

Abstract
Huntington's disease (HD) is caused by polyglutamine (polyQ) expansion in huntingtin (htt), a large (350 kDa) protein that localizes predominantly to the cytoplasm. Proteolytic cleavage of mutant htt yields polyQ-containing N-terminal fragments that are prone to misfolding and aggregation. Disease progression in HD transgenic models correlates with age-related accumulation of soluble and aggregated forms of N-terminal mutant htt fragments, suggesting that multiple forms of mutant htt are involved in the selective neurodegeneration in HD. Although mitochondrial dysfunction is implicated in the pathogenesis of HD, it remains unclear which forms of cytoplasmic mutant htt associate with mitochondria to affect their function. Here we demonstrate that specific N-terminal mutant htt fragments associate with mitochondria in Hdh(CAG)150 knock-in mouse brain and that this association increases with age. The interaction between soluble N-terminal mutant htt and mitochondria interferes with the in vitro association of microtubule-based transport proteins with mitochondria. Mutant htt reduces the distribution and transport rate of mitochondria in the processes of cultured neuronal cells. Reduced ATP level was also found in the synaptosomal fraction isolated from Hdh(CAG)150 knock-in mouse brain. These findings suggest that specific N-terminal mutant htt fragments, before the formation of aggregates, can impair mitochondrial function directly and that this interaction may be a novel target for therapeutic strategies in HD.
Keywords: polyglutamine, mitochondria, trafficking, neurodegeneration, Huntington's disease, proteolysis
Introduction
Huntington's disease (HD) is a fatal, inherited neurological disorder characterized by selective neuropathology, progressive motor deficits, cognitive impairment, and psychiatric abnormalities. HD is caused by a polyglutamine (polyQ) expansion (>36 glutamines) in the N-terminal region of huntingtin (htt) (The Huntington's Disease Collaborative Research Group, 1993). Htt is a large (350 kDa), predominantly cytoplasmic protein that is a substrate for various proteolytic enzymes (Fryer and Zoghbi, 2006). Proteolytic cleavage of full-length mutant htt yields small N-terminal fragments containing the polyQ domain that readily misfold and aggregate in both neuronal nuclei and processes (DiFiglia et al., 1997; Gutekunst et al., 1999). The strong toxic property of proteolytically processed mutant htt is highlighted by the more rapid disease progression of HD mice expressing smaller N-terminal mutant htt fragments than that of mice expressing full-length mutant htt (Li and Li, 2006). This phenomenon has led to extensive study of the proteolysis of htt and the identification of a number cleavage sites in the N-terminal region of htt (Kim et al., 2001; Gafni and Ellerby, 2002; Wellington et al., 2002; Lunkes et al., 2002; Zhou et al., 2003; Graham et al., 2006).
HD neuropathology is characterized by the preferential neurodegeneration of striatal medium spiny neurons, which extends to other brain regions as the disease progresses (Vonsattel et al., 1985; Martin and Gusella, 1986). In postmortem HD brain tissue, activity of complexes II and III of the mitochondrial electron transport chain is impaired, and systemic administration of complex II inhibitors in rodents causes striatal pathology and behavioral abnormalities reminiscent of HD (Lin and Beal, 2006). More recent studies using genetic HD models have identified deficits in mitochondrial energy production, calcium-handling, and trafficking (Trushina and McMurray, 2007). Although increasing evidence indicates that mutant htt can affect mitochondria directly in vitro (Panov et al., 2002; Choo et al., 2004; Rockabrand et al., 2007), it remains unclear whether full-length mutant htt or its proteolytic fragments have a direct effect on mitochondria in vivo. It has been reported that mutant htt can sequester and deplete soluble levels of trafficking proteins (Trushina et al., 2004) and physically impede the trafficking of mitochondria in neuronal processes (Chang et al., 2006). Because changes in mitochondrial function are also observed in HD models in the absence of htt aggregates (Panov et al., 2002; Choo et al., 2004; Chang et al., 2006), it is necessary to characterize the association of soluble forms of htt with mitochondria in vivo to identify the early pathological events in HD.
In this study, we provide evidence for a preferred association of specific N-terminal htt fragments with mitochondria in HD knock-in mouse brain. This association increases with age and correlates with disease progression. We show that soluble mutant htt fragments interfere with the interaction between trafficking proteins and mitochondria and affect the transport and distribution of mitochondria in neuronal processes. Our findings suggest that the htt-mitochondria interaction could be a unique target for HD therapeutics.
Materials and Methods
Mice.
Hdh(CAG)150 knock-in mice (Lin et al., 2001) were bred and maintained in the animal facility at Emory University in accordance with institutional guidelines.
Plasmids.
cDNA constructs encoding untagged or GFP-tagged wild-type (20–23Q) or mutant (120–140Q) N-terminal htt fragments (67, 208, or 508 aa) in the pRK5 eukaryotic expression vector were generated in our previous studies (Zhou et al., 2003; Shin et al., 2005; Cornett et al., 2006). The vector for mitochondrially targeted DsRed2 (pDsRed2-Mito) was obtained from Clontech (Mountain View, CA).
Antibodies and reagents.
Antibodies against N-terminal human htt (EM48; 1:100), huntingtin-associated protein 1 (HAP1; 1:2000), dynactin P150 (P150; 1:2000), and kinesin light chain-2 (KLC; 1:2000) were generated in our previous studies (Li and Li, 1998; Li et al., 2000; McGuire et al., 2006). Antibodies against an internal epitope of htt (2166; 1:1000), expanded polyQ (1C2; 1:5000), kinesin heavy chain (KHC; 1:1000), glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 1:3000), manganese superoxide dismutase (MnSOD; 1:500), and neuronal nuclear protein (NeuN; 1:500) were obtained from Millipore (Billerica, MA). Other antibodies used are against the following proteins: 70 and 30 kDa subunits of succinate dehydrogenase (SDH; Invitrogen, Carlsbad, CA; 1:5000 and 1:200, respectively); sodium/potassium ATPase α1 (Na/K-ATPase; 1:1000) and early endosomal antigen 1 (EEA1; Millipore; 1:1000); α-tubulin (Sigma, St. Louis, MO; 1:20,000); aspartate amino transferase 2 (AAT; Abnova, Taipei, Taiwan; 1:1000); BiP (Stressgen, Ann Arbor, MI; 1:4000); synaptophysin (Abcam, Cambridge, MA; 1:500); and glucose-6-phosphate dehydrogenase (G6PD; Novus Biologicals, Littleton, CO; 1:1000). Secondary antibodies were peroxidase-conjugated donkey anti-mouse, -rabbit, -rat, -guinea pig, -goat, or -sheep IgG (H+L) from Jackson ImmunoResearch (West Grove, PA; all used at 1:5000). Other reagents are from Sigma unless specified otherwise.
Electron microscopy.
Hdh(CAG)150 knock-in mouse brain tissue was labeled with EM48 according to previously published procedures from our laboratory (Li et al., 2003).
Cell culture.
