

Abstract
 Carbocyclic nucleosides are of considerable interest for the development of new therapeutic agents. A key reaction in the preparation of many such nucleoside analogs is dihydroxylation of appropriately substituted cyclopentenes. While often considered a routine reaction, in this paper we report the dramatic influence of substituents on the facial selectivity of dihydroxylations. The substituted cyclopentene substrates are derived from acylnitroso cycloaddition reactions of cyclopentadiene, followed by N-O reduction and efficient enzymatic resolution. The results are directly utilized in a very efficient asymmetric synthesis of an antiviral carbocyclic nucleoside, noraristeromycin 5. Extensions towards synthesis of carbocyclic sinefungin 7 document the importance of realizing the substituent dependence of the dihydroxylation reaction.
Carbocyclic nucleosides are of considerable interest for the development of new therapeutic agents. A key reaction in the preparation of many such nucleoside analogs is dihydroxylation of appropriately substituted cyclopentenes. While often considered a routine reaction, in this paper we report the dramatic influence of substituents on the facial selectivity of dihydroxylations. The substituted cyclopentene substrates are derived from acylnitroso cycloaddition reactions of cyclopentadiene, followed by N-O reduction and efficient enzymatic resolution. The results are directly utilized in a very efficient asymmetric synthesis of an antiviral carbocyclic nucleoside, noraristeromycin 5. Extensions towards synthesis of carbocyclic sinefungin 7 document the importance of realizing the substituent dependence of the dihydroxylation reaction.
Infectious diseases are posing increasingly severe health risks, as evidenced by the recent SARS flu epidemic and the rapid spread of AIDS in developing countries. Accordingly, extensive research has been directed at finding effective therapeutic agents for the treatment of viral infections as well as cancers. To that end, carbocyclic nucleosides have received considerable attention.1 As with normal nucleosides, many carbocyclic nucleosides contain syn -2',3'-dihydroxyl groups. Most often this important functionality is introduced by osmium-mediated reactions of the corresponding cyclopentene precursors (eq 1). For steric reasons it might be anticipated that such syn-dihydroxylations would occur trans relative to the other substituents on the cyclopentene ring. However, the electrophilic nature of OsO4 may alter the facial selectivity of this reaction. Trost and others have reported on the competition of stereoelectronic and steric effects in related important dihydroxylation reactions.2 In this paper we report that the facial selectivity of key dihydroxylation reactions of cyclopentene precursors of carbocyclic nucleoside analogs depends markedly on the additional substituents. The results dramatically influence the efficiency of syntheses of several biologically important carbocyclic nucleosides, including carbocyclic sinefungin and noraristeromycin.
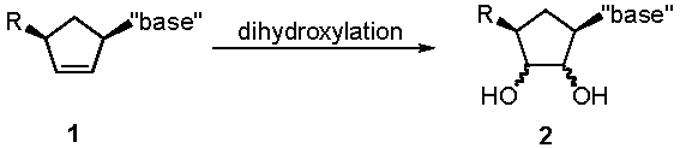 |
(eq 1) |
The natural carbocyclic nucleosides, neplanocin A 3 and aristeromycin 4 have potent antiviral activities (Figure 1).3 Aristeromycin (-)-4 is a carbocyclic analog of adenosine that terminates viral growth by inhibiting S-adenosyl-L-homocysteine (AdoHcy) hydrolase.4 However, the high cytotoxicity of aristeromycin, presumably caused by the metabolism of 4 to its 5'-phosphates, has greatly hindered its therapeutic application.5 In the search for a less toxic analog of aristeromycin, Schneller and coworkers found that the 5'-nor compound (±)-5 had improved antiviral activity with no cytotoxicity.6 Subsequently, the same workers found the (-)-enantiomer of 5 was more active than the (+)-enantiomer.7 Sinefungin 6 is a natural nucleoside first isolated from Streptomyces griseolus in 1973 and S. incarnatus in 1976.8 Since its isolation from natural sources, sinefungin has been synthesized by several groups.9 Preliminary bioassays showed that it inhibited the growth of several fungi8 and viruses10, and that it showed significant antiparasitic11 activity in vitro. There are two key features of the biological activity profile of sinefungin that stand out, one of which is its antiviral activity due to inhibition of methyltransferase.12 Chemotaxis, neurosecretions, membrane receptor interactions, DNA modification-restriction, gene expression and cellular differentiation are among the diverse processes that methylation of biomolecules affects.13 Sinefungin and derivatives have been tested against both vaccinia- and Newcastle disease virus- (guanine-7) methyltransferases.10 The elucidation of structure-activity-relationships for sinefungin and its derivatives was attempted with these studies. Several features were found to be necessary in order to retain methyltransferase inhibition: (1) the L configuration of the side chain (2) both terminal amino and carboxyl groups as well as a three-carbon linker between the S-atom and the terminal amino/carboxyl groups and (3) either the 2' or 3' hydroxyl groups on the ribosyl unit. Few modifications of the heterocyclic base were tolerated. Isosteric replacement of the ribosyl oxygen for a methylene unit was tolerated. Sinefungin has also been co-crystallized into the active site of thermus aquaticus DNA methyltransferase by Schluckebier.14 The structural requirements that appear to be necessary for biological activity from this study (based on binding within the active site) generally agree with the SAR study published by Pugh & Borchardt.10 One point of disparity between the studies is the necessity of both hydroxyl groups for biological activity.
