio-Rad buffer exchange chromatography columns (Bio-Rad Laboratories, Hercules, CA 94547). The wild type protein was incubated with a 4−6 M excess of inhibitor for 24 h prior to crystallization. The F223A mutant was diluted to ∼6 mg/mL and incubated with a 4−6 M excess of MeAdoMet for 24 h prior to crystallization. Crystals of both the native and the mutant complexes were grown using the hanging drop method at 22 °C in 13−16% PEG 8000, 100 mM Tris, pH 8.0−9.0, and 10 mM DTT. Crystals appeared overnight and were stable for 1−2 weeks.
The data for the 12a complex were collected at a home source with a Rigaku R-AxisIV++ image plate detector using Cu Kα radiation from a Rigaku RU-300 rotating anode generator. The data for the 14e complex were collected at NE-CAT beamline 8-BM at the Advanced Photon Source (APS) using a ADSC Q315 detector. Data for the 17f complex were collected at NE-CAT beamline 24-ID-C using an ADSC Q315 detector. The data for the AdoMetDC F223A mutant with MeAdoMet and the complexes with 17d and 21c were collected at the F2, A1, and A1 stations of CHESS, respectively, using an ADSC Q210 detector. The diffraction quality of the crystals strongly depended on cryoprotection conditions. The crystals were sequentially transferred to a solution containing the well solution with 2, 5, 8, 15, and 18% glycerol with 1−2 min equilibration between each step. The data for all of the complexes were indexed, integrated, and scaled using the HKL2000(43) program suite. The data collection statistics are summarized in Table 2.
Table 2. Data Collection Statistics for hAdoMetDC Complexes.
| F223A + MeAdoMet | 12a | 14e | 17d | 17f | 21c | |
|---|---|---|---|---|---|---|
| wavelength | 0.9795 | 1.5418 | 0.9795 | 0.9790 | 0.9792 | 0.9771 |
| space group (Å) | C2 | C2 | C2 | C2 | C2 | C2 |
| a (Å) | 95.98 | 96.78 | 94.43 | 99.82 | 99.65 | 100.08 |
| b (Å) | 44.25 | 44.46 | 50.04 | 50.95 | 50.75 | 50.75 |
| c (Å) | 70.83 | 70.55 | 70.41 | 68.98 | 68.90 | 69.04 |
| β | 104.52 | 104.17 | 105.34 | 105.52 | 105.34 | 105.56 |
| resolution (Å) | 2.62 | 2.43 | 1.83 | 1.84 | 1.91 | 1.86 |
| total/unique reflections | 23532/8160 | 26010/10403 | 83134/26894 | 89749/28243 | 97188/25449 | 77769/27505 |
| redundancy | 2.9(2.6)a | 2.5 (1.9) | 3.1(3.1) | 3.2(2.6) | 3.8(2.6) | 2.8(2.5) |
| % complete | 92.9(91.2) | 93.6(86.8) | 95.6(95.5) | 97.6(94.1) | 98.8(91.0) | 98.7(96.8) |
| I/σ | 13.3(2.0) | 10.9(2.9) | 13.5(2.7) | 17.4(8.0) | 16.6(3.9) | 14.2(2.2) |
| Rsymb | 7.7(45.2) | 9.0(33.8) | 7.2(54.8) | 6.0(14.0) | 7.6(25.0) | 7.1(39.1) |
| Matthews no. | 1.90 | 1.92 | 2.09 | 2.21 | 2.19 | 2.21 |
| solvent content (%) | 34.1 | 34.8 | 39.7 | 43.2 | 42.9 | 43.2 |
Values in parenthesis are for the highest resolution shell.
Rsym = ∑∑i∥Ii − ⟨I⟩∥/∑ ⟨I⟩, where ⟨I⟩ is the mean intensity of the N reflections with intensities Ii and common indices h,k,l.
The structures of all of the complexes were determined by molecular replacement using the structure of native AdoMetDC with MeAdoMet bound (PDB 1I7B) as the search model, and the CNS program suite.(44) The model building was done using the program O(45) or Coot.(46) The conformations of the ligand molecules were determined using difference Fo − Fc and composite omit maps. The parameters and the topology files for the ligands were generated using the HIC-Up server.(47) The difference maps also showed density for a molecule of putrescine bound in all of the structures. The refinement statistics of the complexes are given in Table 3.
Table 3. Refinement Statistics for AdoMetDC Complexes.
| F223A + MeAdoMet | 12a | 14e | 17d | 17f | 21c | |
|---|---|---|---|---|---|---|
| resolution (Å) | 2.62 | 2.43 | 1.83 | 1.84 | 1.91 | 1.86 |
| R factora | 0.203 | 0.199 | 0.208 | 0.204 | 0.197 | 0.200 |
| R-freeb | 0.280 | 0.247 | 0.231 | 0.237 | 0.208 | 0.232 |
| no. of non-H atoms | ||||||
| protein | 2473 | 2419 | 2381 | 2489 | 2454 | 2470 |
| ligand | 28 | 25 | 28 | 26 | 25 | 25 |
| water | 79 | 73 | 137 | 222 | 212 | 217 |
| B factors | ||||||
| protein (Å2) | 41.3 | 31.5 | 29.6 | 26.8 | 28.2 | 32.4 |
| ligand (Å2) | 63.4 | 42.1 | 32.3 | 26.0 | 43.9 | 39.9 |
| putrescine (Å2) | 32.4 | 27.9 | 40.0 | 22.4 | 24.7 | 29.8 |
| rms deviations | ||||||
| bonds (Å) | 0.010 | 0.011 | 0.007 | 0.006 | 0.012 | 0.008 |
| angles (deg) | 1.4 | 1.4 | 1.3 | 1.3 | 1.4 | 1.3 |
| dihedrals (deg) | 24.9 | 25.2 | 25.3 | 25.3 | 25.8 | 25.2 |
| Ramachandran plot | ||||||
| most favored region (%) | 84.2 | 89.3 | 91.4 | 91.8 | 92.1 | 92.5 |
| additional favored region (%) | 14.7 | 9.5 | 7.8 | 7.8 | 7.5 | 7.5 |
| generously allowed region (%) | 0.8 | 0.8 | 0.4 | 0.4 | 0.4 | 0.0 |
| disallowed region (%) | 0.4 | 0.4 | 0.4 | 0.0 | 0.0 | 0.0 |
R factor = ∑hkl∥Fobs| − k|Fcal∥/∑hkl|Fobs|, where Fobs and Fcal are observed and calculated structure factors, respectively.
In R-free, the sum is extended over a subset of reflections that were excluded from all stages of refinement.
Molecular Modeling
Determination of the conformational preference of ligands in the active site of AdoMetDC was carried out with Macromodel version 7.2,(36) available from Schrödinger, LLC. To make the computational studies tractable, the protein was truncated to a shell of atoms that included any residue that contained an atom within 20.0 Å of MeAdoMet located in the active site of AdoMetDC (from PDB 1I7B) and was used as the starting model for conformational searching/energy minimization. Removal of water molecules from this “docking shell” was followed by appropriate hydrogen treatment using Schrödinger’s protein preparation utility that aids in the generation of appropriate ionic states and histidine tautomers for active site amino acids and minimizes the protein’s potential energy gradient through a series of constrained energy minimizations. For the conformational searches, the appropriate ligand was added to the active site and, where appropriate, the covalent bond between the amino terminus of the ligand and the pyruvoyl group was formed.
The resulting structures were subjected to 50,000 mixed Monte Carlo MCMM/low mode conformational search steps,34,35 allowing residues within a 5 Å shell surrounding the active site to freely move during each Monte Carlo/low mode step and subsequent energy minimization step of the search. All other protein atoms were constrained to their starting position. Residues His5, Glu67, Cys226, and Glu247 were also constrained to their starting position. The energy minimization step was considered to have converged when the energy gradient was less than 0.05 kJ/mol. The AMBER* force field,48,49 with a distance dependent dielectric “constant” further attenuated by a factor of 4, was employed for the calculations, and the energy minimizations relied upon the TNCG minimization technique.(50) The global minimum and low energy ensemble of structures within 15 kJ/mol of the global minimum (after convergence) were further refined by energy minimization until a gradient less than 0.01 kJ/mol was obtained with just the ligand allowed to move during this subsequent energy minimization procedure. All protein atoms during this process were constrained to their starting position. The jobs were run with the nucleoside starting in both the syn and anti conformations for completeness. The AMBER* parameters for the sulfonium ion were adapted from the work done by Markham et al.(33)
The modeling of the terminal three atoms of 14e was done using conformational searching with Macromodel version 7.2 as described above. Because the position of the rest of the ligand and the protein was determined to high accuracy by fitting to the electron density determined by X-ray diffraction, all of the protein and the ligand atoms except the last three non-hydrogen atoms and their attached hydrogens were fixed during the conformational search. Torsional rotation was allowed around the last two bonds of the C-5′ extension during the conformational search. A visual survey of the five lowest energy structures, which spanned an energy range of 6.5 kJ/mol, showed that they were similar and the global minimum of the search was utilized to obtain the coordinates of the disordered terminal atoms of 14e.
AdoMetDC Activity and Inhibition
AdoMetDC was assayed by measuring the release of 14CO2 from S-adenosyl-l-[carboxy-14C]methionine (Amersham Pharmacia Biotech, ∼60 mCi/mmol).(51) Assay of 30 ng of C-terminal his-tagged AdoMetDC under these conditions results in ∼7000 cpm with a background of 30 and an activity of ∼1.5 pmol/min/ng protein. For determination of the abilities of compounds to inhibit AdoMetDC, the enzyme activity was determined in the presence of no inhibitor and at least five concentrations of each potential inhibitor. The enzyme concentation was 1 nM. The IC50 values were determined from curve fitting to plots of the inhibitor concentration versus the % inhibition of AdoMetDC.
Target Synthesis
TLC analysis was performed on Analtech precoated (250 μm) silica gel GF plates. Melting points were determined on a Mel-Temp apparatus and are uncorrected. Purifications by flash chromatography were carried out on Merck silica gel (230−400 mesh). Evaporations were performed with a rotary evaporator, higher boiling solvents (dimethylformamide (DMF), pyridine) were removed in vacuo (<1 mm, bath to 35 °C). Products were dried in vacuo (<1 mm) at 22−25 °C over P2O5. The mass spectral data were obtained with a Varian-MAT 311A mass spectrometer in the fast atom bombardment (FAB) mode or with a Bruker BIOTOF II by electrospray ionization (ESI). 1H NMR spectra were recorded on a Nicolet NT-300 NB spectrometer operating at 300.635 MHz. Chemical shifts in CDCl3 and Me2SO-d6 are expressed in parts per million downfield from tetramethylsilane (TMS) and in D2O chemical shifts are expressed in parts per million downfield from sodium 3-(trimethylsilyl)propionate-2,2,3.3-d4 (TMSP). Chemical shifts (δ) listed for multiplets were measured from the approximate centers, and relative integrals of peak areas agreed with those expected for the assigned structures. UV absorption spectra were determined on a Perkin-Elmer Lambda 19 spectrometer by dissolving each compound in MeOH or EtOH, and diluting 10-fold with 0.1 N HCl, pH 7 buffer, or 0.1 N NaOH. Numbers in parentheses are extinction coefficients (ε × 10−3). Microanalyses were performed by Atlantic Microlab, Inc. (Atlanta, GA) or the Spectroscopic and Analytical Department of Southern Research Institute. Analytical results indicated by element symbols were within ±0.4% of the theoretical values, and where solvents are indicated in the formula, their presence was confirmed by 1H NMR.
5′-Chloro-5′-deoxy-8-methyladenosine (8a)
To a stirred suspension of 7a(38) (892 mg, 3.17 mmol) in anhydrous pyridine (501 mg, 0.51 mL, 6.33 mmol) and CH3CN (2.5 mL) cooled in an ice bath was slowly added SOCl2 (1.88 g, 1.15 mL, 15.80 mmol). Stirring was continued at 0−5 °C for 3−4 h, with subsequent warming to ambient temperature overnight. The resulting suspension was concentrated in vacuo. To this reaction mixture was added methanol (20 mL), water (2 mL), and NH4OH (4 mL), followed by stirring for 0.5 h at room temperature. The reaction mixture was concentrated to dryness. The compound was dissolved in MeOH, silica gel (3 g) was added and then solvent was removed. The mixture on silica gel was poured onto a column filled with silica gel and eluted with chloroform:methanol (7:1) to yield 661 mg (70%). MS m/z 300 (M + H)+. 1H NMR (DMSO-d6) δ 8.09 (bs, 1H, H-2), 7.15 (bs, 2H, 6-NH2), 5.81 (d, 1H, H-1′, J1′,2′ = 5.7 Hz), 5.49 (d, 1H, 2′-OH, J2′−2′OH = 6.1 Hz), 5.45 (d, 1H, 3′-OH, J3′−3′OH = 5.3 Hz), 5.13 (ddd, 1H, H-2′, J1′,2′ = 5.7 Hz, J2′,3′ = 4.8 Hz, J2′−2′OH = 6.1 Hz), 4.31 (ddd, 1H, H-3′, J2′,3′ = 4.8 Hz, J3′,4′ = 4.0 Hz, J3′−3′OH = 5.3 Hz), 4.03−4.10 (bm, 1H, H-4′), 3.93−3.99 (m, 1H, 5′-CH2), 3.82−3.88 (m, 1H, 5′-CH2), 2.55 (s, 3H, 8-CH3).
5′-Chloro-5′-deoxy-8-ethyladenosine (8b)
The procedure was the same as reported above for 8a using 7b(38) (1.28 g, 4.33 mmol), pyridine (685 mg, 0.70 mL, 8.65 mmol), CH3CN (10 mL), and SOCl2 (2.57 g, 1.58 mL, 21.60 mmol): yield 498 mg (37%). MS m/z 314 (M + H)+. 1H NMR (DMSO-d6) δ 8.09 (bs, 1H, H-2), 7.14 (bs, 2H, 6-NH2), 5.80 (d, 1H, H-1′, J1′,2′ = 5.7 Hz), 5.48 (d, 1H, 2′-OH, J2′−2′OH = 6.1 Hz), 5.45 (d, 1H, 3′-OH, J3′−3′OH = 5.4 Hz), 5.20 (ddd, 1H, H-2′, J1′,2′ = 5.7 Hz, J2′,3′ = 4.6 Hz, J2′−2′OH = 6.1 Hz), 4.30−4.37 (bm, 1H, H-3′), 4.03−4.10 (bm, 1H, H-4′), 3.94−4.0 (m, 1H, 5′-CH2), 3.83−3.89 (m, 1H, 5′-CH2), 2.89 (q, 2H, 8-CH2CH3), 1.31 (t, 3H, 8-CH2CH3).
5′-Chloro-5′-deoxy-8-(methylamino)adenosine (8c)
Compound 8c was prepared by the same procedure as described for the preparation of 8a using 7c(39,41) (2.9 g, 9.78 mmol), pyridine (1.54 g, 1.57 mL, 19.46 mmol), CH3CN (5 mL), and SOCl2 (5.82 g, 3.56 mL, 48.91 mmol): yield 1.95 g (63%). MS m/z 315 (M + H)+. 1H NMR (DMSO-d6) δ 7.91 (bs, 1H, H-2),), 6.78 (q, 1H, 8CH3−NH), 6.50 (bs, 2H, 6-NH2), 5.70 (d, 1H, H-1′, J1′,2′ = 5.0 Hz), 5.41 (d, 1H, 2′-OH, J2′−2′OH = 5.6 Hz), 5.32 (d, 1H, 3′-OH, J3′−3′OH = 5.3 Hz), 5.18 (ddd, 1H, H-2′, J1′,2′ = 5.0 Hz, J2′,3′ = 5.4 Hz, J2′−2′OH = 5.6 Hz), 4.33 (ddd, 1H, H-3′, J2′,3′ = 5.4 Hz, J3′,4′ = 4.4 Hz, J3′−3′OH = 5.3 Hz),), 3.91−4.02 (bm, 2H, H-4′, 5′-CH2), 3.76−3.82 (m, 1H, 5′-CH2), 2.88 (d, 3H, 8NH-CH3, J = 4.5 Hz).