HEK293 cells were stably transfected with full-length human htt with 23 (293-23Q-FL) or 120 (293-120Q-FL) glutamine repeats (Zhou et al., 2003) or transiently transfected with N-terminal mutant htt. Primary rat cortical and striatal cultures were prepared separately using embryonic day 17–18 fetuses. Newborn pups from matings between heterozygous (150Q/7Q) knock-in animals were used to obtain HD knock-in striatal cultures. For culturing mouse primary neurons, striata from each mouse pup were dissected and plated separately. Viable cells were plated in B27-supplemented Neurobasal media (Invitrogen) at 1 × 106 cells/ml on acid-washed and poly-d-lysine-coated 25 mm round German coverglass in 6-well plates. Expression of mutant htt in HD knock-in neurons was confirmed by Western blotting for mutant htt with the 1C2 antibody. This antibody is raised against expanded polyQ and specifically detects mutant forms of htt.
Analysis of mitochondrial transport in live neurons.
Mitochondrial transport was monitored by imaging primary neurons on a confocal microscope 24 h after transfection with GFP-htt, untagged-htt, and/or DsRed2Mito. Cultured rat brain cortical [4 d in vitro (DIV)] or striatal (5 DIV) neurons were cotransfected with 0.1 μg of DsRed2Mito and 5 μg of GFP-htt DNA. For monitoring mitochondrial transport in the presence of untagged htt, rat striatal neurons were cotransfected with 0.08 μg of DsRed2Mito and 8 μg of either wild-type or mutant 508 aa N-terminal htt. Using this 1:100 DNA ratio, immunofluorescent staining confirmed that >95% of DsRed2Mito+ neurons were also coexpressing 508 aa htt. Cultured HD knock-in mouse neurons (6 DIV) were transfected with 0.2 μg of DsRed2Mito only. Neurons were transfected with Lipofectamine 2000 (Invitrogen) for 5 h and imaged after 24 h. Coverslips were transferred to an Attofluor imaging chamber (Invitrogen) with 1.5 ml of HBSS, and maintained at 37°C with a stage heater for the duration of imaging. Images were acquired using a 63×/1.4 numerical aperture oil-immersion objective on a Zeiss (Oberkochen, Germany) LSM 510 META confocal microscope. Excitation for GFP-htt was 488 nm with detection after a 500–550 nm bandpass filter. DsRed2Mito excitation was 543 nm with detection after a 565–615 nm bandpass filter. Neurons coexpressing GFP-htt and DsRed2Mito were identified by an initial dual scan. Only neurons with low levels of GFP-htt expression and no visible aggregation were imaged further. Mitochondrial transport in both GFP-htt-transfected and HD knock-in neurons was assessed by acquiring three-dimensional stacks (1 μm between z-planes; 3 μm total coverage) of DsRed2-Mito fluorescence every 8 s for a total of 5 min. Frame-by-frame analysis of distance, duration, and directionality of fast transport of individual mitochondria was subsequently performed using both Zeiss LSM and ImageJ (NIH, Bethesda, MD) software. Further calculations were performed on Microsoft (Redmond, WA) Excel. Four separate rat cultures and four separate HD knock-in litters were used in the imaging experiments. Nine to 24 neurons were analyzed for each condition tested. Transport parameters were averaged for each neuron, and group averages were compared for statistically significant differences by Student's t test for GFP-htt or 508 aa htt-transfected neurons or ANOVA with post hoc analysis for HD knock-in neurons.
Analysis of mitochondrial distribution in neurons.
Cultured primary striatal neurons were cotransfected with 0.1 μg of DsRed2Mito and 5 μg of wild-type or mutant 508 aa htt. After 24 h, neurons were fixed and stained with EM48 for transfected htt. Process length, mitochondrial length, and number of mitochondria in neurons expressing 508 aa htt and DsRed2Mito were measured using Zeiss LSM software. Further calculations were performed on Microsoft Excel. Distribution parameters were averaged for each neuron, and group averages were compared for statistically significant differences by Student's t test (n = 19 and 12 neurons for 23Q and 120Q-508 htt, respectively, from 4 separate experiments).
Subcellular fractionation.
Mice were killed by cervical dislocation. A modified version of Sims' Method A was used to isolate subcellular fractions from forebrain tissue (Sims, 1990; Garcia et al., 2005) with only minor changes. Whole-cell lysates were obtained by solubilizing 1/20 of the original homogenate with three volumes of RIPA buffer for 1 h at 4°C. Mitochondrial fractions were purified using Percoll density gradient centrifugation. The supernatant from the 13,000 × g centrifugation was saved as the cytosolic lysate. Synaptosomal fractions of mouse brain tissues were prepared using the previously described method (Phillips et al., 2001). Protein content of brain fractions, as well as all other samples described, was determined using a detergent compatible protein assay (Bio-Rad, Hercules, CA). The purity of the brain fractions (20–30 μg) and mutant htt were assessed by Western blotting with antibodies to mutant forms of htt (1C2), full-length wild-type and mutant htt (2166), and subcellular markers as described above. HEK293 cells expressing full-length or N-terminal 508 aa of htt were fractioned using a method similar to Sims' Method B (Sims, 1990; Garcia et al., 2005). To estimate the size of htt associated with mitochondria, purified brain mitochondria were resolved by SDS-PAGE with whole-cell lysates from cells transiently expressing mutant htt of varying lengths. The blot was then probed with 1C2.
In vitro binding assays.
To examine the effect of mutant htt on the association of mitochondria with trafficking proteins, fresh cytosolic lysates were isolated from 6- to 7-month-old wild-type and 150Q/150Q knock-in mice. Mitochondria were purified from the brain cortex of wild-type mouse brains. To ensure removal of residual Percoll and loosely associated proteins, purified mitochondria were washed twice with isolation buffer. Mitochondria were diluted to 2 μg/μl with isolation buffer and aliquoted (50 μg) to Microfuge tubes on ice. Cytosolic lysates of wild-type or HD mouse brains (200 μl at 0.125 μg/μl) were added to aliquots of purified mitochondria and incubated on ice for 30 min with gentle mixing every 5 min and then were centrifuged for 15 min at 10,000 × g at 4°C. After washes, mitochondrial pellets were subjected to Western blotting with antibodies for mutant htt (1C2) and trafficking proteins. To determine relative amounts of proteins bound to mitochondria, the baseline signal in the absence of cytosolic lysates was subtracted from the signal measured from mitochondria incubated with cytosolic lysates. The relative binding of trafficking proteins to mitochondria in the presence of mutant htt was expressed as a percentage of binding in the presence of wild-type htt.
ATP content assays.
ATP levels were measured using a method described previously (Milakovic and Johnson, 2005). ATP content was measured using a FLUOstar Galaxy microplate reader (BMG Labtech, Offenburg, Germany).
Statistical analysis.
Data are expressed as mean ± SEM and were analyzed for statistical significance (p < 0.05) with Prism software (version 4; GraphPad Software, San Diego, CA). The specific tests used are noted in the text and figure legends.
Results
N-terminal mutant htt fragments associate with knock-in mouse brain mitochondria
Electron microscopy has shown that htt associates with brain mitochondria in wild-type and transgenic HD rodents (Gutekunst et al., 1998; Panov et al., 2002; Yu et al., 2003; Petrasch-Parwez et al., 2007). To verify the association of mitochondria with mutant htt that was expressed at the endogenous level, we evaluated EM48 immunogold labeling of htt in brain tissue from 9-month-old HD knock-in mice that express full-length mutant htt driven by the endogenous mouse Hdh promoter (Lin et al., 2001). Although these HD mice have a lifespan similar to wild-type mice, they show behavioral abnormalities starting at the age of 25 weeks (Lin et al., 2001). EM48 specifically labels mutant htt in sections of HD brains (Gutekunst et al., 1999; Lin et al., 2001). We observed a widespread subcellular distribution of mutant htt and that the surface of some mitochondria contained mutant htt in HD knock-in brain (Fig. 1A, arrows). Because EM48 selectively labeled mutant htt, no EM48 immunogold labeling was seen in wild-type mouse brain (Fig. 1B).