Figure 1.

Although sinefungin showed strong bioactivity against viruses and parasites, it caused fatalities, probably resulting from its nephrotoxic side effects, when tested in vivo with larger mammals.15 Methyltransferases are important for many biological functions in humans and other animals. Inhibition of this important enzyme in order to affect some desirable therapeutic activity may be difficult because of the toxicity. Thus, the development of selective methyltransferase inhibitors continues to be of considerable interest.16 However, it has also been noted in earlier literature that sinefungin possesses antiviral activity that is, partly due to an alternate unknown biological mechanism.10 It may be prudent then to pursue sinefungin derivatives that are not good methyltransferase inhibitors in the hope that the other biological mechanism(s) responsible for its antiviral activity will lead to new antiviral therapeutic agents without toxic side effects to humans.
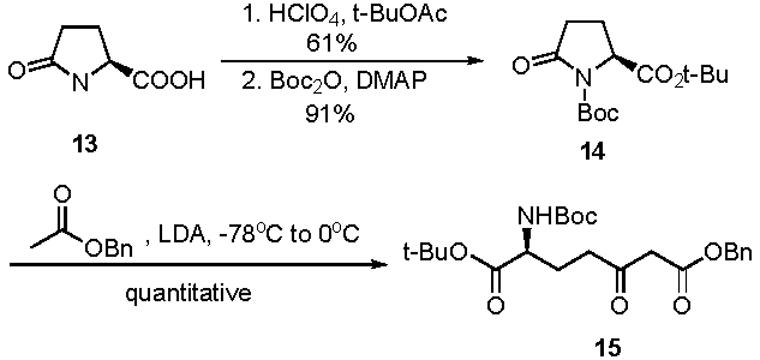
As a starting point for the synthesis of sinefungin analogs for biological testing, we set as a goal the synthesis of carbocyclic sinefungin 7 which contains an isosteric replacement of the ribosyl oxygen for a methylene unit. The synthesis of carboyclic sinefungin 7 is envisioned to arise from functionalized cyclopentene 8 after the key stereoselective dihydroxylation followed by decarboxylation, diastereoselective reductive amination and full deprotection (Scheme 1). Highly functionalized 8 would be available via a Pd(0)-mediated nucleophilic addition of β-keto ester 10 to the carbocyclic core 9 containing an allylic acetate. The carbocyclic core 9 would be synthesized from enantiomerically pure aminocyclopentenol derivative 11 and compound 11 is derived from a hetero Diels-Alder cycloadduct 12 after N-O bond reduction followed by enzymatic resolution as previously described.17 β-Keto ester side chain 10 can be synthesized from L-pyroglutamic acid 13.
Scheme 1.

The synthesis of the β-keto ester 15, a specific example of 10, is shown in Scheme 2. L-Pyroglutamic acid 13 was first reacted with t-butyl acetate in 70% perchloric acid and the resulting t-butyl pyroglutamate was treated with Boc2O to give fully protected pyroglutamate 14.18 The nucleophilic attack of carbonyl of compound 14 by the in situ generated anion of benzyl acetate followed by ring opening gave the linear β-keto ester 15 in quantitative yield.
Scheme 2.
Previously we have reported17 that enantiomerically pure compound 11 can be derived from nitroso-Diels-Alder cycloadduct after N-O bond reduction followed by enzymatic resolution. With compound 11 in hand, the adenine base and side chain 15 were installed sequentially as shown in Scheme 3. After compound 11 was treated with TFA, the resulting amine salt was coupled with 5-amino-4,6-dichloropyrimidine in n-butanol and NEt3 at 110 °C to give compound 16 in 79% yield for the two steps. The cyclized product 17 was obtained in 91% yield by the reaction of compound 16 with CH(OEt)3 in the presence of a catalytic amount of 10-camphorsulfonic acid. Side chain 15 was successfully added to the carbocyclic core 17 by an intermolecular Pd(0) catalyzed π-allyl nucleophilic reaction to give 18 in 80% yield. Treatment of compound 18 with Na2S•9H2O in aqueous solution gave decarboxylated product 19 in 71% yield.