5′-Chloro-5′-deoxy-8-phenyladenosine (8d)
The procedure described for 8a was used to prepare 8d from 7d(40)(4.5 g, 13.10 mmol), pyridine (2.07 g, 2.12 mL, 26.2 mmol), CH3CN (6 mL), and SOCl2 (7.79 g, 4.78 mL, 65.47 mmol): yield 2.21 g (47%). MS m/z 362 (M + H)+. 1H NMR (DMSO-d6) δ 8.20 (s, 1H, H-2), 7.71−7.76 (m, 2H, 8-phenyl o-H’s), 7.59−7.64 (m, 3H, 8-phenyl m- and p-H’s), 7.40 (bs, 2H, 6-NH2), 5.75 (d, 1H, H-1′, J1′,2′ = 6.0 Hz), 5.52 (d, 1H, 2′-OH, J2′−2′OH = 5.6 Hz), 5.39−5.44 (m, 2H, H-2′, 3′-OH), 4.33 (bs, 1H, H-3′), 3.98−4.06 (bm, 2H, H-4′, 5′-CH2), 3.88−3.94 (m, 2H, 5′-CH2).
5′-Deoxy-5′-methylamino-8-methyladenosine (9a)
A mixture of 8a (660 mg, 2.20 mmol) in 33% methylamine/ethanol solution (30 mL) in a steel bomb was heated for two days at 90 °C. The reaction mixture was concentrated to dryness and purified by column chromatography (elution with 4:1:0.3 chloroform:methanol:NH4OH to yield 294 mg (45%). MS m/z 295 (M + H)+. 1H NMR (DMSO-d6) δ 8.08 (bs, 1H, H-2), 7.14 (bs, 2H, 6-NH2), 5.72 (d, 1H, H-1′, J1′,2′ = 6.5 Hz), 5.30 (d, 1H, 2′-OH, J2′−2′OH = 6.3 Hz), 5.17 (bd, 1H, 3′-OH, J3′−3′OH = 3.5 Hz), 5.01 (ddd, 1H, H-2′, J1′,2′ = 6.5 Hz, J2′,3′ = 5.5 Hz, J2′−2′OH = 6.3 Hz), 4.16 (bs, 1H, H-3′), 3.96−4.0 (m, 1H, H-4′), 2.64−2.77 (bm, 2H, 5′-CH2), 2.53 (s, 3H, 8-CH3), 2.29 (s, 3H, 5′NH-CH3).
5′-Deoxy-5′-methylamino-8-ethyladenosine (9b)
The procedure was the same as reported above for 9a using 8b (1.00 g, 3.18 mmol) and 33% methylamine/ethanol solution (30 mL). After column chromatography (elution with 5:1:0.3 chloroform:methanol:NH4OH), a yellow glassy solid was obtained: 498 mg (50%). MS m/z 309 (M + H)+. 1H NMR (DMSO-d6) δ 8.08 (bs, 1H, H-2), 7.13 (bs, 2H, 6-NH2), 5.70 (d, 1H, H-1′, J1′,2′ = 6.6 Hz), 5.30 (d, 1H, 2′-OH, J2′−2′OH = 6.4 Hz), 5.16 (d, 1H, 3′-OH, J3′−3′OH = 4.5 Hz), 5.08 (ddd, 1H, H-2′, J1′,2′ = 6.6 Hz, J2′,3′ = 5.4 Hz, J2′−2′OH = 6.4 Hz), 4.14−4.18 (m, 1H, H-3′), 3.96−4.0 (m, 1H, H-4′), 2.88 (q, 2H, 8-CH2CH3), 2.64−2.78 (m, 2H, 5′-CH2), 2.29 (s, 3H, 5′NH-CH3), 1.30 (t, 3H, 8CH2CH3).
5′-Deoxy-5′,8-bis(methylamino)adenosine (9c)
Compound 9c was prepared by the same procedure as described for the preparation of 9a using 8c (1.00 g, 3.17 mmol) and 33% methylamine/ethanol solution (30 mL). After column chromatography (elution with 7:1:0.4 chloroform:methanol:NH4OH), a yellow glassy solid was obtained: 505 mg (51%). MS m/z 310 (M + H)+. 1H NMR (DMSO-d6) δ 7.89 (bs and q, 2H, H-2 and 8CH3−NH), 6.44 (bs, 2H, 6-NH2), 5.84 (d, 1H, H-1′, J1′,2′ = 7.0 Hz), 5.22 (d, 1H, 2′-OH, J2′−2′OH = 6.4 Hz), 5.12 (d, 1H, 3′-OH, J3′−3′OH = 4.0 Hz), 4.66 (ddd, 1H, H-2′, J1′,2′ = 7.0 Hz, J2′,3′ = 5.4 Hz, J2′−2′OH = 6.4 Hz), 4.14 (bm, 1H, H-3′), 3.94−4.0 (bm, 1H, H-4′), 2.90 (d, 3H, 8NH-CH3, J = 4.4 Hz), 2.77−2.83 (m, 1H, 5′-CH2), 2.57−2.62 (m, 1H, 5′-CH2), 2.35 (s, 3H, 5′NH-CH3).
5′-Deoxy-5′-methylamino-8-phenyladenosine (9d)
Compound 9d was prepared by the same procedure as described for the preparation of 9a using 8d (2.00 g, 5.52 mmol) and 33% methylamine/ethanol solution (40 mL). After column chromatography (elution with 4:1:0.2 chloroform:methanol:NH4OH), a yellow glassy solid was obtained: 963 mg (49%). MS m/z 357 (M + H)+. 1H NMR (DMSO-d6) δ 8.19 (s, 1H, H-2), 7.72−7.76 (m, 2H, 8-phenyl o-H’s), 7.58−7.61 (m, 3H, 8-phenyl m- and p-H’s), 7.40 (bs, 2H, 6-NH2), 5.70 (d, 1H, H-1′, J1′,2′ = 6.4 Hz), 5.38 (d, 1H, 2′-OH, J2′−2′OH = 6.2 Hz), 5.29 (ddd, 1H, H-2′, J1′,2′ = 6.4 Hz, J2′,3′ = 5.1 Hz, J2′−2′OH = 6.2 Hz), 5.14 (bs, 1H, 3′-OH), 4.19 (bs, 1H, H-3′), 3.95−4.0 (m, 1H, H-4′), 2.82 (d, 2H, 5′-CH2,J = 5.2 Hz), 2.34 (s, 3H, 5′NH-CH3).
5′-Deoxy-5′-[[2-[[(1-ethoxyethylidene)amino]oxy]ethyl]methylamino]-8-methyladenosine (11a)
A mixture of compound 9a (416 mg, 1.41 mmol), ethyl N-(2-bromoethoxy)ethanimidate(28) (350 mg, 1.66 mmol), and diisopropylethylamine (DIEA) (11 mg, 0.014 mL, 0.085 mmol) in DMF (5 mL) was heated at 50 °C overnight under nitrogen. The reaction mixture was concentrated to dryness. The resulting syrup was purified by column chromatography (elution with 7:1:0.1 chloroform:methanol:NH4OH) to yield 50 mg (8%) of a yellow glassy sticky solid. MS m/z 424 (M + H)+. 1H NMR (DMSO-d6) δ 8.07 (s, 1H, H-2), 7.11 (bs, 2H, 6-NH2), 5.73 (d, 1H, H-1′, J1′,2′ = 5.5 Hz), 4.31 (d, 1H, 2′-OH, J2′−2′OH = 6.3 Hz), 5.15 (d, 1H, 3′-OH, J3′−3′OH = 5.5 Hz), 5.04 (ddd, 1H, H-2′, J1′,2′ = 5.5 Hz, J2′,3′ = 5.1 Hz, J2′−2′OH = 6.3 Hz), 4.13 (ddd, 1H, H-3′, J2′,3′ = 5.1 Hz, J3′,4′ = 4.7 Hz, J3′−3′OH = 5.5 Hz), 3.92 (q, 2H, CH2CH3), 3.88 (t, 2H, NO-CH2), 2.72−2.78 (m, 1H, 5′-CH2), 2.56−2.62 (m, 4H, 5′-CH2, H-4′, N(CH3)-CH2), 2.53 (s, 3H, 8-CH3), 2.21 (s, 3H, N−CH3), 1.83 (s, 3H, C-CH3), 1.19 (t, 3H, OCH2CH3).
5′-Deoxy-5′-[[2-[[(1-ethoxyethylidene)amino]oxy]ethyl]methylamino]-8-(methylamino)adenosine (11b)
Compound 11b was prepared by the same procedure as reported for 11a using 9c (500 mg, 1.61 mmol), ethyl N-(2-bromoethoxy)ethanimidate(28) (407 mg, 1.93 mmol), DIEA (104 mg, 0.14 mL, 0.80 mmol), and DMF (5 mL). After column chromatography (elution with 7:1:0.3 chloroform:methanol:NH4OH), a yellow glassy sticky solid was obtained: yield 209 mg (30%). MS: m/z 439 (M + H)+. 1H NMR (DMSO-d6) δ 7.89 (s, 1H, H-2), 6.85 (q, 1H, 8CH3−NH), 6.47 (bs, 2H, 6-NH2), 5.69 (d, 1H, H-1′, J1′,2′ = 5.0 Hz), 5.27 (d, 1H, 2′-OH, J2′−2′OH = 5.5 Hz), 5.06 (d, 1H, 3′-OH, J3′−3′OH = 5.0 Hz), 4.88 (ddd, 1H, H-2′, J1′,2′ = 5.0 Hz, J2′,3′ = 5.8 Hz, J2′−2′OH = 5.5 Hz), 4.17 (ddd, 1H, H-3′, J2′,3′ = 5.8 Hz, J3′,4′ = 4.7 Hz, J3′−3′OH = 5.0 Hz), 3.86−3.95 (m, 3H, H-4′, NO-CH2), 3.92 (q, 2H, CH2CH3), 2.88 (d, 3H, 8NH-CH3,J = 4.6 Hz), 2.71−2.77 (m, 1H, 5′-CH2), 2.56−2.67 (m, 3H, 5′-CH2, N(CH3)-CH2), 2.24 (s, 3H, N-CH3), 1.83 (s, 3H, C−CH3), 1.19 (t, 3H, OCH2CH3).
5′-Deoxy-5′-[[2-[[(1-ethoxyethylidene)amino]oxy]ethyl]methylamino]-8-phenyladenosine (11c)
The procedure described for 11a was used to prepare 11c from 9d (400 mg, 1.12 mmol), ethyl N-(2-bromoethoxy)ethanimidate(28) (283 mg, 1.34 mmol), and DIEA (72 mg, 0.10 mL, 0.55 mmol). After column chromatography (elution with 7:1 chloroform:methanol), a yellow glassy sticky solid was obtained: yield 105 mg (20%). MS: m/z 486 (M + H)+. 1H NMR (DMSO-d6) δ 8.17 (s, 1H, H-2), 7.72−7.78 (m, 2H, 8-phenyl o-H’s), 7.58−7.63 (m, 3H, 8-phenyl m- and p-H’s), 7.36 (bs, 2H, 6-NH2), 5.67 (d, 1H, H-1′, J1′,2′ = 5.2 Hz), 5.29−5.34 (m, 2H, 2′-OH, H-2′), 5.13 (d, 1H, 3′-OH, J3′−3′OH = 5.3 Hz), 4.14−4.18 (m, 1H, H-3′), 3.88−3.97 (m, 3H, H-4′, NO-CH2), 3.92 (q, 2H, CH2CH3), 2.78−2.84 (m, 1H, 5′-CH2), 2.60−2.70 (m, 3H, 5′-CH2, N(CH3)-CH2), 2.24 (bs, 3H, N-CH3), 1.83 (s, 3H, C-CH3), 1.19 (t, 3H, OCH2CH3).
5′-[(2-Aminooxyethyl)methylamino]-5′-deoxy-8-methyladenosine sulfate (2.2:1 salt) (12a)
Compound 11a (50 mg, 0.11 mmol) was dissolved in 2 mL of 2 N H2SO4 and stirred for two days at room temperature. The reaction mixture was neutralized with NaHCO3 and lyophilized. The compound was extracted with EtOH (2 × 10 mL) and concentrated to dryness. The residue was purified by column chromatography (silica gel 230−400 mesh, elution with 7:1:0.3 chloroform:methanol:NH4OH). The desired fractions were combined, concentrated, and dried in vacuo. The product was dissolved in 3 mL of EtOH and 2 N H2SO4 was added dropwise. The resulting sulfate salt that precipitated out was filtered and then washed with EtOH. This product, which was hygroscopic in nature, was dissolved in water (2 mL) and lyophilized to give a white solid: yield 20 mg (29%). MS: m/z 354 (M + H)+. 1H NMR (DMSO-d6) δ 8.25 (s, 1H, H-2), 7.80 (bs, 2H, O-NH2), 5.87 (d, 1H, H-1′, J1′,2′ = 5.7 Hz), 4.88 (t, 1H, H-2′, J2′,3′ = 5.2 Hz), 4.35−4.40 (bm, 1H, H-4′), 4.23 (t, 1H, H-3′, J2′,3′ = 3.2 Hz), 4.10 (t, 2H, NH2O−CH2), 3.50−3.57 (m, 1H, 5′-CH2), 3.65−3.72 (m, 1H, 5′-CH2), 3.45 (bm, 2H, N(CH3)-CH2), 2.85 (s, 3H, N-CH3), 2.58 (s, 3H, 8-CH3). UV λmax, nm, pH 1, 274 (ε 15200), pH 7, 276 (ε 15500), pH 13, 277 (ε 15900). Anal. (C14H23N7O4·2.2H2SO4·0.1C2H5OH·0.5H2O) C, H, N.
5′-[(2-Aminooxyethyl)methylamino]-5′-deoxy-8-(methylamino)adenosine sulfate (2.1:1 salt) (12b)
The procedure described for 12a was used to prepare 12b from 11b (200 mg, 0.45 mmol): yield 125 mg (46%). MS: m/z 369 (M + H)+. 1H NMR (DMSO-d6) δ 8.16 (s, 1H, H-2), 7.50−7.65 (bm, 2H, O-NH2), 5.83 (d, 1H, H-1′, J1′,2′ = 5.3 Hz), 6.56 (bs, 2H, 6-NH2), 4.96 (t, 1H, H-2′, J2′,3′ = 4.8 Hz), 4.28−4.35 (bm, 1H, H-4′), 4.25 (t, 1H, H-3′, J3′,4′ = 4.1 Hz), 3.96 (t, 2H, NH2O-CH2), 3.59−3.66 (m, 1H, 5′-CH2), 3.49−3.57 (m, 1H, 5′-CH2), 3.36−3.40 (bm, 2H, N(CH3)-CH2), 2.94 (s, 3H, 8NH-CH3), 2.81 (s, 3H, N-CH3). UV λmax, nm, pH 1, 274 (ε 14300), pH 7, 276.7 (ε 17100), pH 13, 276.1 (ε 17500). Anal. (C14H24N8O4·2.1H2SO4·0.3C2H5OH·0.2H2O) C, H, N, S.
5′-[(2-Aminooxyethyl)methylamino]-5′-deoxy-8-phenyladenosine sulfate (2:1 salt) (12c)
Compound 12c was prepared by the same procedure as described for the preparation of 12a using 11c (99 mg, 0.20 mmol): yield 57 mg (42%). MS: m/z 416 (M + H)+. 1H NMR (D2O) δ 8.37 (s, 1H, H-2), 7.73−7.76 (m, 2H, 8-phenyl o-H’s), 7.60−7.70 (m, 3H, 8-phenyl m- and p-H’s), 6.02 (d, 1H, H-1′, J1′,2′ = 5.7 Hz), 5.25 (t, 1H, H-2′, J2′,3′ = 4.9 Hz), 4.46−4.54 (bm, 2H, H-3′, 4′), 4.03 (t, 2H, NH2O-CH2), 3.87−4.0 (m, 1H, 5′-CH2), 3.61−3.67 (m, 1H, 5′-CH2), 3.50−3.55 (m, 2H, N(CH3)-CH2), 3.0 (s, 3H, N-CH3). UV λmax, nm, pH 1, 275 (ε 21600), pH 7, 275 (ε 17100), pH 13, 274.4 (ε 16800). Anal. (C19H25N7O4·2.0H2SO4·3H2O) C, H, N, S.