Figure 1.
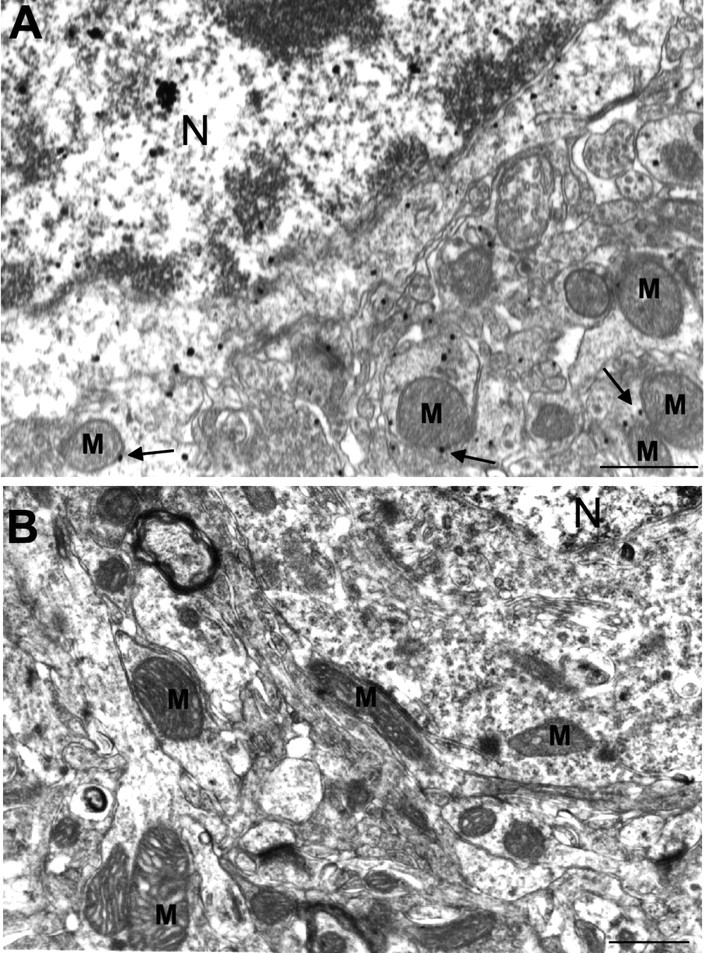
Mutant htt associates with mitochondria in HD knock-in mice. A, EM48 immunogold labeling of the cerebral cortex of HD knock-in brain tissue at 9 months of age showing the association of mutant htt with mitochondria (M; arrows). B, EM48 immunogold labeling of the cerebral cortex of wild-type mice at 12 month of age. N, Nucleus. Scale bars, 500 nm.
Although EM48 electron microscopy offers ultrastructural evidence for the subcellular localization of mutant htt to mitochondria, it cannot readily distinguish between full-length and cleaved mutant htt. Therefore, we used Western blotting to analyze the association of mutant htt with mitochondria that were isolated by Percoll density gradient centrifugation from the forebrain of heterozygous (150Q/7Q) mice. The purity and enrichment of isolated mitochondria were verified by immunoblotting for various subcellular marker proteins (Fig. 2A). To reveal the full profile of mutant htt forms present on purified brain mitochondria (Fig. 2B, top), we probed blots with 1C2, an antibody that was raised against expanded polyQ and can more readily distinguish mutant htt fragments on Western blots than does EM48. The specificity of the 1C2 antibody for mutant forms of htt is demonstrated by the lack of reactivity in the fractions from wild-type mice (7Q/7Q) (Fig. 2B, two leftmost lanes). Levels of full-length wild-type and mutant htt were detected with the monoclonal antibody 2166 (Fig. 2B, bottom). Because polyQ expansion retards the migration of proteins in the gel, we compared mitochondrial htt with transfected htt of known repeats and amino acids on the same blot (Fig. 2C). This comparison suggests that mutant htt fragments with 150Q (120–130 kDa) enriched in the mitochondrial fraction from brain tissue may contain ∼500 aa of N-terminal htt.
Figure 2.

Specific N-terminal mutant htt fragments are enriched in brain mitochondria from HD knock-in mice. A, Forebrains from wild-type (7Q/7Q) or HD (150Q/7Q) knock-in mice were fractioned to purify mitochondria on a Percoll-based gradient. Whole-cell (Whole), cytosolic (Cyto), or mitochondrial (Mito) fractions (20 μg/each) were immunoblotted for various subcellular markers to confirm the purity of the isolated mitochondrial fraction. All mitochondrial proteins (30 and 70 kDa SDH, MnSOD, and AAT) showed significant enrichment in mitochondrial fractions. The blots were also probed with antibodies for plasma membrane (Na/K-ATPase), cytosolic (α-tubulin, GAPDH, G6PD, and NeuN), nuclear (NeuN), ER (BiP), endosomal (EEA1), or synaptosomal (synaptophysin) proteins. B, Subsequent blotting for mutant htt and full-length htt demonstrated a preferential association of N-terminal mutant-htt fragments with brain mitochondria. Note that the 1C2 antibody only reacts with mutant htt, whereas the 2166 antibody detects the full-length forms of both wild-type and mutant htt (bottom). C, Comparison of the sizes of N-terminal htt fragments in mitochondria isolated from HD knock-in mice with mutant htt of known lengths.
Whereas similar low levels of full-length mutant htt were present in the mitochondrial fraction from 3- and 10-month-old HD mouse brains, the amount of mutant htt fragments associated with mitochondria was increased dramatically in mitochondria from older HD mouse brain (Fig. 2B, rightmost lane). HD knock-in mice show age-dependent behavioral phenotypes that correlate with the accumulation of N-terminal htt fragments and the formation of htt aggregates (Lin et al., 2001; Zhou et al., 2003). To further investigate the age-related accumulation of N-terminal mutant htt fragments on mitochondria, we performed immunoblotting of subcellular fractions from wild-type (7Q/7Q) and homozygous (150Q/150Q) HD knock-in mice ranging in age from 3 to 25 months. Blots of whole-cell and cytosolic fractions confirmed that endogenously expressed mutant htt is largely associated with the cytosolic compartment and that N-terminal mutant htt fragments accumulate with age (Fig. 3A). To detect the full profile of mutant htt fragments present on mitochondria at different ages, blots of mitochondrial fractions were subjected to longer exposure times (Fig. 3B). Although mitochondria from homozygous HD knock-in mice show stronger signals of full-length mutant htt than those from heterozygous HD knock-in mice (Fig. 3B), N-terminal htt fragments of 120–130 kDa were also enriched in mitochondria from homozygous HD mouse brains. The results reveal two important phenomena: the age-dependent accumulation of the mutant htt fragments and the appearance of poorly resolved fragments in older animals (>18 months). The age-dependent accumulation of mutant htt fragments on mitochondria may be relevant to the delayed onset phenotypes observed in these mice. The poorly resolved bands may represent either larger N-terminal htt fragments that begin to accumulate on mitochondria in older animals or the same 120–130 kDa N-terminal fragment that has become misfolded and permanently conjugated to other mitochondrial proteins. Although the exact size of these fragments is difficult to determine, it is apparent that specific N-terminal mutant htt fragments accumulate on mitochondria in an age-dependent manner despite the presence of a wide range of mutant htt fragments in the cytosolic compartment (Figs. 2B, 3A).