Scheme 3.

The dihydroxylation reaction was first attempted on compound 18 (Scheme 4). The reaction was carried out in THF with a catalytic amount of OsO4 and excess NMO at room temperature for 30 minutes to give unexpected β-dihydroxylated product 20 in 70% yield. Using 2D NMR, attempts were made to determine the stereochemistry of the newly introduced diol relative to the other substituents of 20, but it was found that the compound was not stable in CDCl3 and it slowly converted to compound 21. Thus, compound 21 was fully characterized and its structure was confirmed by X-ray analysis.
Scheme 4.

Though examples of non-selective dihydroxylation reactions of substituted cyclopentenes have been discussed,2 exclusive β face dihydroxylation reaction of compound 18 to give β-dihydroxylated product 20 was unexpected since many reported syntheses of dihydroxylated carbocyclic nucleosides have employed similar conditions to give predominantly the opposite facial selectivity.19,20 Suspecting that the unusually high β-face selectivity might be due to this specific substrate, we decided to examine a series of substitutionally related cyclopentenes. Thus, the dihydroxylation reaction was next attempted on substrate 19, which was derived from decarboxylation of compound 18 (Scheme 4). The same facial selectivity was observed in this reaction and only β-dihydroxylated product 22 was obtained in 59% yield. Product 22 also was not stable and was slowly transformed to compound 23 upon standing.
Since both advanced intermediates 18 and 19 which had the core carbon structure of carbocyclic sinefungin gave only β-dihydroxylated products 20 and 22, respectively, the dihydroxylation reaction was attempted on the simpler intermediate 17 in which the side chain 15 was not yet installed (Scheme 5). The reaction was carried out under similar conditions and this time the desired α face dihydroxylated product was obtained. After the protection of the two hydroxyl groups, product 24 was obtained as a single stereoisomer in 64% yield over two steps. 1D NOE and 2D ROSEY spectra of compound 24 confirmed the stereochemistry of these two newly generated OH groups. The ultimate proof of exclusive α dihydroxylation of compound 17 came from the X-ray analysis of product 25, which was derived from the removal of the acetyl group from compound 24. As a consequence of the exclusive α face dihydroxylation selectivity of 17 to give 26, the asymmetric synthesis of noraristeromycin could be achieved by replacement of the chloro group with ammonia and removal of the acetate (Scheme 6). Aminolysis of 26 accomplished both steps and afforded (-)-5'-noraristeromycin 5 in 82% yield.
Scheme 5.

Scheme 6.

Compounds 18 and 19 that had side chains installed and compound 17 that had no side chain installed gave completely different face selected dihydroxylation products, which suggested that the side chain might play some role in the face selectivity of dihydroxylation reactions. To further test the effect of the side chain on the face selectivity of dihydroxylation reactions, compound 28 which had the side chain installed but an incompletely formed nucleobase, was synthesized for further studies (Scheme 7). The synthesis of compound 28 started from enantiomerically pure compound 11 which was directly accessible from our enzymatic resolution process. Compound 28 was obtained after the side chain 15 was added to compound 11 by Pd(0) catalyzed intermolecular nucleophilic addition followed by decarboxylation. The dihydroxylation reaction was carried out under similar conditions and only a mixture of hemiacetals 29 derived from β-dihydroxylated product were obtained in 87% yield.
Scheme 7.

It was clear that the side chain 15 had a significant influence on the facial selectivity of the dihydroxylation process. While more detailed studies are warranted, these studies further illustrate the need for considerable attention to the effects of peripheral substitution on the core carbocycle during syntheses of important dihydroxylated carbocyclic nucleosides. One possible explanation for the dramatic effects observed is that the compounds may adopt favored conformations in which large lipophilic side chains block the approach of the dihydroxylation reagent. Support for this is provided by the X-ray structure of compound 21 which indicated that one of the methyl groups from the NHBoc substituent was only 3.6 Å away from one of the carbons of the cyclopentene double bond. The investigation of the diastereoselectivity of related dihydroxylation reactions merits further study because of the importance of this reaction for the syntheses of carbocyclic nucleosides and other biologically useful compounds.