5′-Deoxy-5′-[[4-[[(1-ethoxyethylidene)amino]oxy]butyl]methylamino]-8-(methylamino)adenosine (13a)
Compound 13a was prepared by the same procedure as reported for 11a using 9c (1.00 g, 3.23 mmol), ethyl N-(4-bromobutoxy)ethanimidate(29) (924 mg, 3.87 mmol), and DIEA (209 mg, 0.28 mL, 1.6 mmol): yield 635 mg (42%). MS: m/z 467 (M + H)+. 1H NMR (DMSO-d6) δ 7.89 (s, 1H, H-2), 6.87 (q, 1H, 8CH3-NH), 6.46 (bs, 2H, 6-NH2), 5.69 (d, 1H, H-1′, J1′,2′ = 4.8 Hz), 5.25 (d, 1H, 2′-OH, J2′−2′OH = 5.6 Hz), 5.06 (d, 1H, 3′-OH, J3′−3′OH = 5.4 Hz), 4.91 (ddd, 1H, H-2′, J1′,2′ = 4.8 Hz, J2′,3′ = 5.4 Hz, J2′−2′OH = 5.6 Hz), 4.16 (ddd, 1H, H-3′, J2′,3′ = 5.4 Hz, J3′,4′ = 4.9 Hz, J3′−3′OH = 5.4 Hz), 3.85−3.94 (m, 1H, H-4′), 3.92 (q, 2H, OCH2CH3), 3.80 (t, 2H, NO-CH2), 2.88 (d, 3H, 8NH-CH3,J = 4.6 Hz), 2.65−2.74 (m, 1H, 5′-CH2), 2.46−2.58 (m, 1H, 5′-CH2), 2.34 (t, 2H, N(CH3)-CH2), 2.17 (s, 3H, N-CH3), 1.83 (s, 3H, C-CH3), 1.37−1.61 (bm, 4H, NOCH2−CH2CH2), 1.19 (t, 3H, OCH2CH3).
5′-Deoxy-5′-[[4-[[(1-ethoxyethylidene)amino]oxy]butyl]methylamino]-8-phenyladenosine (13b)
The same procedure as described for 6a was used to prepare 13b from 9d (450 mg, 1.26 mmol), ethyl N-(4-bromobutoxy)ethanimidate(29) (360 mg, 1.51 mmol), and DIEA (81 mg, 0.10 mL, 0.62 mmol). After column chromatography (elution with 7:1 chloroform:methanol), a yellow glassy sticky solid was obtained: yield 312 mg (48%). MS: m/z 514 (M + H)+. 1H NMR (DMSO-d6) δ 8.17 (s, 1H, H-2), 7.71−7.78 (m, 2H, 8-phenyl o-H’s), 7.58−7.64 (m, 3H, 8-phenyl m- and p-H’s), 7.36 (bs, 2H, 6-NH2), 5.67 (d, 1H, H-1′, J1′,2′ = 5.7 Hz), 5.32 (bs, 1H, 2′-OH), 5.31 (t, 1H, H-2′, J1′,2′ = 5.7 Hz, J2′,3′ = 5.4 Hz), 5.11 (d, 1H, 3′-OH, J3′−3′OH = 4.8 Hz), 4.16 (bddd, 1H, H-3′, J2′,3′ = 5.4 Hz, J3′,4′ = 4.0 Hz), 3.92−3.97 (m, 1H, H-4′), 3.91 (q, 2H, OCH2CH3), 3.79 (t, 2H, NO-CH2), 2.72−2.80 (m, 1H, 5′-CH2), 2.54−2.59 (m, 1H, 5′-CH2), 2.34 (bt, 2H, N(CH3)-CH2), 2.17 (bs, 3H, N-CH3), 1.83 (s, 3H, C-CH3), 1.39−1.60 (bm, 4H, NOCH2-CH2CH2), 1.18 (t, 3H, OCH2CH3).
5′-Deoxy-5′-[[4-[[(1-ethoxyethylidene)amino]oxy]butyl]methylamino]-8-oxoadenosine (13c)
The procedure described for 11a was used to prepare 13c from 9i(52) (500 mg, 1.68 mmol), ethyl N-(4-bromobutoxy)ethanimidate(29) (481 mg, 2.01 mmol), DIEA (109 mg, 0.14 mL, 0.84 mmol), and DMF (5 mL). After column chromatography (elution with 4:1:0.2 chloroform:methanol:NH4OH), a yellow glassy sticky solid was obtained: yield 200 mg (26%). MS: m/z 454 (M + H)+. 1H NMR (DMSO-d6) δ 10.34 (bs, 1H, 8-OH), 8.02 (s, 1H, H-2), 6.49 (bs, 2H, 6-NH2), 5.62 (d, 1H, H-1′, J1′,2′ = 5.0 Hz), 4.99 (bs, 1H, 3′-OH), 5.19 (bs, 1H, 2′-OH), 4.90 (t, 1H, H-2′, J2′,3′ = 5.4 Hz), 4.16−4.24 (bm, 1H, H-3′), 3.83−3.89 (m, 1H, H-4′), 3.92 (q, 2H, OCH2CH3), 3.77 (t, 2H, NO-CH2), 2.62−3.68 (m, 1H, 5′-CH2), 2.40−2.46 (m, 1H, 5′-CH2), 2.30 (t, 2H, N(CH3)-CH2), 2.13 (s, 3H, N-CH3), 1.84 (s, 3H, C-CH3), 1.35−1.60 (bm, 4H, NOCH2-CH2CH2), 1.21 (t, 3H, OCH2CH3).
5′-Deoxy-2′,3′-isopropylidene-5′-[[4-[[(1-ethoxyethylidene)amino]oxy]butyl]methylamino]-8-methyladenosine (13d)
Compound 13d was prepared by the same procedure as reported for 11a using 9e (1.00 g, 3.11 mmol), MsCl (392 mg, 0.26 mL, 3.42 mmol), methylamine (25 mL), ethyl N-(4-bromobutoxy)ethanimidate (853 mg, 3.58 mmol), DIEA (200 mg, 0.27 mL, 1.54 mmol), and DMF (8 mL). After column chromatography (95:5 chloroform:methanol), a glassy solid was obtained: yield 176 mg (12%). MS: m/z 492 (M + H)+. 1H NMR (CDCl3) δ 8.27 (s, 1H, H-2), 5.99 (d, 1H, H-1′, J1′,2′ = 1.8 Hz), 5.75 (dd, 1H, H-2′, J1′,2′ = 1.8 Hz, J2′,3′ = 6.4 Hz), 5.39 (bs, 2H, 6-NH2), 5.08 (dd, 1H, H-3′, J2′,3′ = 6.4 Hz, J3′,4′ = 3.5 Hz), 4.27−4.34 (m, 1H, H-4′), 4.0 (q, 2H, OCH2CH3), 3.84 (t, 2H, NO-CH2), 2.64 (s, 3H, 8-CH3), 2.55−2.61 (m, 1H, 5′-CH2), 2.45−2.55 (m, 1H, 5′-CH2), 2.29−2.34 (m, 2H, N(CH3)-CH2), 2.21 (s, 3H, N-CH3), 1.91 (s, 3H, C-CH3), 1.61 and 1.40 (2s, 6H, C(CH3)2), 1.51−1.60 (m, 2H, NOCH2-CH2), 1.37−1.45 (m, 2H, N(CH3)CH2−CH2), 1.27 (t, 3H, OCH2CH3).
5′-Deoxy-2′,3′-isopropylidene-5′-[[4-[[(1-ethoxyethylidene)amino]oxy]butyl]methylamino]-8-ethyladenosine (13e)
The procedure described for 11a was used to prepare 13e from 9f (1.00 g, 2.98 mmol), MsCl (375 mg, 0.25 mL, 3.27 mmol), methylamine (25 mL), ethyl N-(4-bromobutoxy)ethanimidate (852 mg, 3.57 mmol), DIEA (192 mg, 0.25 mL, 1.48 mmol), and DMF (10 mL). After column chromatography (95:5 chloroform:methanol), a glassy solid was obtained: yield 159 mg (11%). MS: m/z 506 (M + H)+. 1H NMR (CDCl3) δ 8.27 (s, 1H, H-2), 5.99 (d, 1H, H-1′, J1′,2′ = 2.0 Hz), 5.73 (dd, 1H, H-2′, J1′,2′ = 2.0 Hz, J2′,3′ = 6.4 Hz), 5.40 (bs, 2H, 6-NH2), 5. 09 (dd, 1H, H-3′, J2′,3′ = 6.4 Hz, J3′,4′ = 3.6 Hz), 4.26−4.33 (m, 1H, H-4′), 4.0 (q, 2H, O CH2CH3), 3.84 (t, 2H, NO-CH2), 2.91−2.99 (m, 2H, CH2 of 8-Et), 2.59−2.65 (m, 1H, 5′-CH2), 2.46−2.53 (m, 1H, 5′-CH2), 2.30−2.35 (m, 2H, N(CH3)-CH2), 2.21 (s, 3H, N-CH3), 1.91 (s, 3H, C-CH3), 1.61 and 1.40 (2s, 6H, C(CH3)2), 1.51−1.59 (m, 2H, NOCH2-CH2), 1.49−1.38 (m, 2H, N(CH3)CH2−CH2), 1.43 (s, 3H, CH3 of 8-Et), 1.27 (t, 3H, OCH2CH3).
5′-Deoxy-2′,3′-isopropylidene-5′-[[4-[[(1-ethoxyethylidene)amino]oxy]butyl]methylamino]-adenosine (13f)
Compound 13f was prepared by the same procedure as reported for 11a using 9g(32) (1.00 g, 3.25 mmol), MsCl (447 mg, 0.30 mL, 3.90 mmol), methylamine (25 mL), ethyl N-(4-bromobutoxy)ethanimidate (511 mg, 2.15 mmol), DIEA (125 mg, 0.17 mL, 0.96 mmol), and DMF (5 mL). After column chromatography (95:5 chloroform:methanol), a pale-yellow syrup was obtained: yield 839 mg (86%). MS: m/z 478 (M + H)+. 1H NMR (CDCl3) δ 8.36 (s, 1H, H-2), 7.96 (s, 1H, H-8), 6.07 (d, 1H, H-1′, J1′,2′ = 2.2 Hz), 5.60 (bs, 2H, 6-NH2), 5.49 (dd, 1H, H-2′, J1′,2′ = 2.2 Hz, J2′,3′ = 6.4 Hz), 4.95 (dd, 1H, H-3′, J2′,3′ = 6.4 Hz, J3′,4′ = 3.4 Hz), 4.36−4.40 (m, 1H, H-4′), 4.0 (q, 2H, OCH2CH3), 3.86 (t, 2H, NO-CH2), 2.61 (dd, 1H, 5′-CH2), 2.55 (dd, 1H, 5′-CH2), 2.38 (bt, 2H, N(CH3)-CH2), 2.26 (s, 3H, N-CH3), 1.91 (s, 3H, C-CH3), 1.61 and 1.40 (2s, 6H, C(CH3)2), 1.55−1.61 (m, 2H, NOCH2-CH2), 1.44−1.52 (m, 2H, N(CH3)CH2−CH2), 1.27 (t, 3H, OCH2CH3).
5′-[(4-Aminooxybutyl)methylamino]-5′-deoxy-8-(methylamino)adenosine sulfate (0.4:1 salt) (14a)
The same procedure used to prepare 12a was used to prepare 14a using 13a (600 mg, 1.28 mmol) and 2 N H2SO4 (10 mL): yield 514 mg (87%). MS: m/z 510 (M + H)+. 1H NMR (DMSO-d6) δ 7.91 (s, 1H, H-2), 6.88 (bq, 1H, 8CH3-NH), 6.50 (bs, 2H, 6-NH2), 5.72 (d, 1H, H-1′, J1′,2′ = 5.1 Hz), 5.08−5.39 (bm, 2H, 2′, 3′-OH), 4.0−5.02 (m, 1H, H-2′), 4.19 (t, 1H, H-3′), 3.94−4.06 (bm, 1H, H-4′), 3.45 (bt, 2H, NH2O-CH2), 3.36−3.54 (m, 2H, 5′-CH2), 2.89 (d, 3H, 8NH-CH3), 2.78−2.95 (bm, 2H, NCH3-CH2), 2.04 (bs, 3H, N-CH3), 1.38−1.52 (bm, 4H, NH2OCH2−CH2CH2). UV λmax, nm, pH 1, 274.8 (ε 14300), pH 7, 276 (ε 16700), pH 13, 277 (ε 17400). Anal. (C16H28N8O4·0.4H2SO4·0.2C2H5OH·0.9H2O) C, H, N, S.
5′-[(4-Aminooxybutyl)methylamino]-5′-deoxy-8-phenyladenosine sulfate (1.75:1 salt) (14b)
Compound 14b was prepared by the same procedure as described for the preparation of 12a using 13b (305 mg, 0.59 mmol) and 2 N H2SO4 (4 mL): yield 252 mg (64%). MS: m/z 444 (M + H)+. 1H NMR (DMSO-d6) δ 8.18 (s, 1H, H-2), 7.20−7.78 (m, 2H, 8-phenyl o-H’s), 7.59−7.61 (m, 3H, 8-phenyl m- and p-H’s), 7.37 (bs, 2H, 6-NH2), 5.85 (s, 2H, O-NH2), 5.68 (d, 1H, H-1′, J1′,2′ = 5.7 Hz), 5.31 (t, 1H, H-2′, J2′,3′ = 5.5 Hz), 5.13 (bd, 1H, 3′-OH), 4.18 (t, 1H, H-3′, J3′,4′ = 3.9 Hz), 3.94−3.99 (bm, 1H, H-4′), 3.49 (t, 2H, NH2O−CH2), 2.73−2.79 (m, 1H, 5′-CH2), 2.54−2.62 (m, 1H, 5′-CH2), 2.27−2.36 (bm, 2H, N(CH3)-CH2), 2.16 (bs, 3H, N-CH3), 1.33−1.53 (bm, 4H, NH2OCH2−CH2CH2). 1H NMR (D2O) δ 8.36 (s, 1H, H-2), 7.72−7.78 (m, 2H, 8-phenyl o-H’s), 7.63−7.71 (m, 3H, 8-phenyl m- and p-H’s), 6.02 (d, 1H, H-1′, J1′,2′ = 5.8 Hz), 5.28−5.41 (bm, 1H, H-2′), 4.43−4.53 (bm, 2H, H-3′, 4′), 3.92−4.03 (m, 1H, 5′-CH2), 3.90 (t, 2H, NH2O-CH2), 3.49−3.59 (m, 1H, 5′-CH2), 3.22− 3.32 (bm, 2H, N(CH3)-CH2), 2.92 (bs, 3H, N-CH3), 1.61−1.83 (bm, 4H, NH2OCH2−CH2CH2). UV λmax, nm, pH 1, 275 (ε 21400), pH 7, 274.5 (ε 16900), pH 13, 274.8 (ε 16,700). Anal. (C21H29N7O4·1.75H2SO4·0.05C2H5OH·2.4H2O) C, H, N, S.
5′-[(4-Aminooxybutyl)methylamino]-5′-deoxy-8-oxoadenosine sulfate (1.9:1 salt) (14c)
The procedure was the same as reported above for 12a using 13c (190 mg, 0.41 mmol) and 2 N H2SO4 (3 mL). The compound was purified by column chromatography (elution with 4:1:0.5 chloroform:methanol:NH4OH): yield 208 mg (82%). MS: m/z 384 (M + H)+. 1H NMR (DMSO-d6) δ 10.45 (bs, 1H, 8-OH), 8.05 (s, 1H, H-2), 6.58 (bs, 2H, 6-NH2), 5.77 (d, 1H, H-1′, J1′,2′ = 5.0 Hz), 5.37−5.71 (bm, 2H, O-NH2), 4.83 (t, 1H, H-2′, J2′,3′ = 4.2 Hz), 4.18−4.29 (m, 2H, H-3′, H-4′), 3.88 (t, 2H, NH2O-CH2), 3.34−3.54 (m, 2H, 5′-CH2), 3.06 (bt, 2H, N(CH3)-CH2), 2.73 (s, 3H, N-CH3), 1.46−1.74 (bm, 4H, NH2OCH2−CH2CH2). UV λmax, nm, pH 1, 263.3 (ε 12200), pH 7, 268.9 (ε 13600), pH 13, 279.9 (ε 15,600). Anal. (C15H25N7O5·1.9H2SO4·0.1C2H5OH·2H2O) C, H, N, S.
5′-[(4-Aminooxybutyl)methylamino]-5′-deoxy-8-methyladenosine sulfate (1.9:1 salt) (14d)
Compound 13d (149 mg, 0.30 mmol) was dissolved in 2.5 mL of 1 N H2SO4 and stirred for 12 days at room temperature. After a work up identical with that used for 7a, the residue was purified by column chromatography (silica gel 230−400 mesh, elution with 4:1:0.2 chloroform:methanol:NH4OH). The product was dissolved in 8 mL of EtOH and 2 N H2SO4 was added dropwise. The sulfate salt that precipitated out was filtered and washed with EtOH. This product was dissolved in water (2 mL) and lyophilized to give a white solid: yield 59 mg (33%). MS: m/z 382 (M + H)+. 1H NMR (D2O) δ 8.39 (s, 1H, H-2), 6.08 (d, 1H, H-1′, J1′,2′ = 5.5 Hz), 5.0−5.19 (bm, 1H, H-2′), 4.51−4.58 (bm, 2H, H-3′, 4′), 4.08 (t, 2H, NH2O-CH2), 3.73−4.0 (m, 1H, 5′-CH2), 3.44−3.69 (m, 1H, 5′-CH2), 3.16−3.36 (bm, 2H, N(CH3)-CH2), 2.92 (bs, 3H, N-CH3), 2.70 (s, 3H, 8-CH3), 1.68−1.88 (bm, 4H, NH2OCH2−CH2CH2). UV λmax, nm, pH 1, 258.2 (ε 15400), pH 7, 259.7 (ε 15500), pH 13, 260.9 (ε 15900). Anal. (C16H27N7O4·1.9H2SO4·0.4C2H5OH) C, H, N.