Figure 3.

N-terminal fragments of wild-type htt associate more readily with mitochondria than full-length htt. A, B, Whole-cell and cytosolic (A) and mitochondrial (B) fractions from wild-type (7Q/7Q) and homozygous HD knock-in (150Q/150Q) mice at the ages of 3–25 months were blotted with 1C2 to specifically detect mutant htt followed by antibodies to either α-tubulin or the mitochondrial protein MnSOD. A nonspecific band (n.s) appears in both 7Q/7Q and 150Q/150Q samples. Longer exposure of blots containing mitochondrial protein revealed an age-dependent accumulation of specific intermediate-sized N-terminal mutant htt fragments on brain mitochondria (bracket). C, Western blotting of whole-cell (W), cytoplasmic (C), and mitochondrial (M) fractions of transfected HEK293 cells expressing full-length htt (23Q-FL and 120Q-FL) or N-terminal htt fragments (23Q-508 and 120Q-508). The blots were probed with EM48 for htt (top) and antibodies to α-tubulin (middle) and the mitochondrial protein MnSOD (bottom).
Association of N-terminal htt with mitochondria in transfected cells
The presence of differing amounts of full-length and N-terminal mutant htt on purified mitochondria led us to compare their association with mitochondria in transfected cells that expressed N-terminal or full-length htt. We have established HEK293 cell lines that express full-length htt with 23Q or 120Q (Zhou et al., 2003). Mitochondrial fractionation assay did not show the presence of full-length htt in the purified mitochondrial fraction (Fig. 3C). However, transfected N-terminal htt fragments (508 aa) containing either 23Q or 120Q were present in the mitochondria fraction. This comparison in cultured cells supports the finding in mouse brain tissue that N-terminal htt associates more readily with mitochondria than does full-length htt. Because some full-length mutant htt is present in the mitochondrial fraction isolated from mouse brain, the association of full-length mutant htt with mitochondria may depend on neuronal specific proteins that are absent in HEK293 cells. Although transfected N-terminal htt with a normal repeat (23Q) is associated with mitochondria in HEK293 cells, Western blotting with 1C2, EM48, or 2166 did not reveal the presence of N-terminal fragments of wild-type htt in purified mitochondrial fraction from mouse brain tissue (data not shown). It is possible that mutant htt fragments are more stable than normal htt fragments such that there is an age-dependent accumulation of mutant N-terminal htt fragments in the mitochondrial fraction from mouse brain.
N-terminal mutant htt interferes with the association of trafficking proteins with mitochondria
It has been reported that polyQ expansion in htt disrupts the formation of complexes formed by htt, HAP1, P150, and microtubules (Gauthier et al., 2004). Thus, it is also likely that mutant htt fragments disrupt the association of trafficking proteins with mitochondria. To test this hypothesis, we assessed the association of trafficking-associated proteins (KHC, KLC, P150, and HAP1) with mitochondria in the presence of either wild-type or mutant htt. Because the association of trafficking proteins with mitochondria in neurons may be regulated by cytoplasmic factors specific to brain tissue, we isolated mitochondria from wild-type mouse brains and then incubated them with the cytosolic fraction from the forebrain of HD knock-in mice. Soluble levels of KHC, KLC, P150, and HAP1 were all unchanged in the forebrain lysates of HD knock-in mice at the age of 6–7 months (Fig. 4A, top panels) and 13 and 18 months of age (data not shown). Only low levels of trafficking proteins remain associated with purified mitochondria (Fig. 4A, top panels, center lane). These purified mitochondria were then incubated with the cytoplasmic fraction of mouse brain lysates containing trafficking proteins and wild-type or mutant htt expressed at endogenous levels. After incubation with wild-type soluble fraction, all four trafficking-associated proteins (HAP1, P150, KLC, and KHC) showed association with mitochondria. However, the association of these proteins with mitochondria was significantly reduced in the presence of mutant htt (Fig. 4A,B). Probing the blots with 1C2 confirmed the association of mutant htt N-terminal fragments with purified mitochondria (Fig. 4A, bottom, arrow).
Figure 4.
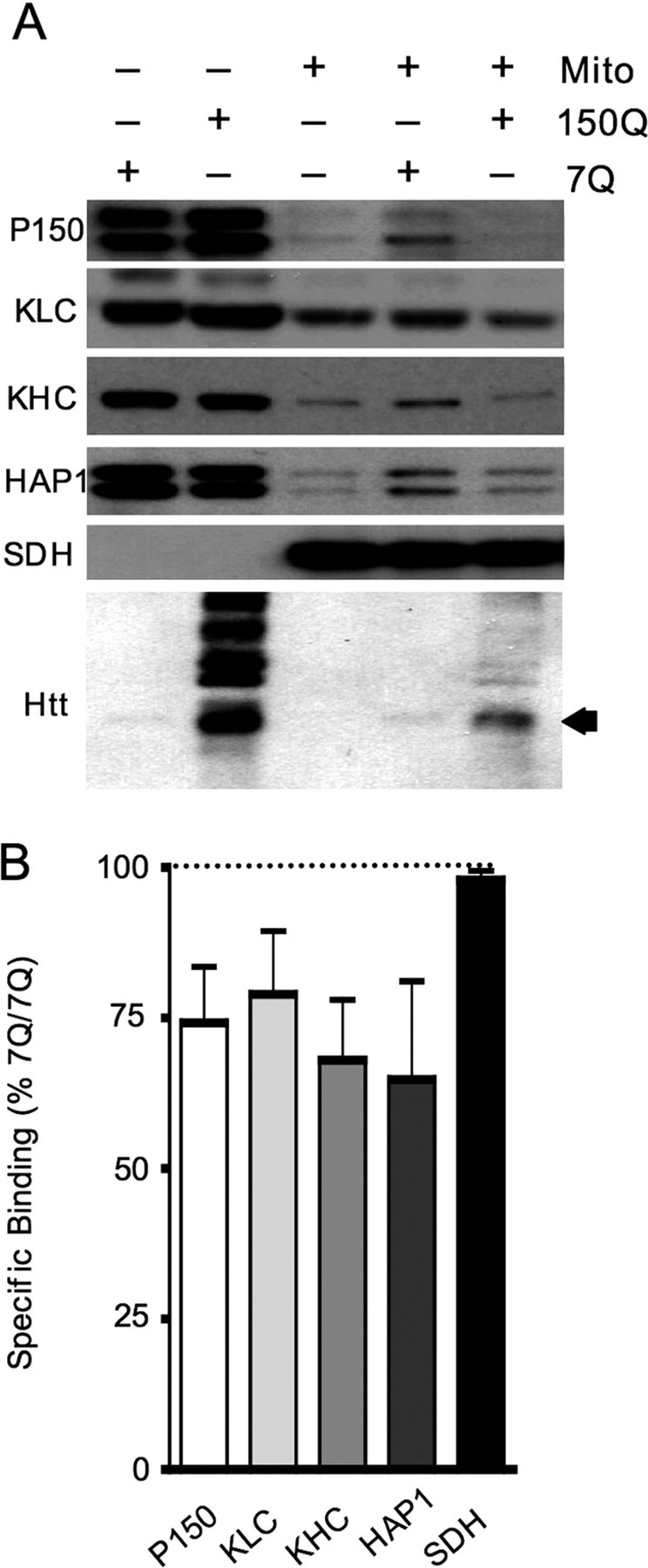
Mutant htt affects the in vitro association of trafficking proteins with mitochondria. A, Cytosolic lysates from 6-month-old 7Q/7Q and 150Q/150Q mouse forebrains were incubated with Percoll-purified mitochondria from wild-type mouse brains (top panel). Mitochondria were pelleted, washed repeatedly, and blotted with antibodies to trafficking proteins (P150, KLC, KHC, and HAP1). The mitochondrial protein (SDH) was also detected as a loading control. B, Relative binding of trafficking proteins to mitochondria in the presence of mutant versus wild-type htt was quantified. Blots probed with 1C2 (A, bottom) show that endogenously expressed N-terminal mutant htt fragments associate with brain mitochondria in this in vitro assay. Preferential binding of intermediate-length mutant htt fragments (arrow) is also confirmed in this assay.