Experimental
(S)-1-Benzyl 7-tert-butyl 6-(tert-butoxycarbonyl)-2-((1S, 4R)-4-(6-chloro-9H-purin-9-yl)cyclopent-2-enyl)-3-oxoheptanedioate (18)
To a solution of keto ester 15 (1.09 g, 2.5 mmol) in THF (4 mL) was added NaH (0.10 g, 2.51 mmol) and the resultant mixture was stirred for 20 min. In a separate flask, palladium acetate (80 mg, 0.35 mmol) was dissolved in THF (2 mL), followed by addition of PPh3 (0.37 g, 1.43 mmol). The resulting canary yellow solution was stirred for 5 min, acetate 17 (0.50 g, 1.79 mmol) was added and the reaction was stirred for another 10 min. The enolate was transferred to the π-allyl complex via cannulation. This mixture was allowed to stir at 40°C for 2h while monitoring the progress of the reaction by TLC. After completion of the reaction, water (5 mL) was added and the biphasic mixture was extracted thoroughly with EtOAc (4×20 mL). The combined organic extracts were dried with NaSO4. After filtration and concentration, the residue was purified by column chromatography eluting with hexanes: EtOAc from 5:1 to 1:1 to afford 1.05 g (80%) compound 18 as a 1:1 mixture of diastereoisomers. 1H NMR (300 MHz, CDCl3) δ 1.31 (s, 9H), 1.39 (s, 18H), 1.41 (s, 9H), 1.52-2.13 (m, 6H), 2.42-2.70 (m, 4H), 2.85-3.03 (m, 2H), 3.55-3.57 (m, 2H), 3.66-3.71 (m, 2H), 4.04-4.06 (m, 2H), 4.96-4.99 (m, 2H), 5.12 (AB, J = 17.4, 12Hz, 1H), 5.124 (AB, J = 13.8, 13.8 Hz, 1H), 5.69-5.76 (m, 2H), 5.83-5.87 (m, 2H), 6.05-6.12 (m, 2H), 7.24-7.33 (m, 10H), 8.10 (s, 1H), 8.13 (s, 1H), 8.69 (s, 2H); 13C NMR (125 MHz, CDCl3) δ 26.2, 26.8, 27.75, 27.78, 28.0, 28.1, 35.6, 36.3, 38.0, 38.6, 43.5, 43.8, 52.5, 53.1, 52.5, 53.1, 59.9, 62.4, 62.6, 67.3, 79.5, 79.6, 82.0, 128.3, 128.4, 128.5, 128.6, 129.0, 129.2, 131.7, 134.66, 134.73, 138.2, 138.5, 143.6, 150.65, 150.68, 151.3, 151.5, 155.3, 155.4, 167.8, 167.9, 171.05, 171.13, 202.1, 202.5; IR (neat) ν 3332, 2886, 1667, 1641, 1245 cm-1; MS (m/e, rel. int.) 654 (25), 326 (20), 189 (25), 136 (100); HRMS calcd. for C33H4135Cl N5O7 (M+H)+: 654.2695, found 654.2670.
(S) -tert-Butyl 2-(tert-butoxycarbonyl)-6-((1R, 4R)-4-(6-chloro-9H-purin-9-yl)cyclopent-2-enyl)-5-oxohexanoate (19)
To a solution of compound 18 (0.37 g, 0.57 mmol) in THF (3 mL)-H2O (3 mL) was added Na2S•9H2O (1.1 g, 4.53 mmol). The mixture was stirred for 60 h and extracted with EtOAc. The combined organic layers were dried over Na2SO4. After filtration and concentration, the residue was purified by column chromatography eluting with 2:3 to 1:2 hexanes: EtOAc to afford 0.21 g (71%) of compound 19 as a colorless oil. [α]D = +3.4° (c = 0.45, CHCl3). 1H NMR (300 MHz, CDCl3) δ 1.37 (s, 9H), 1.42 (s, 9H), 1.50-1.59 (m, 1H), 1.74-1.79 (m, 1H), 2.08-2.13 (m, 1H), 2.37-2.75 (m, 5H), 3.02 (dt, J = 13.8, 8.4 Hz, 1H), 3.26-3.31 (m, 1H), 5.05 (d, J = 7.8 Hz, 1H), 5.73-5.78 (m, 1H), 5.85-5.88 (m, 1H), 6.17-6.21 (m, 1H), 8.15 (s, 1H), 8.72 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 26.8, 27.9, 28.3, 38.5, 38.6, 40.0, 48.0, 53.1, 60.5, 79.7, 82.1, 127.8, 131.9, 141.1, 143.4, 150.9, 151.5, 151.7, 155.4, 171.4, 207.6; IR (CH2Cl2) ν 2980, 2927, 1713, 1590, 158, 1480, 1367, 1260 cm-1; HRMS calcd. for C25H3535ClN5O5 (M+H)+: 520.2327, found 520.2347.