5′-[(4-Aminooxybutyl)methylamino]-5′-deoxy-8-ethyladenosine sulfate (1.9:1 salt) (14e)
The procedure was the same as reported above for 14d using 13e (155 mg, 0.30 mmol): yield 55 mg (30%). MS: m/z 396 (M + H)+. 1H NMR (D2O) δ8.39 (s, 1H, H-2), 6.09 (d, 1H, H-1′, J1′,2′ = 5.6 Hz), 5.07−5.23 (bm, 1H, H-2′), 4.50−4.60 (bm, 2H, H-3′, 4′), 4.06 (t, 2H, NH2O-CH2), 3.82−3.96 (m, 1H, 5′-CH2), 3.45−3.69 (m, 1H, 5′-CH2), 3.27 (bs, 2H, N(CH3)-CH2), 3.0−3.10 (m, 2H, 8CH2CH3), 2.91 (bs, 3H, N-CH3), 1.68−1.86 (bm, 4H, NH2OCH2−CH2CH2), 1.39 (t, 3H, 8CH2CH3). UV λmax, nm, pH 1, 259.1 (ε 16400), pH 7, 260 (ε 15700), pH 13, 260.2 (ε 15900). Anal. (C17H29N7O4·1.9H2SO4·0.2C2H5OH) C, H, N.
5′-[(4-Aminooxybutyl)methylamino]-5′-deoxyadenosine sulfate (2:1 salt) (14f)
The procedure was the same as reported above for 14d using 13f (750 mg, 1.5 mmol): yield 457 mg (48%). MS: m/z 368 (M + H)+. 1H NMR (DMSO-d6) δ 8.43 (s, 1H, H-8), 8.23 (s, 1H, H-2), 7.58 (bs, 2H, 6-NH2), 6.03 (d, 1H, H-1′, J1′,2′ = 5.4 Hz), 4.75 (t, 1H, H-2′, J1′,2′ = 5.4 Hz, J2′,3′ = 4.8 Hz), 4.32−4.40 (bm, 1H, H-4′), 4.23 (t, 1H, H-3′, J3′,4′ = 3.8 Hz), 3.93 (t, 2H, NH2O-CH2), 3.68 (dd, 1H, 5′-CH2), 3.49 (bdd, 1H, 5′-CH2), 3.13 (bt, 2H, N(CH3)-CH2), 2.80 (s, 3H, N-CH3), 1.50−1.78 (bm, 4H, NH2OCH2−CH2CH2). Anal. (C15H25N7O4·2.0H2SO4·0.3C2H5OH·1.5H2O) C, H, N, S.
5′-Deoxy-2′,3′-O-isopropylidene-5′-[(2-hydroxyethyl)methylamino]adenosine (15)
Compound 8i(52) (8.20 g, 17.79 mmol) was dissolved in 2-(methylamino)ethanol (54 mL, 673 mmol) and stirred at room temperature for 41 h. The solvent was evaporated to give a yellow residue. The residue was dissolved in 100 mL of chloroform and washed with NaHCO3 (3 × 50 mL). The organic layer was dried over Na2SO4 and concentrated to dryness to give yellow foam. The residue was purified by column chromatography (silica gel 230−400 mesh, elution with 9:1:0.1 chloroform:methanol:NH4OH) to yield 2.55 g (39%). MS: m/z 365 (M + H)+. 1H NMR (DMSO-d6) δ 8.34 (s, 1H, H-2), 8.18 (s, 1H, H-8), 7.33 (bs, 2H, 6-NH2), 6.13 (d, 1H, H-1′, J1′,2′ = 2.5 Hz), 5.48 (dd, 1H, H-2′, J2′,3′ = 6.3 Hz), 4.96 (dd, 1H, H-3′, J3′,4′ = 3.0 Hz), 4.33 (t, 1H, OH), 4.24 (dt, 1H, H-4′), 3.44 (t, 2H, OH-CH2), 2.64 (dd, 1H, 5′-CH2), 2.35−2.49 (m, 3H, 5′-CH2, N(CH3)-CH2), 2.18 (s, 3H, N-CH3), 1.54 and 1.33 (2s, 6H, C(CH3)2).
5′-Deoxy-2′,3′-O-isopropylidene-5′-[(2-phthalimidooxyethyl)methylamino]-adenosine (16)
To a solution of compound 15 (989 mg, 2.714 mmol), N-hydroxyphthalimide (1.107 g, 6.786 mmol) and P(Ph)3 (1.780 g, 6.787 mmol) in 50 mL of anhydrous THF was added DEAD (1.07 mL, 6.8 mmol) in THF (10 mL) under nitrogen over a period of 3 min at room temperature. After 5 min, 2% of sodium carbonate (75 mL) was added to the reaction mixture followed by dichloromethane (100 mL). The organic layer was washed with 2% Na2CO3 (75 mL) and then with saturated NaCl (2 × 75 mL). The organic layer was dried over Na2SO4 and concentrated to dryness to give a foam. The residue was purified by column chromatography and eluted from the column with 1:3 dichloromethane:acetone to yield 842 mg (61%). MS: m/z 510 (M + H)+)+. 1H NMR (CDCl3) δ 8.36 (s, 1H, H-2), 8.07 (s, 1H, H-8), 7.79−7.83 (m, 2H, phthalimido aromatic H’s), 7.63−7.72 (m, 2H, phthalimido aromatic H’s), 6.12 (d, 1H, H-1′, J1′,2′ = 2.2 Hz), 5.87 (bs, 2H, 6-NH2), 5.49 (dd, 1H, H-2′, J1′,2′ = 2.2 Hz, J2′,3′ = 6.4 Hz), 5.05 (dd, 1H, H-3′, J2′,3′ = 6.4 Hz, J3′,4′ = 3.3 Hz), 4.40−4.45 (m, 1H, H-4′), 4.29 (t, 2H, NO-CH2), 2.89 (t, 2H, NOCH2−CH2), 2.83−2.89 (m, 1H, 5′-CH2), 2.72−2.79 (m, 1H, 5′-CH2), 2.40 (s, 3H, N-CH3), 1.62 and 1.40 (2s, 6H, C(CH3)2).
5′-[(2-Aminooxyethyl)methylamino]-5′-deoxyadenosine sulfate (1:1 salt) (5)
A solution of 16 (373 mg, 0.73 mmol) in 1 N H2SO4 (5 mL) was heated at 60 °C for 3 h. The reaction mixture was neutralized with NaHCO3 and lyophilized. The compound was extracted with EtOH (2 × 20 mL) and concentrated to dryness. The residue was purified by column chromatography, eluting with 77:20:3 chloroform:methanol:NH4OH). The product was dissolved in 10 mL of EtOH and 1 N H2SO4 was added dropwise with cooling to precipitate the salt, which was filtered and washed with EtOH and dried in vacuo: yield 100 mg. MS: m/z 340 (M + H)+. 1H NMR (DMSO-d6) δ 8.42 (s, 1H, H-8), 8.25 (s, 1H, H-2), 7.68 (bs, 2H, 6-NH2), 5.99 (d, 1H, H-1′, J1′,2′ = 5.3 Hz), 5.67 (t, 1H, H-2′, J2′,3′ = 4.6 Hz), 4.33−4.39 (bm, 1H, H-4′), 4.22 (t, 1H, H-3′, J3′,4′ = 4.7 Hz), 4.06 (bt, 2H, NH2O-CH2), 3.63−3.71 (dd, 1H, 5′-CH2), 3.52−3.59 (bdd, 1H, 5′-CH2), 3.43 (bm, 2H, N(CH3)-CH2), 2.84 (s, 3H, N-CH3). UV λmax, nm, pH 1, 258.2 (ε 14,300), pH 7, 259 (ε 14,600), pH 13, 259 (ε 15,500). Anal. (C13H21N7O4·1.0 H2SO4·0.5C2H5OH·1.0H2O) C, H, N.
5′-[(2-Carboethoxyethyl)methylamino]-5′-deoxy-8-methyladenosine (17a)
A mixture of 9a (500 mg, 1.69 mmol), ethyl 3-chloropropionate (270 mg, 1.97 mmol), DIEA (109 mg, 0.14 mL, 0.84 mmol), and DMF (5 mL) was heated at 60 °C for two days. Starting material remained but because the solution was getting darker, heating was stopped. The reaction mixture was concentrated to dryness. The product was purified by column chromatography (7:1:0.1 chloroform:methanol:NH4OH) to give a sticky solid: yield 210 mg (31%). MS m/z 395 (M + H)+. 1H NMR (DMSO-d6) δ 8.08 (s, 1H, H-2), 7.11 (bs, 2H, 6-NH2), 5.74 (d, 1H, H-1′, J1′2′ = 5.6 Hz), 5.33 (bd, 1H, OH-2′), 5.16 (bd, 1H, OH-3′), 5.12 (bdd, 1H, H-2′, J1′,2′ = 5.6 Hz, J2′,3′ = 5.5 Hz), 4.21 (bdd, 1H, H-3′, J2′,3′ = 5.5 Hz, J3′,4′ = 4.3 Hz), 4.01 (q, 2H, OCH2CH3), 3.91−4.00 (m, 1H, H-4′), 2.70−2.77 (m, 1H, 5′-CH2), 2.54−2.66 (m, 3H, 5′-CH2, CO-CH2), 2.53 (s, 3H, 8-CH3), 2.38 (t, 2H, N(CH3)-CH2), 2.16 (bs, 3H, N-CH3), 1.15 (t, 3H, OCH2CH3).
5′-[(2-Carboethoxyethyl)methylamino]-5′-deoxy-8-ethyladenosine (17b)
Compound 17b was prepared by the same procedure as described for the preparation of 17a using 9b (260 mg, 0.84 mmol), ethyl 3-chloropropionate (138 mg, 1.0 mmol), DIEA (53 mg, 0.07 mL, 0.41 mmol), and DMF (4 mL). After column chromatography (elution with 7:1:0.1 chloroform:methanol:NH4OH), a glassy sticky solid was obtained: yield 153 mg (44%). MS: m/z 409 (M + H)+. 1H NMR (DMSO-d6) δ 8.08 (s, 1H, H-2), 7.10 (bs, 2H, 6-NH2), 5.71 (d, 1H, H-1′, J1′2′ = 5.5 Hz), 5.32 (bd, 1H, OH-2′, J2′−2′OH = 5.0 Hz), 5.16 (bd, 1H, OH-3′, J3′−3′OH = 5.1 Hz), 5.12 (ddd, 1H, H-2′, J1′,2′ = 5.5 Hz, J2′,3′ = 5.7 Hz, J2′−2′OH = 5.0 Hz), 4.14 (ddd, 1H, H-3′, J2′,3′ = 5.7 Hz, J3′,4′ = 4.1 Hz, J3′−3′OH = 5.1 Hz), 4.01 (q, 2H, OCH2CH3), 3.91−3.98 (m, 1H, H-4′), 2.87 (q, 2H, CH2 of 8-Et), 2.71−2.79 (m, 1H, 5′-CH2), 2.51−2.65 (m, 3H, 5′-CH2, CO-CH2), 2.38 (t, 2H, N(CH3)-CH2), 2.16 (bs, 3H, N-CH3), 1.30 (t, 3H, CH3 of 8-Et), 1.15 (t, 3H, OCH2CH3).
5′-[(Carboethoxymethyl)methyamino]-5′-deoxy-8-methyladenosine (17c)
Compound 17c was prepared by the same procedure as described for the preparation of 17a using 9a (415 mg, 1.41 mmol), ethyl chloroacetate (207 mg, 0.18 mL, 1.68 mmol), DIEA (91 mg, 0.12 mL, 0.70 mmol), and DMF (5 mL): yield 204 mg (38%). MS: m/z 381 (M + H)+. 1H NMR (DMSO-d6) δ 8.06 (s, 1H, H-2), 7.11 (bs, 2H, 6-NH2), 5.73 (d, 1H, H-1′, J1′2′ = 5.4 Hz), 5.33 (bd, 1H, OH-2′, J2′−2′OH = 4.7 Hz), 5.19 (bd, 1H, OH-3′, J3′−3′OH = 4.9 Hz), 5.03 (ddd, 1H, H-2′, J1′,2′ = 5.4 Hz, J2′,3′ = 5.7 Hz, J2′−2′OH = 4.7 Hz), 4.17 (ddd, 1H, H-3′, J2′,3′ = 5.7 Hz, J3′,4′ = 4.4 Hz, J3′−3′OH = 4.9 Hz), 4.02 (q, 2H, OCH2CH3), 3.92−3.99 (m, 1H, H-4′), 3.27 (bs, 2H, N(CH3)-CH2), 2.83−2.90 (m, 1H, 5′-CH2), 2.70−2.79 (m, 1H, 5′-CH2), 2.53 (s, 3H, 8-CH3), 2.31 (s, 3H, N-CH3), 1.13 (t, 3H, OCH2CH3). UV λmax, nm, pH 1, 258.9 (ε 16100), pH 7, 260 (ε 15900), pH 13, 260.1 (ε 16200). Anal. (C16H24N6O5·0.5CHCl3·0.3CH3OH) C, H, N.
5′-[(2-Carboxamidoethyl)methylamino]-5′-deoxy-8-methyladenosine sulfate (1.5:1 salt) (17d)
Compound 17a (89 mg, 0.22 mmol) was dissolved in 5 mL of methanolic ammonia and stirred for five days at room temperature. The reaction mixture was concentrated to dryness and purified by column chromatography (4:1:0.2 chloroform:methanol:NH4OH). The product was dissolved in 8 mL of EtOH and 2 N H2SO4 was added dropwise. The salt that precipitated out was filtered and washed with EtOH. This product, which was hygroscopic in nature, was dissolved in water (2 mL) and lyophilized to give a white solid: yield 65 mg (55%). MS: m/z 366 (M + H)+. 1H NMR (D2O) δ 8.43 (s, 1H, H-2), 6.09 (d, 1H, H-1′, J1′,2′ = 5.9 Hz), 5.0−5.30 (bm, 1H, H-2′), 4.61−4.70 (bm, 1H, H-4′), 4.51−4.54 (bm, 1H, H-3′), 3.30−3.89 (bm, 5H, N-CH3 and N(CH3)-CH2), 2.96 (bs, 2H, 5′-CH2), 2.77 (bs, 2H, NH2CO-CH2), 2.70 (s, 3H, 8-CH3). UV λmax, nm, pH 1, 258.4 (ε 14900), pH 7, 260.1 (ε 14900), pH 13, 260.1 (ε 15,300). Anal. (C15H23N7O4·1.5H2SO4·0.8H2O) C, H, N, S.
5′-[(2-Carboxamidoethyl)methyamino]-5′-deoxy-8-ethyladenosine sulfate (1.1:1 salt) (17e)
The procedure was the same as reported above for 17d using 17b (149 mg, 0.36 mmol) and methanolic ammonia (5 mL): yield 94 mg (51%). MS: m/z 380 (M + H)+. 1H NMR (DMSO-d6) δ 8.08 (s, 1H, H-2), 7.31 (bs, 1H, CO-NH2), 7.10 (bs, 2H, 6-NH2), 6.71 (bs, 1H, CO-NH2), 5.72 (d, 1H, H-1′, J1′2′ = 5.4 Hz), 5.31 (d, 1H, OH-2′, J2′−2′OH = 6.2 Hz), 5.16 (d, 1H, OH-3′, J3′−3′OH = 5.5 Hz), 5.09 (ddd, 1H, H-2′, J1′,2′ = 5.4 Hz, J2′,3′ = 5.7 Hz, J2′−2′OH = 6.2 Hz), 4.17 (ddd, 1H, H-3′, J2′,3′ = 5.7 Hz, J3′,4′ = 4.3 Hz, J3′−3′OH = 5.5 Hz), 3.92−3.99 (m, 1H, H-4′), 2.87 (q, 2H, 8-CH2CH3), 2.69−2.75 (m, 1H, 5′-CH2), 2.52−2.60 (m, 3H, CO-CH2, 5′-CH2), 2.18 (bs, 2H, N(CH3)-CH2), 2.16 (s, 3H, N-CH3), 1.30 (t, 3H, CH3 of 8-Et). 1H NMR (D2O) δ 8.38 (s, 1H, H-2), 6.09 (d, 1H, H-1′, J1′,2′ = 6.2 Hz), 5.33 (bs, 1H, H-2′), 4.56−4.62 (m, 1H, H-4′), 4.51−4.54 (m, 1H, H-3′), 3.87−3.96 (bm, 2H, NH2CO-CH2), 3.56 (s, 3H, N-CH3), 2.98−3.80 (bm, 2H, 8-CH2CH3), 2.96 (bs, 2H, 5′-CH2), 2.72−2.82 (m, 2H, N(CH3)-CH2), 1.39 (s, 3H, CH3 of 8-Et). UV λmax, nm, pH 1, 259.4 (ε 15200), pH 7, 260.8 (ε 15100), pH 13, 260.6 (ε 15500). Anal. (C16H25N7O4·1.1H2SO4·1.05H2O) C, H, N, S.