Impaired axonal trafficking in HD models is mediated by soluble N-terminal mutant htt fragments
Previous studies indicate that loss of endogenous htt, polyQ expansion, or aggregation of mutant htt can disrupt the transport of mitochondria in neuronal processes (Trushina et al., 2004; Chang et al., 2006). However, it remains unclear whether soluble mutant htt fragments can directly affect the trafficking of mitochondria. To test this hypothesis, we first performed time-lapse confocal imaging of mitochondria in primary striatal neurons that had been transiently transfected with wild-type (GFP-23Q-208) or mutant (GFP-130Q-208) N-terminal 208 aa of htt fused to GFP. The size of GFP-130Q-208 fusion protein is similar to N-terminal htt fragments (∼500 aa) seen in the mitochondria isolated from HD knock-in mice, and use of the GFP-tagged proteins allowed us to test the effects of htt fragments on mitochondrial transport in live neurons. To visualize mitochondrial movement, neurons were cotransfected with DsRed2Mito fused to a mitochondrial targeting sequence. To detect the effects of soluble but not aggregated htt, only neurons without visible aggregates were imaged. Representative images of cotransfected striatal neurons (Fig. 5A,B; supplemental Fig. 1, available at www.jneurosci.org as supplemental material) demonstrate the diffuse distribution of GFP-htt proteins throughout the cytoplasm and processes. Examples of tracked fast-moving mitochondria from transfected neurons are presented in the series of images below (Fig. 5C,D) (full 5 min time series included as supplemental movies 1 and 2, available at www.jneurosci.org as supplemental material). As indicated, mitochondria that moved anterogradely (white triangles) or retrogradely (yellow arrow) in GFP-23Q-208-transfected neurons tended to travel longer distances over shorter periods of time than those in GFP-130Q-208-transfected neurons.
Figure 5.
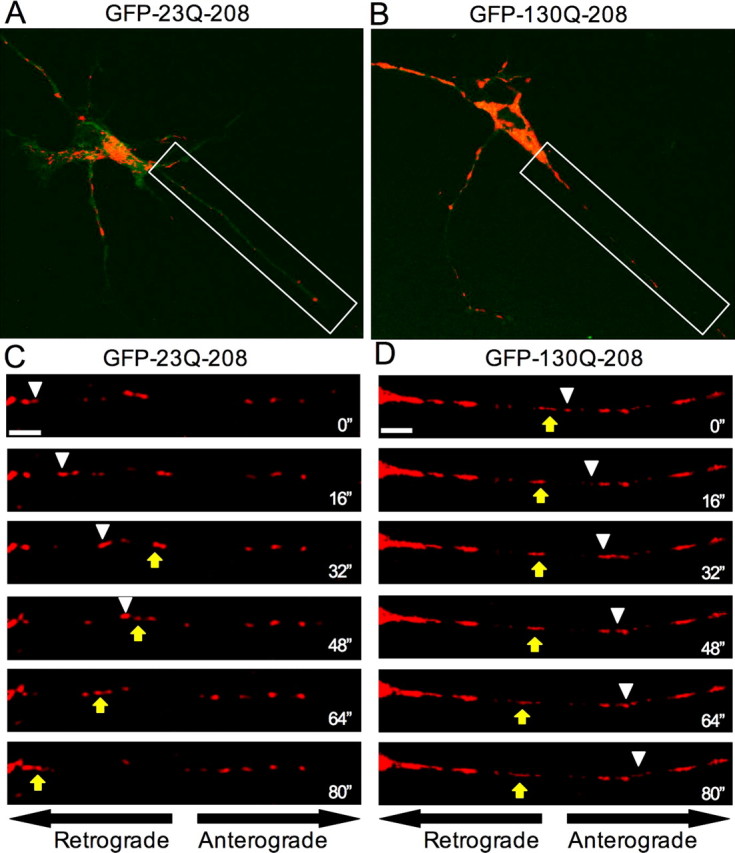
Soluble N-terminal mutant htt impairs mitochondrial trafficking in neurons. A, B, Representative images of DsRed2Mito and GFP-htt expression in striatal neurons transiently transfected with GFP-23Q-208 (A) or GFP-130Q-208 (B). Only neurons showing diffuse htt without visible aggregates were selected for analysis of mitochondrial transport. C, D, White framed rectangles highlight the region presented in the time-series panels below (C, D) and in the full 5 min sequences (supplemental Movies 1, 2, available at www.jneurosci.org as supplemental material). Both anterogradely (white arrowheads) and retrogradely (yellow arrows) moving mitochondria tended to travel longer distances over shorter periods of time in neurons expressing wild-type N-terminal htt fragments compared with those in neurons expressing soluble mutant htt N-terminal fragment. Scale bars, 5 μm.
Next, we quantified the velocity of moving mitochondria in transfected striatal neurons that expressed GFP-tagged mutant htt. The velocity was consistently reduced in the presence of soluble GFP-130Q-208 htt (Fig. 6A). To further examine the direct effect of N-terminal htt on mitochondrial function, we transfected 23Q-508 or 120Q-508 htt into cultured primary neurons. These htt fragments were not tagged with GFP such that the sequences and size of mutant htt (120Q-508) are very close to N-terminal mutant htt fragments associated with mitochondria in HD mouse brain (Fig. 2C). To ensure that DsRed2Mito-positive neurons expressed transfected htt, we cotransfected DsRed2Mito and htt at a 1:100 ratio (DsRed2Mito:htt). A significant decrease in the velocity of moving DsRed-labeled mitochondria was also seen in 120Q-508 transfected neurons (Fig. 6B). We next chose to examine mitochondrial transport in cultured striatal neurons from HD knock-in mice in which mutant htt is expressed at the endogenous level. Significant impairment of mitochondrial trafficking in HD knock-in neurons was observed (Fig. 6C), although the degree of impairment was not as severe as that seen in neurons transfected with N-terminal mutant htt. Full analysis of transport parameters is included in supplemental Table 1 (available at www.jneurosci.org as supplemental material). Importantly, the transport deficit in knock-in neurons was more pronounced in neurons homozygous for the HD mutation, suggesting that this deficit is dependent on the dosage of mutant htt. Indeed, immunoblots of the HD knock-in neurons with 1C2 revealed higher levels of N-terminal mutant htt fragments in homozygous HD knock-in neurons than in heterozygous neurons (Fig. 6D). This result also suggests that the degree of mitochondrial trafficking impairment is associated with the amount of N-terminal mutant htt fragments produced by HD knock-in neurons. Together, these findings support the hypothesis that soluble N-terminal mutant htt fragments can directly inhibit the trafficking of mitochondria in neurons.