(3aR,5R,6R,6aS)-Benzyl 2-((S)-4-tert-butoxy-3-(tert-butoxycarbonyl)-4-oxobutyl)-5-(6-chloro-9H-purin-9-yl)-6-hydroxy-4,5,6,6a-tetrahydro-3aH-cyclopenta[b]furan-3-carboxylate (21)
To a solution of compound 18 (66 mg, 0.1 mmol) in THF (1 mL) was added NMO (24 mg, 0.2 mmol) followed by an OsO4 solution in 2-methyl-2-propanol (0.13 mL, 0.01 mmol) dropwise. The mixture was stirred at room temperature for 4 h and the reaction was monitored by TLC. The reaction was quenched with 10% Na2S2O5 solution (2 mL) and the mixture was stirred for 10 min. It was diluted with H2O, extracted with EtOAc. The organic layer was washed with H2O and brine and dried over Na2SO4. After filtration and concentration, the residue was purified by column chromatography eluting with 1:1 hexanes: EtOAc to afford 48 mg (70%) of compound 20 as an oil. Compound 20 was not stable and it was slowly transformed to compound 21. Compound 21 was characterized. Mp=118-120°C. 1H NMR (500 MHz, CDCl3) δ 1.41 (s, 9H), 1.46 (s, 9H), 1.89-1.96 (m, 1H), 2.14-2.21 (m, 1H), 2.43-2.49 (m, 1H), 2.51-2.59 (m, 1H), 2.65-2.70 (m, 1H), 3.16-3.21 (m, 1H), 3.72-3.78 (m, 1H), 4.24-4.28 (m, 1H), 4.48 (t, J = 4 Hz, 1H), 4.95-5.00 (m, 1H), 5.09-5.24 (m, 4H), 5.28-5.30 (m, 1H), 7.32-7.35 (m, 5H), 8.62 (s, 1H), 8.73 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 24.1, 27.9, 28.3, 29.8, 36.5, 43.0, 52.7, 55.0, 65.7, 71.8, 80.5, 82.5, 85.6, 107.3, 127.9, 128.1, 128.6, 128.7, 136.2, 145.8, 150.7, 151.6, 151.9, 155.9, 164.8, 171.5, 171.6; IR (neat) ν 3332, 2979, 1702, 1639, 1591, 1565, 1368, 1153, 755 cm-1; HRMS calcd. for C33H4135ClN5O8 (M+H)+: 670.2644, found 670.2690. The structure of this compound was confirmed by X-ray analysis.
(S)-tert-Butyl 2-(tert-butoxycarbonyl)-4-(3aR,5R,6R,6aS)-5-(6-chloro-9H-purin-9-yl)-2,6-dihydroxy-hexahydro-2H-cyclopenta[b]furan-2-yl)butanoate (23)
The compound was prepared using the same procedure for the synthesis of compound 21. 1H NMR (300 MHz, CDCl3) δ 1.41 (s, 9H), 1.42 (s, 9H), 1.44 (s, 9H), 1.45(s, 9H), 1.84-2.61 (m, 14H), 2.81-2.93 (m, 1H), 3.06 (dd, J = 13.5, 9.9 Hz, 1H), 4.16-4.27 (m, 4H), 4.51 (dd, J = 9.9, 3.9 Hz, 1H), 4.88-4.97 (m, 2H), 8.50 (s, 1H), 8.67 (s, 1H), 8.68 (s, 1H), 8.82 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 26.0, 26.7, 27.9(2C), 28.21, 28.24, 35.2, 36.3, 37.9, 38.0, 39.3, 40.3, 40.6, 43.2, 57.9, 58.1, 60.9, 61.6, 70.1, 71.3, 80.5, 81.1, 81.2, 81.4, 81.5, 86.1, 105.2, 105.4, 131.1(2C), 145.5, 146.0, 150.56, 150.63, 151.4, 151.6, 151.7, 151.8, 152.8, 153.1, 171.2, 171.7; IR (neat, CH2Cl2) ν 3361, 2978, 2934, 1740, 1697, 1591, 1368, 1158 cm-1; HRMS (FAB) calcd. for C25H3535ClN5O6 (M-H2O), 536.2276, found 536.2270.