5′-[(Carboxamidomethyl)methylamino]-5′-deoxy-8-methyladenosine sulfate (1.45:1 salt) (17f)
The same procedure used to prepare 17d was used to prepare 17f using 17c (200 mg, 0.52 mmol) and methanolic ammonia (5 mL): yield 105 mg (39%). MS: m/z 352 (M + H)+. 1H NMR (DMSO-d6) δ 8.08 (s, 1H, H-2), 7.11 (bs, 2H, CO-NH2), 7.07 (bs, 2H, 6-NH2), 5.74 (d, 1H, H-1′, J1′2′ = 5.2 Hz), 5.34 (d, 1H, OH-2′, J2′−2′OH = 5.9 Hz), 5.21 (d, 1H, OH-3′, J3′−3′OH = 5.7 Hz), 4.97 (ddd, 1H, H-2′, J1′,2′ = 5.2 Hz, J2′,3′ = 5.5 Hz, J2′−2′OH = 5.9 Hz), 4.21 (ddd, 1H, H-3′, J2′,3′ = 5.5 Hz, J3′,4′ = 5.2 Hz, J3′−3′OH = 5.7 Hz), 3.94−4.01 (m, 1H, H-4′), 2.94 (d, 1H, N(CH3)-CH2, J = 15.7 Hz), 2.88 (d, 1H, N(CH3)-CH2, J = 15.7 Hz), 2.75−2.80 (m, 1H, 5′-CH2), 2.63−2.70 (m, 1H, 5′-CH2), 2.53 (s, 3H, 8-CH3), 2.23 (s, 3H, N-CH3). 1H NMR (D2O) δ 8.40 (s, 1H, H-2), 6.08 (d, 1H, H-1′, J1′,2′ = 4.8 Hz), 5.01 (t, 1H, H-2′, J2′,3′ = 5.2 Hz), 4.60 (t, 1H, H-3′, J3′,4′ = 4.9 Hz), 4.50−4.59 (m, 1H, H-4′), 4.03−4.17 (m, 2H, NH2CO-CH2), 3.82−3.92 (m, 1H, 5′-CH2), 3.68−3.76 (m, 1H, 5′-CH2), 3.03 (s, 3H, N-CH3), 2.70 (s, 3H, 8-CH3). UV λmax, nm, pH 1, 259 (ε 15900), pH 7, 259.7 (ε 16100), pH 13, 260.2 (ε 16100). Anal. (C14H21N7O4·1.45H2SO4·0.2C2H5OH·1.3H2O) C, H, N, S.
5′-[(2-Carboethoxyethyl)methylamino]-5′-deoxyadenosine (17g)
The general procedure previously described for 17a was used to prepare 17g using 9h(53) (400 mg, 1.42 mmol), ethyl 3-chloropropionate (214 mg, 1.56 mmol), DIEA (92 mg, 0.12 mL, 0.71 mmol), and DMF (5 mL). The reaction mixture was heated at 60 °C for two days. The product was purified by column chromatography (7:1 chloroform:methanol) to give a solid: yield 102 mg (19%). MS m/z 381 (M + H)+.
5′-Deoxy-5′-[(2-carboethoxyethyl)methylamino]-2′,3′-O-isopropylideneadenosine (17h)
To compound 9g (380 mg, 1.18 mmol) in anhydrous CH3CN (5 mL) was added ethyl 3-chloropropionate (195 mg, 0.18 mL, 1.42 mmol) and diisopropylethylamine (153 mg, 0.2 mL, 1.18 mmol) and the reaction mixture was stirred at 75−80 °C for 72 h under nitrogen. The reaction mixture was evaporated to dryness, and the residue was purified by column chromatography (silica gel 230−400 mesh, elution with 9:1 methylene chloride:methanol) to yield 260 mg (52%), MS: m/z 421 (M + H)+.
5′-[(2-Carboxamidoethyl)methylamino]-5′-deoxy-2′,3′-O-isopropylideneadenosine (17i)
A mixture of 17h (80 mg, 0.19 mmol) in saturated methanolic ammonia (15 mL) was stirred for 12 days at room temperature. The resulting solution was concentrated to dryness, and the residue was purified by column chromatography (elution with 9:1:0.1 methylene chloride:methanol:NH4OH) to yield 40 mg (55%). MS: m/z 392 (M + H)+.
5′-[(2-Carboxamidoethyl)methylamino]-5′-deoxyadenosine sulfate (1.9:1 salt) (17j)
Compound 17i (20 mg) was dissolved in 1 N H2SO4 (2 mL) and the solution was stirred at room temperature for 36 h. The reaction mixture was concentrated to 0.5 mL, absolute ethanol (3 mL) was added to the solution to produce a slight turbidity, and the mixture was chilled at 0 °C. The solid was filtered, washed with ethanol, and dried under high vacuum: yield 15 mg (51%). MS: m/z 352 (M + H)+. 1H NMR (DMSO-d6) δ 8.64 (s, 1H, H-8), 8.43 (s, 1H, H-2), 7.59 (bs, 1H, NH2), 7.17 (bs, 1H, NH2), 6.02 (bdd, 1H, H-1′, J1′,2′ = 3.4 Hz), 4.65 (t, 1H, H-2′, J2′,3′ = 6.4 Hz), 4.17−4.21 (bm, 1H, H-3′), 3.34−3.42 (bm, 1H, H-4′), 3.52−3.67 (bm, 4H, 5′-CH2, N(CH3)-CH2), 2.80 (bs, 3H, N-CH3), 2.54−2.60 (bm, 2H, CO-CH2). UV λmax, nm, pH 1, 256.3 (ε 15200), pH 7, 258.5 (ε 15400), pH 13, 259.6 (ε 16100). Anal. (C14H21N7O4·1.9H2SO4·1.6H2O) C, H, N, S.
5′-Deoxy-5′-[(2-hydrazinocarbonylethyl)methylamino]-8-methyladenosine sulfate (2:1 salt) (17k)
Compound 17a (115 mg, 0.29 mmol) was dissolved in 10 mL of anhydrous ethanol, and hydrazine monohydrate (73 mg, 0.07 mL, 1.46 mmol) was added to the solution. The reaction mixture was heated to reflux overnight. Hydrazine monohydrate (0.07 mL) was added again, and heating was continued overnight. The resulting solution was evaporated to dryness. The crude product was purified by column chromatography (4:1:0.5 chloroform:methanol:NH4OH), affording a sticky solid. The product was dissolved in 8 mL of EtOH, and 2 N H2SO4 was added dropwise. The salt that was precipitated out was filtered and washed with EtOH. This salt, which was hygroscopic in nature, was dissolved in water (2 mL) and lyophilized to give a white solid: yield 71 mg (65%). MS: m/z 381 (M + H)+. 1H NMR (D2O) δ 8.44 (s, 1H, H-2), 6.09 (d, 1H, H-1′, J1′,2′ = 5.6 Hz), 5.10 (dd, 1H, H-2′, J2′,3′ = 4.5 Hz), 4.53−4.62 (bm, 2H, H-3′, H-4′), 3.86−3.94 (m, 1H, 5′-CH2), 3.52−3.66 (m, 3H, 5′-CH2, N(CH3)-CH2), 2.95 (s, 3H, N-CH3), 2.86 (t, 2H, NHCO-CH2), 2.70 (s, 3H, 8-CH3). UV λmax, nm, pH 1, 258.4 (ε 15000), pH 7, 259.3 (ε 15200), pH 13, 260.4 (ε 15700). Anal. (C15H24N8O4·2.0 H2SO4·2.7H2O) C, H, N, S.
5′-Deoxy-5′-[(2-hydrazinocarbonylethyl)methylamino]-adenosine sulfate (2:1 salt) (17l)
The procedure was the same as reported above for 17k using 17g (95 mg, 0.25 mmol) and hydrazine monohydrate (63 mg, 0.06 mL, 1.25 mmol): yield 39 mg (43%). MS: m/z 367 (M + H)+. 1H NMR (D2O) δ 8.47 (s, 1H, H-8), 8.45 (s, 1H, H-2), 6.17 (d, 1H, H-1′, J1′,2′ = 4.7 Hz), 4.90 (dd, 1H, H-2′, J2′,3′ = 5.2 Hz), 4.56−4.60 (bm, 1H, H-4′), 4.48 (dd, 1H, H-3′, J3′,4′ = 5.0 Hz), 3.82 (dd, 1H, 5′-CH2), 3.66 (dd, 1H, 5′-CH2), 3.59 (bt, 2H, N(CH3)-CH2), 2.97 (s, 3H, N-CH3), 2.86 (t, 2H, NHCO-CH2). UV λmax, nm, pH 1, 256.8 (ε 16600), pH 7, 258.5 (ε 16800), pH 13, 259.5 (ε 17,600). Anal. (C14H22N8O4·2.0 H2SO4·2.0H2O) C, H, N.
5′-Deoxy-5′-[(hydrazinocarbonylmethyl)methylamino]-8-methyladenosine (17m)
The same procedure used to prepare 17k was used to prepare 17m using 17c (167 mg, 0.44 mmol) and hydrazine monohydrate (109 mg, 0.11 mL, 2.18 mmol). After the column it yielded a white solid: yield 154 mg (96%). MS: m/z 367 (M + H)+. 1H NMR (DMSO-d6) δ 8.75 (bs, 1H, NH), 8.09 (s, 1H, H-2), 7.10 (bs, 2H, NH-NH2), 5.74 (d, 1H, H-1′, J1′,2′ = 5.2 Hz), 5.31 (d, 1H, OH-2′, J2′−2′OH = 6.0 Hz), 5.18 (d, 1H, OH-3′, J3′−3′OH = 5.6 Hz), 5.0 (ddd, 1H, H-2′, J1′,2′ = 5.2 Hz, J2′,3′ = 5.7 Hz, J2′−2′OH = 6.0 Hz), 4.19 (ddd, 1H, H-3′, J2′,3′ = 5.7 Hz, J3′,4′ = 4.9 Hz, J3′−3′OH = 5.6 Hz), 3.94−3.99 (m, 1H, H-4′), 3.01 (d, 1H, CO-CHa Hb, J = 15.2 Hz), 2.95 (d, 1H, CO-CHa Hb, J = 15.2 Hz), 2.78 (dd, 1H, 5′-CH2), 2.66 (dd, 1H, 5′-CH2), 2.53 (s, 3H, 8-CH3), 2.22 (s, 3H, N-CH3). UV λmax, nm, pH 1, 258.9 (ε 15300), pH 7, 259.3 (ε 15600), pH 13, 260.1 (ε 16100). Anal. (C14H22N8O4·0.2 CH3OH·0.4H2O) C, H, N.
5′-[(3-Aminopropyl)methylamino]-5′-deoxy-8-(methylamino)adenosine sulfate (2.4:1 salt) (18a) and 5′-deoxy-8-(methylamino)-5′-(3-methylaminopropylamino)adenosine sulfate (2.4:1 salt) (19a)
A solution of 8c (175 mg, 0.55 mmol) in 2 mL of N-methyl-1,3-propanediamine was stirred for four days at ambient temperature. The mixture was poured into diethyl ether (20 mL). Decantation of the ether layer left an oil, which was purified by column chromatography. The column was eluted with 4:1:0.5 chloroform:methanol:NH4OH to afford two isomers. These separated isomers were dissolved in 6 mL of EtOH and 5 mL of EtOH respectively, and 2 N H2SO4 was added dropwise. The sulfate salts that precipitated out were filtered and washed with EtOH. These salts, which were hygroscopic in nature, were dissolved in water (2 mL) and lyophilized to give white solids: 18a: yield 50 mg (15%). MS: m/z 367 (M + H)+. 1H NMR (DMSO-d6) δ 7.91 (s, 1H, H-2), 6.90 (bs, 1H, 8CH3−NH), 6.49 (bs, 2H, 6-NH2), 5.70 (d, 1H, H-1′, J1′,2′ = 4.8 Hz), 4.92 (t, 1H, H-2′, J2′,3′ = 5.5 Hz), 4.17 (t, 1H, H-3′, J3′,4′ = 5.2 Hz), 3.89−3.96 (bm, 1H, H-4′), 2.89 (s, 3H, 8NH-CH3), 2.53−2.70 (m, 4H, NH2−CH2, 5′-CH2), 2.38 (t, 2H, N(CH3)-CH2), 2.17 (s, 3H, N-CH3), 1.48−1.60 (m, 2H, NH2CH2-CH2). 1H NMR (D2O) δ 8.28 (s, 1H, H-2), 5.87 (d, 1H, H-1′, J1′,2′ = 5.1 Hz), 5.18 (t, 1H, H-2′, J2′,3′ = 5.1 Hz), 4.54 (t, 1H, H-3′, J3′,4′ = 5.1 Hz), 4.46−4.51 (m, 1H, H-4′), 3.01−3.82 (bm, 6H, 5′-CH2, NH2−CH2, N(CH3)-CH2), 3.05 (s, 3H, 8NH-CH3), 2.94 (s, 3H, N-CH3), 2.05−2.18 (bm, 2H, NH2CH2−CH2). UV λmax, nm, pH 1, 275.5 (ε 14500), pH 7, 275 (ε 17200), pH 13, 277 (ε 17500). Anal. (C15H26N8O3·2.4H2SO4·0.2C2H5OH) C, H, N. 19a: yield 29 mg (7%). MS: m/z 367 (M + H)+. 1H NMR (DMSO-d6) δ 7.96 (s, 1H, H-2), 7.0 (bs, 1H, 8CH3−NH), 6.57 (bs, 2H, 6-NH2), 5.80 (d, 1H, H-1′, J1′,2′ = 5.5 Hz), 4.97 (t, 1H, H-2′, J2′,3′ = 5.5 Hz), 5.4 (bs, 2H, 2′, 3′-OH’s), 4.33 (t, 1H, H-3′, J3′,4′ = 3.7 Hz), 4.17−4.24 (bm, 1H, H-4′), 3.20−3.50 (m, 2H, 5′-CH2), 3.05−3.85 (m, 4H, NH-CH2), 2.88 (s, 3H, 8NH-CH3), 2.49 (s, 3H, NH-CH3), 1.85−2.0 (bm, 2H, NHCH2−CH2). 1H NMR (D2O) δ 8.27 (s, 1H, H-2), 5.86 (d, 1H, H-1′, J1′,2′ = 5.2 Hz), 5.16 (t, 1H, H-2′, J2′,3′ = 5.5 Hz), 4.55 (t, 1H, H-3′, J3′,4′ = 4.5 Hz), 4.36−4.44 (m, 1H, H-4′), 3.47−3.69 (m, 2H, 5′-CH2), 3.22 (t, 2H, NH-CH2), 3.10 (t, 2H, CH3NH-CH2), 3.05 (s, 3H, 8NH-CH3), 2.71 (s, 3H, NH-CH3), 2.04−2.18 (bm, 2H, NHCH2−CH2). UV λmax, nm, pH 1, 274.1 (ε 14300), pH 7, 276 (ε 17100), pH 13, 277.1 (ε 18700). Anal. (C15H26N8O3·2.4H2SO4·0.2C2H5OH) C, H, N.