Figure 6.

Mutant htt reduces the velocity of moving mitochondria in the processes of cultured striatal neurons. A, B, The velocity of moving mitochondria in the processes of cultured striatal neurons that were transfected with GFP-htt-208 (A) or N-terminal htt-508 (B). C, The velocity of moving mitochondria in the processes of cultured striatal neurons of wild-type (7Q/7Q), heterozygous (150Q/7Q), and homozygous (150Q/150Q) HD knock-in mice. *p < 0.05 for 150Q/150Q versus 7Q/7Q or 150Q/7Q. Complete analysis of trafficking parameters is included in supplemental Table 1 (available at www.jneurosci.org as supplemental material). D, 1C2 immunoblotting analysis revealed that higher levels of N-terminal mutant htt fragments (inner bracket) are present in cultured 150Q/150Q neurons than in 150Q/7Q neurons. A nonspecific band (n.s.) in 7Q/7Q cultures indicates equivalent protein loading for each sample.
If mutant htt impairs the transport of mitochondrial in neuronal processes, we may see the reduced distribution of mitochondria in the processes of HD neurons. We first examined the effect of 120Q-508 on mitochondrial distribution in PC12 cells, which extend long neurites after treatment with NGF (nerve growth factor). Immunofluorescent labeling of the mitochondrial chaperone Hsp60 (Liou et al., 2005) was performed to assess the distribution mitochondria in neurites expressing either wild-type or mutant 508 aa htt. We observed a significant reduction in the number and density of Hsp60-positive mitochondria in neurites of 120Q-508-transfected PC12 cells compared with 23Q-508-transfected cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To substantiate this finding, we examined the distribution of DsRed-labeled mitochondria in transfected 120Q-508 primary neurons. Immunofluorescent double labeling confirmed that DsRed2-positive neurons also expressed transfected 23Q-508 or 120Q-508 (Fig. 7A). Importantly, there were reduced DsRed2Mito signals specifically in long neurites of 120Q-508-transfected neurons. Quantitative assessment of the number of DsRed-labeled mitochondria per 10 μm [1.61 ± 0.12 for 120Q-508 (n = 12) vs 2.06 ± 0.12 for 23Q-508 (n = 19); mean ± SEM; p = 0.021] and their occupancy (percentage) in the measured processes [25.05 ± 2.54% for 120Q-508 (n = 12) vs 41.03 ± 1.78% for 23Q-508 (n = 19); mean ± SEM; p = 0.00011] validated that both the number and relative occupancy of mitochondria in neuronal processes were significantly decreased in 120Q-508-transfected neurons compared with control 23Q-508-transfected neurons. Because intranuclear htt can alter gene expression essential to mitochondrial function (Cui et al., 2006; Weydt et al., 2006), we also tagged mutant htt with the nuclear localization sequence to direct transfected htt to the nucleus of cultured neurons. However, nuclear expression of mutant htt led to significant toxicity of cultured primary neurons and retraction of long neurites, preventing us from monitoring mitochondrial distribution or transport in the processes (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 7.

Mutant N-terminal htt fragments decrease the distribution of mitochondria in the processes of cultured neurons. Fluorescent images of cultured rat brain striatal neurons that expressed transfected htt (23Q-508 or 120Q-508, green) and DsRed2Mito (red). Arrowheads indicate neuronal processes. Quantitative assessment of the number of DsRed-labeled mitochondria per 10 μm and their occupancy (percentage) in the measured processes is described in the text.
Next we extended our electron microscopy studies to characterize mitochondria at nerve terminals of HD knock-in mice. We observed that some mitochondrial-like organelles were degenerated in association with mutant htt in both axonal and dendritic terminals of HD mice (Fig. 8A). This evidence suggests that age-related association of N-terminal mutant htt could impair the energetic capacity of mitochondria in neuronal processes. Impaired replacement of dysmorphic/dysfunctional mitochondria may lead to a reduced supply of ATP in nerve terminals. To test this hypothesis, we isolated synaptosomes from the cerebral cortex of HD knock-in mice and measured their ATP content. Although cytoplasmic ATP levels did not show significant differences between wild-type and HD knock-in mice, a significant decrease in ATP was found in the synaptosomal fraction from 14-month-old HD knock-in mice compared with age-matched wild-type mice (Fig. 8B). Importantly, the decrease in synaptic ATP levels was seen only in old animals and thus correlates with the age-dependent association of N-terminal mutant htt with mitochondria in HD knock-in mice (Figs. 2, 3).
Figure 8.

HD mouse brains show degenerated mitochondria in nerve terminals and decreased ATP level in synaptosomal fraction. A, EM48 immunogold labeling of the cerebral cortex of HD knock-in mice at the age of 12 months. Single arrows indicate synapses. Double arrows indicate degenerated, mitochondria-like organelles in axonal (left) and dendritic (right) terminals. Mutant htt is labeled by dark immunogold particles. Scale bars, 250 nm. B, ATP levels of cytoplasmic and synaptosomal fractions isolated from the brain cortex of wild-type (7Q/7Q) and homozygous HD knock-in (150Q/150Q) mice at 4 and 14 months of age. *p < 0.05 compared with 7Q/7Q.
Discussion
Previous studies have demonstrated dysfunction of mitochondria in various HD models, yet there remains a lack of biochemical evidence for the association of endogenous mutant htt with mitochondria in HD brain. Using an HD knock-in mouse model, we show that specific N-terminal fragments of mutant htt preferentially associate with mitochondria in vivo and that N-terminal mutant htt fragments affect the trafficking of mitochondria. The implications of our findings are severalfold. First, our observation that specific cytoplasmic forms of mutant htt associate with mitochondria provides a direct biochemical basis for the mitochondrial dysfunction observed in HD. We also show age-dependent accumulation of mutant htt on mitochondria that directly correlates with disease progression. Finally, our data indicate that mitochondrial function can be disrupted by soluble N-terminal mutant htt fragments independent of their nuclear accumulation or aggregation. Together, these findings suggest that abnormal interaction between mutant htt and mitochondria may represent a cytoplasmic pathological event that can serve as a therapeutic target for HD.
Htt is expressed ubiquitously in the brain, distributed widely throughout cells, and proteolytically processed to produce multiple N-terminal fragments (Gutekunst et al., 1998; Fryer and Zoghbi, 2006). PolyQ-containing N-terminal fragments are prone to aggregation and are associated with more aggressive disease progression in HD mouse models (Li and Li, 2006). Therefore, to evaluate the pathogenic mechanisms of mutant htt-induced neurotoxicity, it is essential to identify htt forms that affect cellular function. Although electron microscopy has demonstrated the localization of mutant htt to mitochondria in brain tissue (Panov et al., 2002; Yu et al., 2003), it is difficult to quantify data and identify different forms of htt with this method. Similarly, experiments on cultured cells using subcellular fractionation (Choo et al., 2004) and fluorescence imaging (Rockabrand et al., 2007) were limited by their in vitro nature, although they provided support for an intrinsic association between htt and mitochondria. Other studies investigating the direct effects of mutant htt on mitochondrial function have also been limited by their reliance on in vitro assays or exogenously expressed htt-fusion proteins (Panov et al., 2002; Choo et al., 2004; Puranam et al., 2006). Therefore, there remains a need for a more complete biochemical analysis of the interaction of mutant htt with mitochondria in vivo.