(3aR,5R,6R,6aS)-6-(6-Chloro-9H-purin-9-yl)-2,2-dimethyl-tetrahydro-3aH-cyclopenta[d][1,3]dioxol-4-yl acetate (24)
To a solution of compound 17 (0.24 g, 0.86 mmol) in THF (8 mL) was added NMO (0.2 g, 1.72 mmol) followed by OsO4 solution in 2-methyl-2-propanol (1.08 mL, 0.086 mmol) dropwise. The mixture was stirred at room temperature for 1 h and was monitored by TLC. The reaction was quenched with 10% Na2S2O5 solution (4 mL) and the mixture was stirred for 10 min. It was diluted with H2O, extracted with EtOAc. The organic layer was washed with H2O and brine and dried over Na2SO4. After filtration and concentration, the residue was dissolved in 2,2-dimethoxypropane followed by pTSA•H2O (16 mg, 0.086 mmol). The reaction was stirred at room temperature overnight. The reaction was quenched with NaHCO3 solution and extracted with EtOAc. The organic layer was washed with H2O and brine and dried over Na2SO4. After filtration and concentration, the residue was purified by column chromatography eluting with 1:1 hexanes: EtOAc to afford 0.13 g (64% for two steps) of compound 24 as a colorless oil. [α]D = - 8.0° (c = 0.5, CHCl3). 1H NMR (300 MHz, CDCl3) δ 1.32 (s, 3H), 1.54 (s, 3H), 2.02 (s, 3H), 2.39-2.44 (m, 1H), 2.85-2.95 (m, 1H), 4.77-4.79 (m, 1H), 5.07-5.10 (m, 2H), 5.28-5.30 (m, 1H), 8.22 (s, 1H), 8.77 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 20.9, 24.2, 26.5, 34.5, 61.2, 77.9, 84.1, 84.2, 112.6, 131.8, 143.9, 151.7, 152.0, 169.4; IR (CH2Cl2) ν 1746, 1591, 1562, 1235 cm-1; HRMS calcd. for C15H1835ClN4O4 (M+H)+: 353.1017, found 353.1015.
(3aR,5R,6R,6aS)-6-(6-Chloro-9H-purin-9-yl)-2,2-dimethyl-tetrahydro-3aH-cyclopenta[d][1,3]dioxol-4-ol (25)
To a solution of compound 24 (0.18 g, 0.51 mmol) in CH3OH (5 mL) was added Bu2SnO (13 mg, 0.051 mmol). The mixture was stirred at 70°C overnight under an Ar atmosphere. The solvent was removed under reduced pressure and the residue was purified by column chromatography eluting with 1:3 hexanes: EtOAc to afford 135 mg (85%) of compound 25 as a white crystalline solid. Mp=152-153°C. [α]D = - 20° (c = 0.75, CH3OH). 1H NMR (300 MHz, CDCl3) δ 1.31 (s, 3H), 1.52 (s, 3H), 2.22-2.28 (m, 1H), 2.85-2.95 (m, 1H), 3.59 (bs, 1H), 4.53-4.54 (m, 1H), 4.71 (d, J = 5.7 Hz, 1H), 4.91 (d, J = 5.7 Hz, 1H), 5.06-5.09 (m, 1H), 8.49 (s, 1H), 8.76 (s, 1H); 13C NMR (75 MHz, CDCl3) δ 24.2, 26.7, 36.9, 62.6, 76.1, 86.2, 87.0, 111.8, 131.5, 145.8, 151.0, 151.3, 151.8; IR (CH2Cl2) ν 3339, 1592, 1562, 1338, 1211 cm-1; HRMS calcd. For C13H1635ClN4O3 (M+H)+: 311.0911, found 311.0926.
Acetic acid 4-(6-chloro-purin-9-yl)-2,3-dihydroxy-cyclopentyl Ester (26)
To a solution of compound 17 (127 mg, 0.456 mmol) in t-BuOH-H2O (1:1, 4 mL) was added K3Fe(CN)6 (570 mg, 1.7 mmol), K2CO3 (328 mg, 2.37 mmol) followed by an OsO4 solution in 2-methyl-2-propanol (99 μL, 0.009 mmol) dropwise. The mixture was stirred at room temperature for 44 h and was monitored by TLC. The reaction was quenched with 10% Na2S2O5 solution (4 mL) and the mixture was stirred for 10 min. It was diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O and brine and dried over Na2SO4. After filtration, removal of solvent under reduced pressure gave 106 mg (74%) of 26 as a colorless oil. [α]D = + 53.2° (c = 3.6, MeOH). 1H NMR (500 MHz, CDCl3) δ 2.14 (s, 3H), 2.39 (m, 1H), 3.07 (m, 1H), 3.39 (m, 1H), 4.30 (m, 1H), 4.56 (d, J = 5 Hz, 1H), 4.66 (m, 1H), 4.85 (q, J = 8 Hz, 1H), 5.10 (m, 1H), 8.18 (s, 1H), 8.76 (s, 1H); 13C NMR (125 MHz, CDCl3) δ 20.9, 32.5, 60.7, 74.8, 75.2, 77.1, 132.2, 144.3, 151.5, 151.6, 151.7, 170.9; HRMS calcd. for C12H1435ClN4O4 (M+H)+: 313.0704, found 313.0681.