5′-[(3-Aminopropyl)methylamino]-5′-deoxy-8-phenyladenosine sulfate (2.2:1 salt) (18b) and 5′-deoxy-5′-(3-methylaminopropylamino)-8-phenyladenosine sulfate (1.7:1 salt) (19b)
The same procedure as described above for 18a was used to prepare 18b and 19b from 8d (200 mg, 0.55 mmol), and N-methyl-1,3-propanediamine (3 mL) except in this case, after one day of stirring at room temperature, the reaction mixture was heated at 65 °C for two days. The column was eluted with 4:1:0.2 chloroform:methanol:NH4OH. After the same work up, two isomers were obtained. 18b: yield 124 mg (34%). MS: m/z 414 (M + H)+. 1H NMR (DMSO-d6) δ 8.38 (s, 1H, H-2), 7.61−7.84 (m, 7H, 8-phenyl, 6-NH2), 5.88 (d, 1H, H-1′, J1′,2′ = 6.3 Hz), 5.17−5.30 (bm, 1H, H-2′), 4.34−4.38 (bm, 1H, H-3′), 4.25−4.31 (bm, 1H, H-4′), 3.10−3.85 (bm, 4H, N(CH3)-CH2, 5′-CH2), 2.80−2.94 (m, 2H, NH2−CH2), 2.78 (s, 3H, N-CH3), 1.81−2.0 (bm, 2H, NH2CH2−CH2). UV λmax, nm, pH 1, 274.2 (ε 20500), pH 7, 275.8 (ε 16300), pH 13, 274.5 (ε 16400). Anal. (C20H27N7O3·2.2H2SO4·0.1C2H5OH·2.5H2O) C, H, N, S. 19b: yield 151 mg (42%). MS: m/z 414 (M + H)+. 1H NMR (DMSO-d6) δ 8.23 (s, 1H, H-2), 7.0−7.74 (m, 2H, 8-phenyl o-H’s), 7.60−7.65 (m, 3H, 8-phenyl m- and p-H’s), 7.46 (bs, 2H, 6-NH2), 5.80 (d, 1H, H-1′, J1′,2′ = 6.2 Hz), 5.25 (t, 1H, H-2′, J2′,3′ = 4.9 Hz), 5.5−6.0 (m, 2H, NH’s), 4.29−4.35 (bm, 1H, H-3′), 4.12−4.20 (m, 1H, H-4′), 3.40−3.47 (m, 1H, 5′-CH2), 3.19−3.25 (m, 1H, 5′-CH2), 2.94 (m, 4H, NH-CH2, CH3NH-CH2), 2.54 (s, 3H, NH-CH3), 1.80−1.95 (bm, 2H, NHCH2−CH2). UV λmax, nm, pH 1, 275.5 (ε 20600), pH 7, 274.9 (ε 16200), pH 13, 274.9 (ε 16,300). Anal. (C20H27N7O3·1.7H2SO4·0.05C2H5OH·3.3H2O) C, H, N, S.
5′-[(3-Aminopropyl)methylamino]-5′-deoxy-2′,3′-O-isopropylideneadenosine (18c)
A mixture of 8h(42) (1.0 g, 2.60 mmol) and N-methyl-1,3-propanediamine (1.35 mL, 13.0 mmol) was stirred overnight under an argon atmosphere. The reaction mixture was concentrated to dryness, and the crude product was purified by column chromatography using chloroform:methanol:NH4OH (6:1:0.1) as eluent to give a semisolid. This material was dissolved in 3 mL of water and lyophilized: yield 361 mg (37%). MS: m/z 378 (M + H)+. 1H NMR (DMSO-d6) δ 8.33 (s, 1H, H-8), 8.17 (s, 1H, H-2), 7.33 (bs, 2H, 6-NH2), 6.13 (d, 1H, H-1′, J1′,2′ = 2.3 Hz), 5.49 (dd, 1H, H-2′, J2′,3′ = 4.0 Hz), 4.94 (dd, 1H, H-3′, J3′,4′ = 2.9 Hz), 4.21−4.27 (m, 1H, H-4′), 2.52−2.58 (m, 2H, NH2−CH2), 2.26−2.38 (m, 4H, 5′-CH2, N(CH3)-CH2), 2.12 (s, 3H, N-CH3), 1.53 and 1.33 (2s, 6H, C(CH3)2), 1.37−1.47 (m, 2H, NH2CH2−CH2).
5′-[(3-Aminopropyl)methylamino]-5′-deoxyadenosine sulfate (2:1 salt) (18d)
The procedure described for 17j was used to prepare 18d from 18c (200 mg, 0.53 mmol): yield 121 mg (41%). MS: m/z 338 (M + H)+. 1H NMR (DMSO-d6) δ 8.47 (bs, 1H, H-8), 8.29 (bs, 1H, H-2), 7.0−10.0 (broad peaks, NH2s + H2SO4), 5.97 (d, 1H, H-1′, J1′,2′ = 4.7 Hz), 5.70 (bs, 1H, 2′-OH), 5.56 (bs, 1H, 3′-OH), 4.72 (bt, 1H, H-2′), 4.30 (bm, 1H, H-4′), 4.21 (bt, 1H, H-3′), 3.30−3.70 (bm, 2H, 5′-CH2), 3.08 (bs, 2H, N(CH3)-CH2), 2.83 (bm, 2H, NH2−CH2), 2.70 (bs, 3H, N-CH3), 1.84−1.92 (m, 2H, NH2CH2−CH2). UV λmax, nm, pH 1, 257(ε 14700), pH 7, 259.2 (ε 15000), pH 13, 260 (ε 15300). Anal. (C14H23N7O3·2.0H2SO4·0.25C2H5OH·0.7H2O) C, H, N.
5′-[(2-Aminoethyl)methylamino]-5′-deoxyadenosine (18e) and 5′-deoxy-5′-(2-methylaminoethylamino)adenosine (19c)
A mixture of 8g(32) (1.0 g, 3.5 mmol) and N-methylethylenediamine (8 mL) was stirred at room temperature for 12 days. The reaction mixture was poured into diethyl ether (50 mL). The ether layer was decanted and the resulting syrup was purified by column chromatography (silica gel 230−400 mesh, elution with 4:1:0.5 chloroform:methanol:NH4OH) to give the two isomers. 18e: yield 576 mg (51%). MS: m/z 324 (M + H)+. 1H NMR (DMSO-d6) δ 8.35 (s, 1H, H-8), 8.15 (s, 1H, H-2), 7.27 (bs, 2H, 6-NH2), 5.85 (d, 1H, H-1′, J1′,2′ = 5.3 Hz), 4.64 (dd, 1H, H-2′, J2′,3′ = 5.3 Hz), 4.11 (dd, 1H, H-3′, J3′,4′ = 4.3 Hz), 3.95−4.0 (m, 1H, H-4′), 2.90−3.60 (bs, 4H, 2′, 3′-OHs + NHs), 2.70 (dd, 1H, 5′-CH2), 2.50−2.60 (bm, 3H, 5′-CH2, N(CH3)-CH2), 2.36 (t, 2H, NH2−CH2), 2.19 (s, 3H, N-CH3). UV λmax, nm, pH 1, 256.9 (ε 14100), pH 7, 259.5 (ε 14700), pH 13, 259.2 (ε 15300). Anal. (C13H21N7O3·0.25CHCl3·0.5H2O) C, H, N. 19c: yield 323 mg (29%). MS: m/z 324 (M + H)+. 1H NMR (DMSO-d6) δ 8.35 (s, 1H, H-8), 8.14 (s, 1H, H-2), 7.28 (bs, 2H, 6-NH2), 5.84 (d, 1H, H-1′, J1′,2′ = 5.9 Hz), 4.69 (dd, 1H, H-2′, J2′,3′ = 5.3 Hz), 4.12 (dd, 1H, H-3′, J3′,4′ = 3.5 Hz), 3.94−3.99 (m, 1H, H-4′), 2.90−3.60 (bs, 4H, 2′, 3′-OHs + NHs), 2.80 (dd, 1H, 5′-CH2), 2.73 (dd, 1H, 5′-CH2), 2.50−2.62 (bm, 4H, NHCH3−CH2, NH-CH2), 2.25 (s, 3H, NH-CH3). UV λmax, nm, pH 1, 256.4 (ε 13900), pH 7, 259 (ε 13900), pH 13, 259.8 (ε 14000). Anal. (C13H21N7O3·0.05CH3OH·0.1H2O) C, H, N.
5′-[(2-Aminoethyl)methylamino]-5′-deoxy-8-methyladenosine (18f) and 5′-deoxy-8-methyl-5′-(2-methylaminoethylamino)adenosine (19d)
The procedure described above for 18e/19c was used to prepare 18f/19d from 8a (300 mg, 1.0 mmol) and N-methylethylenediamine (3 mL). 18f: yield ( 69 mg (18.3%). MS: m/z 338 (M + H)+. 1H NMR (DMSO-d6) δ 8.08 (s, 1H, H-2), 7.11 (bs, 2H, 6-NH2), 5.74 (d, 1H, H-1′, J1′,2′ = 5.4 Hz), 5.31 (bs, 1H, 2′-OH), 5.04 (dd, 1H, H-2′, J2′,3′ = 5.6 Hz), 4.17 (dd, 1H, H-3′, J3′,4′ = 4.5 Hz), 4.08 (bs, 1H, 3′-OH), 3.93−3.98 (m, 1H, H-4′), 2.70 (dd, 1H, 5′-CH2), 2.53−2.57 (bm, 3H, 5′-CH2, N(CH3)-CH2), 2.53 (s, 3H, 8-CH3), 2.30−2.35 (m, 2H, NH2−CH2), 2.16 (s, 3H, N-CH3). UV λmax, nm, pH 1, 258.5 (ε 15600), pH 7, 259.1 (ε 16000), pH 13, 260 (ε 16200). Anal. (C14H23N7O3·0.5CH3OH·0.3H2O) C, H, N. 19d: yield ( 58 mg (15.4%). MS: m/z 338 (M + H)+. 1H NMR (DMSO-d6) δ 8.07 (s, 1H, H-2), 7.12 (bs, 2H, 6-NH2), 5.72 (d, 1H, H-1′, J1′,2′ = 6.4 Hz), 5.27 (bs, 1H, 2′-OH), 5.02 (dd, 1H, H-2′, J2′,3′ = 5.4 Hz), 4.17 (dd, 1H, H-3′, J3′,4′ = 3.1 Hz), 3.94−3.99 (m, 1H, H-4′), 2.80 (dd, 1H, 5′-CH2), 2.74 (dd, 1H, 5′-CH2), 2.54−2.63 (bm, 4H, NHCH3−CH2, NH-CH2), 2.54 (s, 3H, 8-CH3), 2.24 (s, 3H, NH-CH3). UV λmax, nm, pH 1, 258.4 (ε 15400), pH 7, 259.5 (ε 15400), pH 13, 260 (ε 15900). Anal. (C14H23N7O3·0.4CH3OH·0.7H2O) C, H, N.
5′-Deoxy-2′,3′-O-isopropylidene-5′-[(3-phthalimidopropyl)methylamino]-8-methyladenosine (20a)
To a cold solution of compound 7e (500 mg, 1.5 mmol) in anhydrous pyridine (2 mL) was added methanesulfonyl chloride (196 mg, 0.13 mL, 1.7 mmol) and the solution was stirred for 2 h at 0 °C. The reaction mixture was concentrated to dryness to afford crude 8e. Methylamine (33% solution in EtOH, 12 mL) was added to this crude mixture, and the solution was stirred for three days at room temperature. The reaction mixture was evaporated to dryness. The resulting crude 9e was dissolved in anhydrous DMF (3 mL), DIEA (0.07 mL), and N-(3-bromopropyl)phthalimide (502 mg, 1.87 mmol) were added, and the reaction mixture was heated overnight at 60 °C. The solution was evaporated to dryness, and the residue was dissolved in CHCl3 (10 mL), washed with water, dried over Na2SO4, and concentrated to dryness. The resulting syrup was purified by column chromatography. The column was eluted with 97:3 chloroform:methanol to yield 108 mg (13%). MS m/z 522 (M + H)+. 1H NMR (CDCl3) δ 8.26 (s, 1H, H-2), 7.82−7.86 (m, 2H, phthalimido aromatic H’s), 7.69−7.73 (m, 2H, phthalimido aromatic H’s), 5.98 (d, 1H, H-1′, J1′,2′ = 1.8 Hz), 5.76 (dd, 1H, H-2′, J1′,2′ = 1.8 Hz, J2′,3′ = 6.5 Hz), 5.38 (bs, 2H, 6-NH2), 5.10 (dd, 1H, H-3′, J2′,3′ = 6.5 Hz, J3′,4′ = 3.5 Hz), 4.26−4.32 (m, 1H, H-4′), 3.60−3.76 (m, 2H, N-CH2), 2.64 (s, 3H, 8-CH3), 2.58−2.63 (m, 1H, 5′-CH2), 241−2.48 (m, 1H, 5′-CH2), 2.38 (t, 2H, N(CH3)-CH2), 2.21 (s, 3H, N-CH3), 1.68−1.80 (m, 2H, NCH2−CH2), 1.61 and 1.40 (2s, 6H, C(CH3)2).
5′-Deoxy-2′,3′-O-isopropylidene-5′-[(3-phthalimidopropyl)methylamino]-8-ethyladenosine (20b)
The same procedure as described for 20a was used to prepare 20b from 7f (600 mg, 1.78 mmol), MsCl (225 mg,0.15 mL, 1.96 mmol), methylamine (12 mL), and N-(3-bromopropyl)phthalimide (553 mg, 2.06 mmol): yield 81 mg (8.5%). MS: m/z 536 (M + H)+. 1H NMR (CDCl3) δ 8.26 (s, 1H, H-2), 7.81−7.86 (m, 2H, phthalimido aromatic H’s), 7.67−7.72 (m, 2H, phthalimido aromatic H’s), 5.98 (d, 1H, H-1′, J1′,2′ = 2.0 Hz), 5.75 (dd, 1H, H-2′, J1′,2′ = 2.0 Hz, J2′,3′ = 6.5 Hz), 5.37 (bs, 2H, 6-NH2), 5.12 (dd, 1H, H-3′, J2′,3′ = 6.5 Hz, J3′,4′ = 3.5 Hz), 4.25−4.33 (m, 1H, H-4′), 3.61−3.75 (m, 2H, N-CH2), 2.94 (q, 2H, CH2 of 8-Et), 2.62−2.68 (m, 1H, 5′-CH2), 243−2.49 (m, 1H, 5′-CH2), 2.38 (bt, 2H, N(CH3)-CH2), 2.22 (s, 3H, N-CH3), 1.70−1.80 (m, 2H, NCH2−CH2), 1.60 and 1.40 (2s, 6H, C(CH3)2), 1.42 (t, 3H, CH3 of 8-Et).
5′-Deoxy-2′,3′-O-isopropylidene-5′-[(3-aminopropyl)methylamino]-8-methyladenosine (21a)
To a boiling solution of 20a (100 mg, 0.19 mmol) in 3 mL of ethanol was added hydrazine monohydrate (50 mg, 0.048 mL, 0.99 mmol) and the solution was heated to reflux for 1 h. The reaction mixture was cooled down to room temperature, and the solid was filtered and washed with ethanol. The filtrate was evaporated to dryness. This crude product was purified by column chromatography using chloroform:methanol:NH4OH (7:1:0.2) for elution: yield 69 mg (92%). MS: m/z 392 (M + H)+. 1H NMR (DMSO-d6) δ 8.12 (s, 1H, H-2), 7.18 (bs, 2H, 6-NH2), 6.06 (d, 1H, H-1′, J1′,2′ = 1.9 Hz), 5.79 (dd, 1H, H-2′, J1′,2′ = 1.9 Hz, J2′,3′ = 6.3 Hz), 5.0 (dd, 1H, H-3′, J2′,3′ = 6.3 Hz, J3′,4′ = 3.1 Hz), 4.15−4.21 (m, 1H, H-4′), 2.56 (s, 3H, 8-CH3), 2.40−2.50 (m, 2H, N(CH3)-CH2), 2.13−2.31 (bm, 4H, NH2−CH2, 5′-CH2), 2.07 (s, 3H, N-CH3), 1.53 and 1.33 (2s, 6H, C(CH3)2), 1.22−1.32 (m, 2H, NH2CH2−CH2).