To this end, we incorporated a variety of techniques with a focus on HD knock-in mice to characterize the association of htt with mitochondria. HD knock-in mice express full-length mutant htt under the control of the endogenous mouse Hdh promoter (Lin et al., 2001). Because these mice do not show overt neurological pathology, they are a good model system for investigating early pathological events in the progression of HD. In addition, although full-length htt is expressed, subsequent proteolytic processing results in an age-related accumulation of multiple N-terminal fragments and their aggregation in brain tissue (Zhou et al., 2003), a phenomenon that is also observed in human HD patients (Kim et al., 2001). These features of HD knock-in mice more accurately reflect the human disease condition and avoid limitations imposed by overexpression of specific transgenic forms of mutant htt. Using subcellular fractionation of HD knock-in brain tissue, we found that intermediate fragments (∼500 aa) of mutant htt associate more readily with brain mitochondria than full-length htt. This association was confirmed in transfected cells that express different forms of htt. Interestingly, we observed an age-related increase in the association of N-terminal mutant htt fragments with mitochondria, which correlates with disease progression and supports an important role for this association in HD pathogenesis.
There are several unique findings in our study that deserve more extensive discussion. First, why does an intermediate form of mutant htt associate most readily with mitochondria? This preferred interaction cannot be explained by the presence of a unique binding sequence in these intermediate fragments, because full-length mutant htt displays a weaker association with mitochondria. In addition, recent evidence suggests that the first 17 aa of htt are required for localization of exon1 htt to mitochondria (Rockabrand et al., 2007). Thus, a more likely explanation is that a conformational change caused by the expanded polyQ domain in the context of these intermediate sized fragments influences their association with mitochondria.
The second issue is whether the interactions of N-terminal htt fragments with mitochondria are physiologically relevant to mitochondrial function. Decreasing htt expression is known to impair mitochondrial transport in cultured primary neurons (Trushina et al., 2004), but whether full-length or truncated htt plays a role in this transport remains unknown. There are no consistent estimates for the size of N-terminal htt fragments generated in the brain, and available antibodies do not reliably identify wild-type htt fragments in mouse brain. Although our study shows that transfected wild-type htt fragments associate with mitochondria in cultured cells, we have not been able to reveal stable fragments of wild-type htt in the brain or verify their in vivo association with isolated brain mitochondria using EM48 and other commercially available htt antibodies. It is possible that wild-type htt fragments are more rapidly cleared or less stable than mutant htt in the brain. If so, the association of stable mutant htt fragments with mitochondria would likely have a greater impact on mitochondrial function. Indeed, the association of mutant htt fragments with brain mitochondria increases in an age-dependent manner and correlates with disease progression.
Previous reports show that mutant htt can affect a variety of functional aspects of mitochondria (Panov et al., 2002; Trushina et al., 2004; Brustovetsky et al., 2005; Milakovic and Johnson, 2005; Benchoua et al., 2006; Chang et al., 2006; Oliveira et al., 2006, 2007). Impaired axonal trafficking is known to contribute to HD pathogenesis (Gunawardena et al., 2003; Szebenyi et al., 2003; Li and Li, 2006); however, it is still unclear which form of mutant htt (full-length vs truncated) is the primary cause of impaired axonal trafficking. Several mechanisms have been hypothesized to explain the inhibitory effect of polyQ expanded proteins on axonal transport, including direct blockage of axoplasmic flow by large aggregates, sequestration of soluble motor proteins by insoluble aggregates, and direct disruption of the formation of trafficking complexes and their binding to microtubules by mutant htt (Gunawardena et al., 2003; Szebenyi et al., 2003; Gauthier et al., 2004; Trushina et al., 2004, Chang et al., 2006). Our studies provide evidence for the direct and toxic effect of N-terminal mutant htt on mitochondria in the processes of neuronal cells. Trafficking deficits were observed in the absence of mutant htt aggregation, supporting the hypothesis that early impairment of axonal transport of mitochondria is likely caused by altered interactions of trafficking complexes. Because of unique conformational changes, intermediate mutant htt fragments may preferentially bind mitochondria without any change in the preference for binding trafficking proteins. On the other hand, other forms of htt that associate weakly with mitochondria may still maintain their strong associations with trafficking proteins. Together, these changes would reduce the overall association of trafficking proteins with mitochondria. In addition, we did not find that the total soluble pool of the trafficking proteins is reduced in HD knock-in mouse brain lysates. Instead, our observation of impaired mitochondrial trafficking and decreased ATP level in synaptosomes by mutant htt suggests that impaired trafficking of mitochondria in neuronal processes can decrease mitochondrial ATP supply in nerve terminals. Thus, the decreased trafficking of mitochondria highlights a specific and direct effect of mutant htt on mitochondrial function.
Footnotes
This work was supported by National Institutes of Health Grants AG19206 (X.-J.L.), NS41669 (S.L.), National Natural Science Foundation of China Grant 30225024 (H.L.), and by the Jim Newman Fund for Huntington's Disease Research through the Emory Huntington's Disease Center of Excellence (A.L.O.). We thank Gary Bassell for providing an antibody to MAP2 and Ranjita Betarbet for antibodies against SDH and synaptophysin.