(-)-5' Noraristeromycin (5)
A solution of compound 26 (21.8 mg, 0.0698 mmol) in MeOH (1 mL) in a sealable tube was cooled to -78°C. Liquid ammonia (1 mL) was added via a condenser. The clear solution was then sealed and heated to 43°C for 2 d. Solvent was removed under reduced pressure. Flash chromatography (CHCl3: MeOH, 10:1, 2:3) gave 14.4 mg (82%) of compound 5 as a white solid. [α]D = -45.7° (c = 0.18, DMF) [lit.21 [α]D = -40.7° (c = 1.16, DMF)]. Mp = 251-252°C [lit.21 250°C (dec.)]; 1H NMR (500 MHz, DMSO-d6) δ 1.80 (m, 1H), 2.59 (m, 1H), 3.76 (m, 1H), 3.89 (m, 1H), 4.50 (m, 1H), 4.68 (q, J = 9.5 Hz, 1H), 4.94 (d, J = 3 Hz, 1H), 5.07 (d, J = 6.5 Hz, 1H), 5.41 (d, J = 4.5 Hz, 1H), 7.24 (bs, 2H), 8.11 (s, 1H), 8.18 (s, 1H); 13C NMR (125 MHz, DMSO-d6) δ 36.6, 58.4, 73.7, 75.4, 76.8, 119.2, 140.0, 149.4, 152.0, 156.0; HRMS calcd. for C10H14N5O3 (M+H)+: 252.1097, found 252.1078.
Supplementary Material
Acknowledgment
The authors thank Eli Lilly and Co. and NIH (R01 GM68012) for the support of this research. We acknowledge Dr. Jaroslav Zajicek for NMR assistance and Dr. Bruce Noll for X-ray analysis as well as Nonka Sevova for Mass Spectroscopy. We thank Theresa Bollinger for help with manuscript preparation.
Footnotes
Supporting Information Available: General methods and procedures for compounds 15, 16, 17, 28, 29 and spectral data for all new compounds, and details for the crystal structures of 21 and 25. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).For a discussion of inhibitors of enzymes involved with purine and pyrimidine metabolism, see: Hobbs JB. In: Comprehensive Medicinal Chemistry. Hansch C, editor. vol. 2. Pergamon; New York: 1990. p. 306..
- (2).(a) Trost BM, Madsen R, Guile SD, Brown B. J. Am. Chem. Soc. 2000;122:5947. [Google Scholar]; (b) Palmer CF, McCague R, Ruecroft G, Savage S, Taylor SJC, Ries C. Tetrahedron Lett. 1996;37:4601. [Google Scholar]; (c) Li F, Brogan JB, Gage JL, Zhang D, Miller MJ. J. Org. Chem. 2004;69:4538. doi: 10.1021/jo0496796. [DOI] [PubMed] [Google Scholar]
- (3).Goodchild J. In: Topics in Antibiotic Chemistry. Sammes PG, editor. vol. 6B. Wiley; New York: 1982. p. 99. [Google Scholar]
- (4).(a) De Clercq E. Biochem. Pharmacol. 1987;36:2567. doi: 10.1016/0006-2952(87)90533-8. [DOI] [PubMed] [Google Scholar]; (b) Cools M, De Clercq E. Biochem. Pharmacol. 1989;38:1061. doi: 10.1016/0006-2952(89)90249-9. [DOI] [PubMed] [Google Scholar]
- (5).Bennett LL, Jr., Brockman RW, Rose LM, Allan PW, Shaddix SC, Shealy YF, Clayton JD. Mol. Pharmacol. 1985;27:666. [PubMed] [Google Scholar]
- (6).Patil SD, Schneller SW, Hosoya M, Snoeck R, Andrei G, Balzarini J, De Clercq E. J. Med. Chem. 1992;35:3372. doi: 10.1021/jm00096a012. [DOI] [PubMed] [Google Scholar]
- (7).Siddiqi SM, Chen X, Schneller SW, Ikeda S, Snoeck R, Andrei G, Balzarini J, De Clercq E. J. Med. Chem. 1994;37:551. doi: 10.1021/jm00030a014. [DOI] [PubMed] [Google Scholar]
- (8).(a) Hamill RL, Nagarajan R. US Patent. 4 087 603.; (b) Florent J, Lunel J, Mancy D. US Patent. 4 189 349.