5′-Deoxy-2′,3′-O-isopropylidene-5′-[(3-aminopropyl)methylamino]-8-ethyladenosine (21b)
Compound 21b was prepared by the same procedure as reported for 21a using 20b (76 mg, 0.14 mmol) and hydrazine monohydrate (38 mg, 0.036 mL, 0.76 mmol): yield 47 mg (82%). MS: m/z 406 (M + H)+. 1H NMR (DMSO-d6) δ 8.12 (s, 1H, H-2), 7.16 (bs, 2H, 6-NH2), 6.03 (d, 1H, H-1′, J1′,2′ = 2.0 Hz), 5.76 (dd, 1H, H-2′, J1′,2′ = 2.0 Hz, J2′,3′ = 6.4 Hz), 5.01 (dd, 1H, H-3′, J2′,3′ = 6.4 Hz, J3′,4′ = 3.0 Hz), 4.15−4.22 (m, 1H, H-4′), 2.87−2.94 (bm, 2H, 5′-CH2), 2.38−2.53 (m, 2H, CH2 of 8-Et), 2.13−2.34 (m, 4H, N(CH3)-CH2, NH2−CH2), 2.08 (s, 3H, N-CH3), 1.53 and 1.33 (2s, 6H, C(CH3)2), 1.22−1.39 (m, 2H, NH2CH2−CH2), 1.31 (t, 3H, CH3 of 8-Et).
5′-[(3-Aminopropyl)methylamino]-5′-deoxy-8-methyladenosine sulfate (2:1 salt) (21c)
Compound 21a (66 mg, 0.168 mmol) was dissolved in 2 mL of 1 N H2SO4 and stirred overnight. To this solution was added ethanol (10 mL), causing a fine solid to separate. The solvent was decanted, the compound was dissolved in water (1 mL), and 15 mL of ethanol was added. The resulting solid was dissolved in water (2 mL) and lyophilized to give a white solid: yield 66 mg (67%). MS: m/z 352 (M + H)+. 1H NMR (DMSO-d6) δ 8.16 (s, 1H, H-2), 7.74 (bs, 2H, CH2−NH2), 7.24 (bs, 2H, 6-NH2), 5.84 (d, 1H, H-1′, J1′,2′ = 6.0 Hz), 5.62 (bs, 2H, 2′, 3′-OH’s), 4.98 (t, 1H, H-2′, J2′,3′ = 4.4 Hz), 4.29−4.36 (bm, 1H, H-4′), 4.23 (t, 1H, H-3′), 3.40−3.68 (bm, 2H, 5′-CH2), 2.99−3.19 (bm, 2H, N(CH3)-CH2), 2.83 (t, 2H, NH2−CH2), 2.70 (bs, 3H, N-CH3), 2.55 (s, 3H, 8-CH3), 1.80−1.93 (m, 2H, NH2CH2−CH2). UV λmax, nm, pH 1, 258.7 (ε 14900), pH 7, 259.5 (ε 15100), pH 13, 260.7 (ε 15300). Anal. (C15H25N7O3·2.0H2SO4·2.5H2O) C, H, N, S.
5′-[(3-Aminopropyl)methylamino]-5′-deoxy-8-ethyladenosine sulfate (2.5:1 salt) (21d)
The procedure described for 21c was used to prepare 21d from 21b (44 mg, 0.108 mmol). In this case after the addition of EtOH a fine solid came out, which was collected by centrifugation. It was then dissolved in water (2 mL) and lyophilized: yield 30 mg (69%). MS: m/z 366 (M + H)+. 1H NMR (DMSO-d6) δ; 8.15 (s, 1H, H-2), 7.68 (bs, 2H, CH2−NH2), 7.21 (bs, 2H, 6-NH2), 5.82 (d, 1H, H-1′, J1′,2′ = 6.0 Hz), 5.61 (bs, 2H, 2′, 3′-OH’s), 5.04 (t, 1H, H-2′), 4.19−4.27 (bm, 2H, H-3′, 4′), 3.22−3.43 (bm, 4H, 5′-CH2, N(CH3)-CH2), 2.90 (q, 2H, 8CH2CH3), 2.81 (t, 2H, NH2−CH2), 2.50 (bs, 3H, N-CH3), 1.73−1.88 (bm, 2H, NH2CH2−CH2), 1.32 (t, 3H, 8CH2CH3). UV λmax, nm, pH 1, 259.3 (ε 15100), pH 7, 260.5 (ε 15100), pH 13, 260.3 (ε 15100). Anal. (C16H27N7O3·2.5H2SO4·2.5H2O) C, H, N.
5′-Deoxy 5′-[(2-guanidinoethyl)methylamino]adenosine (22a)
To a stirred solution of 18e (218 mg, 0.67 mmol) and 1H-pyrazole-1-carboxamidine hydrochloride(30) (196 mg, 1.34 mmol) in anhydrous DMF (5 mL) was added DIEA (479 mg, 0.65 mL, 3.7 mmol) under nitrogen at 5 °C. Stirring was continued at room temperature overnight. The reaction mixture was concentrated to dryness, and the product was purified by column chromatography (silica gel 230−400 mesh, elution with 4:1:0.3 chloroform:methanol:NH4OH) to afford 219 mg (89%). MS: m/z 366 (M + H)+. 1H NMR (DMSO-d6) δ 8.34 (s, 1H, H-8), 8.15 (s, 1H, H-2), 7.48, 7.37, 7.28 (bs, NHs), 5.87 (d, 1H, H-1′, J1′,2′ = 5.2 Hz), 5.50 (d, 1H, 2′-OH, J2′−2′OH = 5.7 Hz), 5.28 (d, 1H, 3′-OH, J3′−3′OH = 4.5 Hz), 4.65 (ddd, 1H, H-2′, J1′,2′ = 5.2 Hz, J2′,3′ = 5.1 Hz, J2′−2′OH = 5.7 Hz), 4.12 (ddd, 1H, H-3′, J2′,3′ = 5.1 Hz, J3′,4′ = 4.6 Hz, J3′−3′OH = 4.5 Hz), 4.0−4.10 (bm, 1H, H-4′), 3.30−3.50 (bm, 2H, 5′-CH2), 3.12−3.28 (bm, 2H, NH-CH2), 2.55−2.65 (bm, 2H, N(CH3)-CH2), 2.25 (bs, 3H, N-CH3). UV λmax, nm, pH 1, 256.3 (ε 10900), pH 7, 259 (ε 11300), pH 13, 259 (ε 11000). Anal. (C14H23N9O3·0.05CHCl3·3.5H2O) C, H, N.
5′-[(2-Cyanoethyl)methylamino]-5′-deoxyadenosine (22b)
A solution of 8j(54) (1.0 g, 2.37 mmol) in 10 mL of 3-(methylamino)propionitrile was stirred at room temperature for five days. The reaction mixture was poured into diethyl ether (50 mL). The ether layer was decanted, and the resulting syrup was purified by column chromatography (silica gel, elution with 7:1:0.1 chloroform:methanol:NH4OH) to yield 685 mg (87%). MS: m/z 324 (M + H)+. 1H NMR (DMSO-d6) δ 8.34 (s, 1H, H-8), 8.15 (s, 1H, H-2), 7.29 (bs, 2H, 6-NH2), 5.87 (d, 1H, H-1′, J1′,2′ = 5.4 Hz), 5.46 (bd, 1H, 2′-OH), 5.22 (bd, 1H, 3′-OH), 4.65 (m, 1H, H-2′), 4.13 (m, 1H, H-3′), 3.97−4.03 (m, 1H, H-4′), 2.78 (dd, 1H, 5′-CH2), 2.58−2.68 (bm, 5H, 5′-CH2, NC-CH2CH2), 2.24 (s, 3H, N-CH3).
5′-Deoxy-5′-[(2-hydroxyamidinoethyl)methylamino]adenosine (22c)
To a solution of 22b (470 mg, 1.4 mmol) in 20 mL of anhydrous MeOH and 4 mL of anhydrous DMF under nitrogen was added hydroxylamine hydrochloride (258 mg, 3.7 mmol) and potassium hydroxide (206 mg, 3.7 mmol) and the resulting suspension was stirred at room temperature for two days. The reaction mixture was concentrated to dryness, and the crude product was extracted with EtOAc (2 × 40 mL) and washed with brine solution (20 mL). The organic layer was dried over Na2SO4, filtered, washed with EtOAc, and concentrated to dryness. The product was purified by column chromatography. The column was eluted with 4:1:0.5 chloroform:methanol:NH4OH to yield 165 mg (32%). MS: m/z 367 (M + H)+. 1H NMR (DMSO-d6) δ 8.70 (bs, 1H, NOH), 8.33 (s, 1H, H-8), 8.15 (s, 1H, H-2), 7.27 (bs, 2H, 6-NH2), 5.86 (d, 1H, H-1′, J1′,2′ = 5.3 Hz), 5.43 (d, 1H, 2′-OH, J2′−2′OH = 6.0 Hz), 5.35 (bs, 2H, C-NH2), 5.20 (d, 1H, 3′-OH, J3′−3′OH = 5.2 Hz), 4.63 (ddd, 1H, H-2′, J1′,2′ = 5.3 Hz, J2′,3′ = 4.9 Hz, J2′−2′OH = 6.0 Hz), 4.10 (ddd, 1H, H-3′, J2′,3′ = 4.9 Hz, J3′,4′ = 3.5 Hz, J3′−3′OH = 5.2 Hz), 3.96−4.0 (m, 1H, H-4′), 2.64−2.75 (bm, 2H, 5′-CH2), 2.56 (t, 2H, N(CH3)-CH2), 2.20 (s, 3H, N-CH3), 2.09 (t, 2H, C-CH2). Anal. (C14H22N8O4·1.2C2H5OH·0.2CH3OH) C, H, N.
5′-Deoxy-5′-(N,N-dimethylamino)-8-methyladenosine (23a)
A mixture of 8a (150 mg, 0.50 mmol) and a 2 M solution of dimethylamine in methanol (10 mL) in a steel bomb was heated for two days at 90 °C. The reaction mixture was concentrated to dryness and purified by column chromatography (elution with 4:1:0.15 chloroform:methanol:NH4OH) to afford 38 mg (25%). MS m/z 309 (M + H)+. 1H NMR (DMSO-d6) δ 8.08 (s, 1H, H-2), 7.11 (bs, 2H, 6-NH2), 5.74 (d, 1H, H-1′, J1′,2′ = 5.5 Hz), 5.31 (d, 1H, OH-2′, J2′−2′OH = 5.9 Hz), 5.18 (d, 1H, OH-3′, J3′−3′OH = 5.4 Hz), 5.03 (ddd, 1H, H-2′, J1′,2′ = 5.5 Hz, J2′,3′ = 4.6 Hz, J2′−2′OH = 5.9 Hz), 4.15 (ddd, 1H, H-3′, J2′,3′ = 4.6 Hz, J3′,4′ = 5.5 Hz, J3′−3′OH = 5.4 Hz), 3.91−3.96 (m, 1H, H-4′), 2.55−2.61 (m, 1H, 5′-CH2), 2.53 (s, 3H, 8-CH3), 2.42−2.48 (m, 1H, 5′-CH2), 2.14 (bs, 6H, N-(CH3)2). UV λmax, nm, pH 1, 258.5 (ε 15300), pH 7, 259.3 (ε 15300), pH 13, 260.1 (ε 15500). Anal. (C13H20N6O3·0.35CHCl3·0.5C2H5OH) C, H, N.
5′-Deoxy-5′-(N,N-dimethylamino)adenosine (23b)
Compound 23b was prepared by the same procedure as described for the preparation of 23a using 8g(32) (500 mg, 1.75 mmol) and a 2 M solution of dimethylamine in methanol (20 mL): yield 238 mg (46%). MS m/z 295 (M + H)+. 1H NMR (DMSO-d6) δ 8.33 (s, 1H, H-8), 8.15 (s, 1H, H-2), 7.29 (bs, 2H, 6-NH2), 5.86 (d, 1H, H-1′, J1′,2′ = 5.4 Hz), 5.45 (d, 1H, OH-2′, J2′−2′OH = 5.9 Hz), 5.22 (bd, 1H, OH-3′, J3′−3′OH = 3.9 Hz), 4.65 (ddd, 1H, H-2′, J1′,2′ = 5.4 Hz, J2′,3′ = 5.5 Hz, J2′−2′OH = 5.9 Hz), 4.10 (ddd, 1H, H-3′, J2′,3′ = 5.5 Hz, J3′,4′ = 4.5 Hz, J3′−3′OH = 3.9 Hz), 3.94−4.0 (m, 1H, H-4′), 2.62 (dd, 1H, 5′-CH2), 2.48 (dd, 1H, 5′-CH2), 2.18 (bs, 6H, N-(CH3)2). UV λmax, nm, pH 1, 256.3 (ε 15100), pH 7, 259.2 (ε 15500), pH 13, 259.7 (ε 15600). Anal. (C12H18N6O3·0.35CH3OH) C, H, N.
5′-Deoxy-5′-methylthio-8-methyladenosine (24a)
A solution of 8a (200 mg, 0.66 mmol) and sodium thiomethoxide (47 mg, 0.67 mmol) in 2 mL of anhydrous DMF was stirred for two days at room temperature and then concentrated to dryness. The crude product was purified by column chromatography using chloroform:methanol (7:1) as eluent to yield 102 mg (49%). MS m/z 312 (M + H)+. 1H NMR (DMSO-d6) δ 8.09 (s, 1H, H-2), 7.12 (bs, 2H, 6-NH2), 5.77 (d, 1H, H-1′, J1′,2′ = 5.7 Hz), 5.38 (bd, 1H, OH-2′, J2′−2′OH = 4.4 Hz), 5.31 (d, 1H, OH-3′, J3′−3′OH = 4.2 Hz), 5.15 (bddd, 1H, H-2′, J1′,2′ = 5.7 Hz, J2′,3′ = 5.7 Hz, J2′-2′OH = 4.4 Hz), 4.20 (ddd, 1H, H-3′, J2′,3′ = 5.7 Hz, J3′,4′ = 3.6 Hz, J3′−3′OH = 4.2 Hz), 3.97−4.05 (m, 1H, H-4′), 2.74−2.92 (m, 2H, 5′-CH2), 2.54 (s, 3H, 8-CH3), 2.03 (s, 3H, S-CH3).
5′-Deoxy-5′-dimethylsulfonio-8-methyladenosine bromide (25a)
Compound 24a (78 mg, 0.25 mmol) in a 2:1 mixture (4 mL) of formic and acetic acid was treated with a 2 M solution of bromomethane in diethyl ether (5 mL) and stirred for six days in darkness at room temperature. Solvents were removed in vacuo, and a solution of the residue in water (10 mL) was extracted with (3 × 10 mL) ether. The aqueous layer was concentrated to dryness. The resulting product was dissolved in MeOH (10 mL), filtered, and treated with diethyl ether to precipitate out the salt. The salt was filtered, washed with ether, and dried in vacuo to give white solid: yield 79 mg (78%). MS m/z 326 (M)+. 1H NMR (D2O) δ 8.24 (s, 1H, H-2), 6.03 (d, 1H, H-1′, J1′,2′ = 5.4 Hz), 5.29 (t, 1H, H-2′, J2′,3′ = 5.7 Hz), 4.80 (t, 1H, H-3′, J3′,4′ = 4.8 Hz), 4.52−4.60 (m, 1H, H-4′), 4.11−4.20 (m, 1H, 5′-CH2), 3.81−3.90 (m, 1H, 5′-CH2), 2.92 and 2.89 (2s, 6H, S-(CH3)2), 2.67 (s, 3H, 8-CH3).
5′-Deoxy-5′-dimethylsulfonio-8-methyladenosine chloride (25b)
Ion exchange resin (IRA-400, Cl− form) was washed repeatedly with water and loaded into the column. The column was left overnight and washed again repeatedly with water. The bromide salt 25a (50 mg) was dissolved in water (1 mL) and put on the column. The column was eluted with water very slowly in the dark. The desired fractions were combined and lyophilized to yield 30 mg (68%). MS m/z 326 (M)+. 1H NMR (D2O) δ 8.24 (s, 1H, H-2), 6.03 (d, 1H, H-1′, J1′,2′ = 5.4 Hz), 5.29 (t, 1H, H-2′, J2′,3′ = 5.6 Hz), 4.80 (t, 1H, H-3′, J3′,4′ = 4.9 Hz), 4.52−4.60 (m, 1H, H-4′), 3.82−4.11 (m, 1H, 5′-CH2), 3.75−3.83 (m, 1H, 5′-CH2), 2.95 and 2.92 (2s, 6H, S-(CH3)2), 2.69 (s, 3H, 8-CH3). UV λmax, nm, pH 1, 258.8 (ε 15000), pH 7, 259.1 (ε 14700), pH 13, 263.5 (ε 13000). Anal. (C13H20ClN5O3S·2H2O) C, H, N.
5′-Deoxy-5′-dimethylsulfonioadenosine bromide (25c)
The procedure described for 25a was used to prepare 25c from 24b (58 mg, 0.19 mmol): yield 49 mg (65%). MS m/z 312 (M)+.