References
- Benchoua A, Trioulier Y, Zala D, Gaillard MC, Lefort N, Dufour N, Saudou F, Elalouf JM, Hirsch E, Hantraye P, Deglon N, Brouillet E. Involvement of mitochondrial complex II defects in neuronal death produced by N-terminus fragment of mutated huntingtin. Mol Biol Cell. 2006;17:1652–1663. doi: 10.1091/mbc.E05-07-0607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, LaFrance R, Purl KJ, Brustovetsky T, Keene CD, Low WC, Dubinsky JM. Age-dependent changes in the calcium sensitivity of striatal mitochondria in mouse models of Huntington's Disease. J Neurochem. 2005;93:1361–1370. doi: 10.1111/j.1471-4159.2005.03036.x. [DOI] [PubMed] [Google Scholar]
- Chang DT, Rintoul GL, Pandipati S, Reynolds IJ. Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons. Neurobiol Dis. 2006;22:388–400. doi: 10.1016/j.nbd.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004;13:1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- Cornett J, Smith L, Friedman M, Shin JY, Li XJ, Li SH. Context-dependent dysregulation of transcription by mutant huntingtin. J Biol Chem. 2006;281:36198–36204. doi: 10.1074/jbc.M607839200. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- Fryer JD, Zoghbi HY. Huntingtin's critical cleavage. Nat Neurosci. 2006;9:1088–1089. doi: 10.1038/nn0906-1088. [DOI] [PubMed] [Google Scholar]
- Gafni J, Ellerby LM. Calpain activation in Huntington's disease. J Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia M, Bondada V, Geddes JW. Mitochondrial localization of mu-calpain. Biochem Biophys Res Commun. 2005;338:1241–1247. doi: 10.1016/j.bbrc.2005.10.081. [DOI] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Graham RK, Deng Y, Slow EJ, Haigh B, Bissada N, Lu G, Pearson J, Shehadeh J, Bertram L, Murphy Z, Warby SC, Doty CN, Roy S, Wellington CL, Leavitt BR, Raymond LA, Nicholson DW, Hayden MR. Cleavage at the caspase-6 site is required for neuronal dysfunction and degeneration due to mutant huntingtin. Cell. 2006;125:1179–1191. doi: 10.1016/j.cell.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Gunawardena S, Her LS, Brusch RG, Laymon RA, Niesman IR, Gordesky-Gold B, Sintasath L, Bonini NM, Goldstein LS. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron. 2003;40:25–40. doi: 10.1016/s0896-6273(03)00594-4. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Ferrante RJ, Li XJ, Hersch SM. The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J Neurosci. 1998;18:7674–7686. doi: 10.1523/JNEUROSCI.18-19-07674.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, Qin ZH, Aronin N, DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci USA. 2001;98:12784–12789. doi: 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wyman T, Yu ZX, Li SH, Li XJ. Abnormal association of mutant huntingtin with synaptic vesicles inhibits glutamate release. Hum Mol Genet. 2003;12:2021–2030. doi: 10.1093/hmg/ddg218. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ. Aggregation of N-terminal huntingtin is dependent on the length of its glutamine repeats. Hum Mol Genet. 1998;7:777–782. doi: 10.1093/hmg/7.5.777. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ. Multiple pathways contribute to the pathogenesis of Huntington disease. Mol Neurodegener. 2006;1:19. doi: 10.1186/1750-1326-1-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Li H, Torre ER, Li XJ. Expression of huntingtin-associated protein-1 in neuronal cells implicates a role in neuritic growth. Mol Cell Neurosci. 2000;16:168–183. doi: 10.1006/mcne.2000.0858. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Liou JY, Aleksic N, Chen SF, Han TJ, Shyue SK, Wu KK. Mitochondrial localization of cyclooxygenase-2 and calcium-independent phospholipase A2 in human cancer cells: implication in apoptosis resistance. Exp Cell Res. 2005;306:75–84. doi: 10.1016/j.yexcr.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Lunkes A, Lindenberg KS, Ben-Haiem L, Weber C, Devys D, Landwehrmeyer GB, Mandel JL, Trottier Y. Proteases acting on mutant huntingtin generate cleaved products that differentially build up cytoplasmic and nuclear inclusions. Mol Cell. 2002;10:259–269. doi: 10.1016/s1097-2765(02)00602-0. [DOI] [PubMed] [Google Scholar]
- Martin JB, Gusella JF. Huntington's disease. Pathogenesis and management. N Engl J Med. 1986;315:1267–1276. doi: 10.1056/NEJM198611133152006. [DOI] [PubMed] [Google Scholar]
- McGuire JR, Rong J, Li SH, Li XJ. Interaction of Huntingtin-associated protein-1 with kinesin light chain: implications in intracellular trafficking in neurons. J Biol Chem. 2006;281:3552–3559. doi: 10.1074/jbc.M509806200. [DOI] [PubMed] [Google Scholar]
- Milakovic T, Johnson GV. Mitochondrial respiration and ATP production are significantly impaired in striatal cells expressing mutant huntingtin. J Biol Chem. 2005;280:30773–30782. doi: 10.1074/jbc.M504749200. [DOI] [PubMed] [Google Scholar]
- Oliveira JM, Chen S, Almeida S, Riley R, Goncalves J, Oliveira CR, Hayden MR, Nicholls DG, Ellerby LM, Rego AC. Mitochondrial-dependent Ca2+ handling in Huntington's disease striatal cells: effect of histone deacetylase inhibitors. J Neurosci. 2006;26:11174–11186. doi: 10.1523/JNEUROSCI.3004-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira JM, Jekabsons MB, Chen S, Lin A, Rego AC, Goncalves J, Ellerby LM, Nicholls DG. Mitochondrial dysfunction in Huntington's disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem. 2007;101:241–249. doi: 10.1111/j.1471-4159.2006.04361.x. [DOI] [PubMed] [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Petrasch-Parwez E, Nguyen HP, Lobbecke-Schumacher M, Habbes HW, Wieczorek S, Riess O, Andres KH, Dermietzel R, Von Horsten S. Cellular and subcellular localization of Huntingtin aggregates in the brain of a rat transgenic for Huntington disease. J Comp Neurol. 2007;501:716–730. doi: 10.1002/cne.21272. [DOI] [PubMed] [Google Scholar]
- Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon RE, Gawinowicz MA, Zhao Y, Colman DR. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- Puranam KL, Wu G, Strittmatter WJ, Burke JR. Polyglutamine expansion inhibits respiration by increasing reactive oxygen species in isolated mitochondria. Biochem Biophys Res Commun. 2006;341:607–613. doi: 10.1016/j.bbrc.2006.01.007. [DOI] [PubMed] [Google Scholar]
- Rockabrand E, Slepko N, Pantalone A, Nukala VN, Kazantsev A, Marsh JL, Sullivan PG, Steffan JS, Sensi SL, Thompson LM. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet. 2007;16:61–77. doi: 10.1093/hmg/ddl440. [DOI] [PubMed] [Google Scholar]
- Shin JY, Fang ZH, Yu ZX, Wang CE, Li SH, Li XJ. Expression of mutant huntingtin in glial cells contributes to neuronal excitotoxicity. J Cell Biol. 2005;171:1001–1012. doi: 10.1083/jcb.200508072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims NR. Rapid isolation of metabolically active mitochondria from rat brain and subregions using Percoll density gradient centrifugation. J Neurochem. 1990;55:698–707. doi: 10.1111/j.1471-4159.1990.tb04189.x. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Morfini GA, Babcock A, Gould M, Selkoe K, Stenoien DL, Young M, Faber PW, MacDonald ME, McPhaul MJ, Brady ST. Neuropathogenic forms of huntingtin and androgen receptor inhibit fast axonal transport. Neuron. 2003;40:41–52. doi: 10.1016/s0896-6273(03)00569-5. [DOI] [PubMed] [Google Scholar]
- Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience. 2007;145:1233–1248. doi: 10.1016/j.neuroscience.2006.10.056. [DOI] [PubMed] [Google Scholar]
- Trushina E, Dyer RB, Badger JD, II, Ure D, Eide L, Tran DD, Vrieze BT, Legendre-Guillemin V, McPherson PS, Mandavilli BS, Van Houten B, Zeitlin S, McNiven M, Aebersold R, Hayden M, Parisi JE, Seeberg E, Dragatsis I, Doyle K, Bender A. Mutant huntingtin impairs axonal trafficking in mammalian neurons in vivo and in vitro. Mol Cell Biol. 2004;24:8195–8209. doi: 10.1128/MCB.24.18.8195-8209.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Wellington CL, Ellerby LM, Gutekunst CA, Rogers D, Warby S, Graham RK, Loubser O, van Raamsdonk J, Singaraja R, Yang YZ, Gafni J, Bredesen D, Hersch SM, Leavitt BR, Roy S, Nicholson DW, Hayden MR. Caspase cleavage of mutant huntingtin precedes neurodegeneration in Huntington's disease. J Neurosci. 2002;22:7862–7872. doi: 10.1523/JNEUROSCI.22-18-07862.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Cao F, Wang Z, Yu ZX, Nguyen HP, Evans J, Li SH, Li XJ. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J Cell Biol. 2003;163:109–118. doi: 10.1083/jcb.200306038. [DOI] [PMC free article] [PubMed] [Google Scholar]