- (9).(a) Ghosh AK, Liu W. J. Org. Chem. 1997;62:2299. doi: 10.1021/jo964030p. [DOI] [PubMed] [Google Scholar]; (b) Ghosh AK, Lu W. J. Org. Chem. 1996;61:6175. doi: 10.1021/jo960670g. [DOI] [PubMed] [Google Scholar]; (c) Peterli-Roth P, Maguire MP, Leon E, Rapoport H. J. Org. Chem. 1994;59:4186. [Google Scholar]; (d) Mouna AM, Blanchard P, Fourrey J-L, Robert-Gero M. Tetrahedron Lett. 1990;31:7003. [Google Scholar]; (e) Maguire MP, Feldman PL, Rapoport H. J. Org. Chem. 1990;55:948. [Google Scholar]; (f) Buchanan JG, Flinn A, Mundill PHC, Wightman RH. Nucleosides Nucleotides. 1986;5:313. [Google Scholar]; (g) Geze M, Blanchard P, Fourrey JL, Robert-Gero M. J. Am. Chem. Soc. 1983;105:7638. [Google Scholar]; (h) Mock GA, Moffatt JG. Nucl. Acids Res. 1982;10:6223. doi: 10.1093/nar/10.20.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Chang C-D, Coward JK. J. Med. Chem. 1976;29:684. doi: 10.1021/jm00227a021. [DOI] [PubMed] [Google Scholar]
- (10).Pugh CSG, Borchardt RT. Biochemistry. 1982;21:1535. doi: 10.1021/bi00536a011. [DOI] [PubMed] [Google Scholar]
- (11).Trager W, Tershakovac M, Chiang PK, Cantoni GL. Exp. Parasitol. 1980;50:83. doi: 10.1016/0014-4894(80)90010-7. [DOI] [PubMed] [Google Scholar]
- (12).Blanchard P, Dodic N, Fourrey JL, Geze M, Lawrence F, Malina H, Paolantonacci P, Vedel M, Tempete C, Robert-Gero M, Lederer E. In: Biological Methylation and Drug Design. Borchardt RT, Creveling CR, Ueland PM, editors. Humana Press; New Jersey: 1986. p. 435. [Google Scholar]
- (13).Lawrence F, Robert-Gero M. J. Euk. Microbiol. 1993;40:581. doi: 10.1111/j.1550-7408.1993.tb06111.x. [DOI] [PubMed] [Google Scholar]
- (14).Schluckebier G, Kozak M, Bleimling N, Weinhold E, Saenger W. J. Mol. Biol. 1997;265:56. doi: 10.1006/jmbi.1996.0711. [DOI] [PubMed] [Google Scholar]
- (15).Zweygarth E, Schillinger D, Kaufmann W, Rottcher D. Trop. Med. Parasit. 1986;37:255. [PubMed] [Google Scholar]
- (16).(a) Wahnon DC, Shier VK, Benkovic SJ. J. Am. Chem. Soc. 2001;123:976. doi: 10.1021/ja003285o. [DOI] [PubMed] [Google Scholar]; (b) Van Lanen SG, Iwata-Reuyl D. Biochemistry. 2003;42:5312. doi: 10.1021/bi034197u. [DOI] [PubMed] [Google Scholar]
- (17).(a) Jiang MX-W, Warshakoon NC, Miller MJ. J. Org. Chem. 2005;70:2824. doi: 10.1021/jo0484070. [DOI] [PubMed] [Google Scholar]; (b) Mulvihill MJ, Gage JL, Miller MJ. J. Org. Chem. 1998;63:3357. [Google Scholar]
- (18).(a) Kolasa T, Miller MJ. J. Org. Chem. 1990;55:1711. [Google Scholar]; (b) August RA, Khan JA, Moody CM, Young DW. J. Chem. Soc., Perkin Trans. 1. 1996:507. [Google Scholar]
- (19).Marquez VE, Lim MI. Med. Res. Rev. 1986;6:1. doi: 10.1002/med.2610060102. [DOI] [PubMed] [Google Scholar]
- (20).Crimmins MT. Tetrahedron. 1998;54:9229. [Google Scholar]
- (21).Kitade Y, Kozaki A, Miwa T, Nakanishi M. Tetrahedron. 2002;58:1271. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.