5′-Deoxy-5′-dimethylsulfonioadenosine chloride (25d)
The procedure used was identical to that for 25b but starting with 25c. The desired fractions were combined together and lyophilized to yield 30 mg (44%). MS m/z 312 (M)+. 1H NMR (D2O) δ 8.29 (s, 1H, H-8), 8.27 (s, 1H, H-2), 6.12 (d, 1H, H-1′, J1′,2′ = 4.4 Hz), 4.99 (t, 1H, H-2′, J2′,3′ = 5.1 Hz), 4.55−4.62 (m, 2H, H-3′, H-4′), 3.84−4.0 (m, 2H, 5′-CH2), 2.95 and 2.93 (2s, 6H, S-(CH3)2). UV λmax, nm, pH 1, 256.4 (ε 14300), pH 7, 259.6 (ε 14400), pH 13, 266 (ε 12300). Anal. (C12H18ClN5O3S·1.5H2O·0.1C2H5OH) C, H, N, S.
Acknowledgments
This work was supported by grant PO1 CA-94000 from the National Cancer Institute. S.E.E. is indebted to the W. M. Keck Foundation and the Lucille P. Markey Charitable Trust. We thank the staffs of NE-CAT beamlines 8-BM and 24-ID-C at the Advanced Photon Source (NIH grant RR-15301) and the F2 beam line of the Cornell High Energy Synchrotron Source (NIH grant RR-01646) for providing beam time. We thank Leslie Kinsland and Stephanie Brannon for assistance in the preparation of this manuscript and thank Rachel Robbins for her assistance in performing some of the conformational searches reported in this manuscript.
Supporting Information Available
Elemental analyses used to determine the degree of purity for all target compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: AdoMetDC, S-adenosylmethionine decarboxylase; AdoMet, S-adenosylmethionine; ODC, ornithine decarboxylase; SSAT, spermidine/spermine N1-acetyltransferase; DFMO, α-difluoromethylornithine; MGBG 1, methylglyoxal bis(guanylhydrazone); MHZPA 3, 5′-deoxy-5′-[(3-hydrazinopropyl)methylamino]adenosine; MAOEA 5, 5′-[(2-aminooxyethyl)methylamino]-5′-deoxyadenosine; MHZEA 4, 5′-deoxy-5′-[(3-hydrazinoethyl)methylamino)adenosine; MeAdoMet, methyl ester of S-adenosylmethionine; hAdoMetDC, human S-adenosylmethionine decarboxylase; DMF, dimethylformamide; DTT, dithiothreitol; Tris, tris(hydroxymethyl)aminomethane; PEG, poly(ethylene glycol); CCD, charge-coupled device; HEPES, N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid; DIEA, diisopropylethylamine.
Supplementary Material
References
- Hackert M. L.; Pegg A. E.. Pyruvoyl-dependent enzymes. In Comprehensive Biological Catalysis; Sinnott M. L., Ed.; Academic Press: London, 1997; pp 201−216. [Google Scholar]
- Pegg A. E.; Xiong H.; Feith D.; Shantz L. M. S-adenosylmethionine decarboxylase: structure, function and regulation by polyamines. Biochem. Soc. Trans. 1998, 26, 580–586. [DOI] [PubMed] [Google Scholar]
- Tabor C. W.; Tabor H. Polyamines. Annu. Rev. Biochem. 1984, 53, 749–790. [DOI] [PubMed] [Google Scholar]
- Tabor C. W.; Tabor H. Methionine adenosyltransferase (S-adenosylmethionine synthetase) and S-adenosylmethionine decarboxylase. Adv. Enzymol. Related Areas Mol. Biol. 1984, 56, 251–282. [DOI] [PubMed] [Google Scholar]
- Casero R. A. Jr.; Celano P.; Ervin S. J.; Applegren N. B.; Wiest L.; Pegg A. E. Isolation and characterization of a cDNA clone that codes for human spermidine/spermine N1-acetyltransferase. J. Biol. Chem. 1991, 266, 810–814. [PubMed] [Google Scholar]
- van Poelje P. D.; Snell E. E. Pyruvoyl-dependent enzymes. Annu. Rev. Biochem. 1990, 59, 29–59. [DOI] [PubMed] [Google Scholar]
- Wallace H. M.; Fraser A. V.; Hughes A. A perspective of polyamine metabolism. Biochem. J. 2003, 376, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerner E. W.; Meyskens F. L. Jr Polyamines and cancer: old molecules, new understanding. Nat. Rev. Cancer 2004, 4, 781–792. [DOI] [PubMed] [Google Scholar]
- Pegg A. E. Polyamine metabolism and its importance in neoplastic growth and as a target for chemotherapy. Cancer Res. 1988, 48, 759–774. [PubMed] [Google Scholar]
- Metcalf B. W.; Bey P.; Danzin C.; Jung M. J.; Casara P.; Vevert J. P. Catalytic irreversible inhibition of mammalian ornithine decarboxylase (EC 4.1.1.17) by substrate and product analogues. J. Am. Chem. Soc. 1978, 100, 2551–2553. [Google Scholar]
- Bewley M. C.; Graziano V.; Jiang J.; Matz E.; Studier F. W.; Pegg A. E.; Coleman C. S.; Flanagan J. M. Structures of wild-type and mutant human spermidine/spermine N1-acetyltransferase, a potential therapeutic drug target. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 2063–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhonen-Hongisto L.; Seppanen P.; Janne J. Intracellular putrescine and spermidine deprivation induces increased uptake of the natural polyamines and methylglyoxal bis(guanylhydrazone). Biochem. J. 1980, 192, 941–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuroy U. K.; Gerner E. W. Emerging concepts in targeting the polyamine metabolic pathway in epithelial cancer chemoprevention and chemotherapy. J. Biochem. 2006, 139, 27–33. [DOI] [PubMed] [Google Scholar]
- Fabian C. J.; Kimler B. F.; Brady D. A.; Mayo M. S.; Chang C. H.; Ferraro J. A.; Zalles C. M.; Stanton A. L.; Masood S.; Grizzle W. E.; Boyd N. F.; Arneson D. W.; Johnson K. A. A phase II breast cancer chemoprevention trial of oral alpha-difluoromethylornithine: breast tissue, imaging, and serum and urine biomarkers. Clin. Cancer Res. 2002, 8, 3105–3117. [PubMed] [Google Scholar]
- Meyskens F. L. Jr.; Gerner E. W. Development of difluoromethylornithine (DFMO) as a chemoprevention agent. Clin. Cancer Res. 1999, 5, 945–951. [PubMed] [Google Scholar]
- Meyskens F. L.; McLaren C. E.; Pelot D.; Fujikawa-Brooks S.; Carpenter P. M.; Hawk E.; Kelloff G.; Lawson M. J.; Kidao J.; McCracken J.; Albers C. G.; Ahnen D. J.; Turgeon D. K.; Goldschmid S.; Lance P.; Hagedorn C. H.; Gillen D. L.; Gerner E. W. Difluoromethylornithine Plus Sulindac for the Prevention of Sporadic Colorectal Adenomas: A Randomized Placebo-Controlled, Double-Blind Trial. Cancer. Prev. Res. (Philadelphia, PA, U.S.) 2008, 1, 9–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raul F.; Gosse F.; Osswald A. B.; Bouhadjar M.; Foltzer-Jourdainne C.; Marescaux J.; Soler L. Follow-up of tumor development in the colons of living rats and implications for chemoprevention trials: assessment of aspirin-difluoromethylornithine combination. Int. J. Oncol. 2007, 31, 89–95. [PubMed] [Google Scholar]
- Williams-Ashman H. G.; Schenone A. Methylglyoxal bis(guanylhydrazone) as a potent inhibitor of mammalian and yeast S-adenosylmethionine decarboxylases. Biochem. Biophys. Res. Commun. 1972, 46, 288–295. [DOI] [PubMed] [Google Scholar]
- Millward M. J.; Joshua A.; Kefford R.; Aamdal S.; Thomson D.; Hersey P.; Toner G.; Lynch K. Multi-centre Phase II trial of the polyamine synthesis inhibitor SAM486A (CGP48664) in patients with metastatic melanoma. Invest. New Drugs 2005, 23, 253–256. [DOI] [PubMed] [Google Scholar]
- Shantz L. M.; Stanley B. A.; Secrist J. A. III; Pegg A. E. Purification of human S-adenosylmethionine decarboxylase expressed in Escherichia coli and use of this protein to investigate the mechanism of inhibition by the irreversible inhibitors, 5′-deoxy-5′-[(3-hydrazinopropyl)methylamino]adenosine and 5′{[(Z)-4-amino-2-butenyl]methylamino-5′-deoxyadenosine. Biochemistry 1992, 31, 6848–6855. [DOI] [PubMed] [Google Scholar]
- Danzin C.; Marchal P.; Casara P. Irreversible inhibition of rat S-adenosylmethionine decarboxylase by 5′-{[(Z)-4-amino-2-butenyl]methyl amino}-5′-deoxyadenosine. Biochem. Pharmacol. 1990, 40, 1499–1503. [DOI] [PubMed] [Google Scholar]
- Ekstrom J. E.; Matthews I. I.; Stanley B. A.; Pegg A. E.; Ealick S. E. The crystal structure of human S-adenosylmethionine decarboxylase at 2.25 Å resolution reveals a novel fold. Structure 1999, 7, 583–595. [DOI] [PubMed] [Google Scholar]
- Ekstrom J. L.; Tolbert W. D.; Xiong H.; Pegg A. E.; Ealick S. E. Structure of a human S-adenosylmethionine decarboxylase self-processing ester intermediate and mechanism of putrescine stimulation of processing as revealed by the H243A mutant. Biochemistry 2001, 40, 9495–9504. [DOI] [PubMed] [Google Scholar]
- Tolbert W. D.; Zhang Y.; Cottet S. E.; Bennett E. M.; Ekstrom J. L.; Pegg A. E.; Ealick S. E. Mechanism of human S-adenosylmethionine decarboxylase proenzyme processing as revealed by the structure of the S68A mutant. Biochemistry 2003, 42, 2386–2395. [DOI] [PubMed] [Google Scholar]
- Tolbert D. W.; Ekstrom J. L.; Mathews I. I.; Secrist J. A. III; Kapoor P.; Pegg A. E.; Ealick S. E. The structural basis for substrate specificity and inhibition of human S-adenosylmethionine decarboxylase. Biochemistry 2001, 40, 9484–9494. [DOI] [PubMed] [Google Scholar]
- Secrist J. A. III New substrate analogues as inhibitors of S-adenosylmethionine decarboxylase. Nucleosides Nucleotides 1987, 6, 73–83. [Google Scholar]
- Mitsunobu O. The use of diethyl azodicarboxylate and triphenylphosphine in synthesis and transformation of natural products. Synthesis 1981, 1–28. [Google Scholar]
- Khomutov A. R.; Vepsalainen J. J.; Shvetsov A. S.; Hyvonen T.; Keinanen T. A.; Pustobaev V. N.; Eloranta T. O.; Khomutov R. M. Synthesis of hydroxylamine analogs of polyamines. Tetrahedron 1996, 52, 13751–13766. [Google Scholar]
- Webb R. R.; Kaneko T. Synthesis of 1-(aminooxy)-4-[(3-nitro-2-pyridyl)dithio]butane and 1-(aminooxy)-4-[(3-nitro-2-pyridyl)dithio]-2-butene, novel heterobifunctional crosslinking reagents. Bioconjugate Chem. 1990, 1, 96–99. [DOI] [PubMed] [Google Scholar]
- Kucznierz R.; Grams F.; Leinert H.; Marzenell K.; Engh R. A.; von der Saal W. Tetrahydro-isoquinoline-based factor Xa inhibitors. J. Med. Chem. 1998, 41, 4983–4994. [DOI] [PubMed] [Google Scholar]
- Pankaskie M.; Abdel-Monem M. M. Inhibitors of polyamine biosynthesis. 8. Irreversible inhibition of mammalian S-adenosyl-l-methionine decarboxylase by substrate analogues. J. Med. Chem. 1980, 23, 121–127. [DOI] [PubMed] [Google Scholar]
- Borchardt R. T.; Huber J. A.; Wu Y. S. Convenient preparation of S-adenosylhomocysteine and related compounds. J. Org. Chem. 1976, 41, 565–567. [DOI] [PubMed] [Google Scholar]
- Markham G. D.; Norrby P. O.; Bock C. W. S-Adenosylmethionine conformations in solution and in protein complexes: conformational influences of the sulfonium group. Biochemistry 2002, 41, 7636–7646. [DOI] [PubMed] [Google Scholar]
- Chang G.; Guida W. C.; Still W. C. An Internal Coordinate Monte Carlo Method for Searching Conformational Space. J. Am. Chem. Soc. 1989, 111, 4379–4386. [Google Scholar]
- Kolossvary I.; Guida W. C. Low-mode conformational search elucidated: application to C39H80 and flexible docking of 9-deazaguanine inhibitors into PNP. J. Comput. Chem. 1999, 20, 1671–1684. [Google Scholar]
- Mohamadi F.; Richards N. G. J.; Guida W. C.; Liskamp R.; Lipton M.; Caufield C.; Chang G.; Hendrickson T.; Still W. C. Macromodel—An Integrated Software System for Modeling Organic and Bioorganic Molecules Using Molecular Mechanics. J. Comput. Chem. 1990, 11, 440–467. [Google Scholar]
- Ikehara M.; Uesugi S.; Kaneko M. Bromination of adenosine nucleosides and nucleotides. Chem. Commun. 1967, 17–18. [Google Scholar]
- Van Aerschot A. A.; Mamos P.; Weyns N. J.; Ikeda S.; De Clercq E.; Herdewijn P. A. Antiviral activity of C-alkylated purine nucleosides obtained by cross-coupling with tetraalkyltin reagents. J. Med. Chem. 1993, 36, 2938–2942. [DOI] [PubMed] [Google Scholar]
- Chattopadhyaya J. B.; Reese C. B. Reaction between 8-bromoadenosine and amines. Chemistry of 8-hydrazinoadenosine. Synthesis 1977, 725–726. [Google Scholar]
- Kohyama N.; Katashima T.; Yamamoto Y. Synthesis of novel 2-aryl AICAR derivatives. Synthesis 2004, 2799–2804. [Google Scholar]
- Long R. A.; Robins R. K.; Townsend L. B. Purine nucleosides. XV. Synthesis of 8-amino- and 8-substituted aminopurine nucleosides. J. Org. Chem. 1967, 32, 2751–2756. [DOI] [PubMed] [Google Scholar]
- Gani D.; Johnson A. W. The base-sugar conformation of certain derivatives of adenosine. J. Chem. Soc., Perkin Trans. 1 1982, 1197–1204. [Google Scholar]
- Otwinowski Z.; Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Brünger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr., Sect. D: Biol Crystallogr. 1998, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Jones T. A.; Zou J.-Y.; Cowan S. W.; Kjeldgaard M. Improved methods for the building of protein models in electron density maps and the location of errors in these models. Acta Crystallogr., Sect. A: Found. Crystallogr. 1991, 47, 110–119. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crytallogr. 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Kleywegt G. J.; Jones T. A. Databases in protein crystallography. Acta Crystallogr., Sect. D: Biol Crystallogr. 1998, 54, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Weiner S. J.; Kollman P. A.; Case D. A.; Singh U. C.; Ghio C.; Alagona G.; Profeta S. Jr.; Weiner P. A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 1984, 106, 765–784. [Google Scholar]
- Weiner S. J.; Kollman P. A.; Nguyen D. T.; Case D. A. An all atom force field for simulations of proteins and nucleic acids. J. Comput. Chem. 1986, 7, 230–252. [DOI] [PubMed] [Google Scholar]
- Ponder J. W.; Richards F. M. An efficient Newton-like method for molecular mechanics energy minimization of large molecules. J. Comput. Chem. 1987, 8, 1016–1024. [Google Scholar]
- Shantz L. M.; Pegg A. E.. Assay of S-adenosylmethionine decarboxylase In Methods in Molecular Biology, Vol 79: Polyamine Protocols; Morgan D. M. L., Ed.; Humana Press Inc.: Totowa, NJ, 1998; pp 45−50. [DOI] [PubMed] [Google Scholar]
- Kuhn R.; Jahn W. Thioethers and sulfoxides derived from adenosine. Chem. Ber. 1965, 98, 1699–1704. [DOI] [PubMed] [Google Scholar]
- Minnick A. A.; Kenyon G. L. General synthetic approach to stable nitrogen analogs of S-adenosylmethionine. J. Org. Chem. 1988, 53, 4952–4961. [Google Scholar]
- Schmidt R. R.; Schloz U.; Schwille D. Synthesis of 5′-modified adenosine derivatives. Chem. Ber. 1968, 101, 590–594. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.