

Abstract
Non-human primates are valuable for modelling human disorders and for developing therapeutic strategies; however, little work has been reported in establishing transgenic non-human primate models of human diseases. Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder characterized by motor impairment, cognitive deterioration and psychiatric disturbances followed by death within 10-15 years of the onset of the symptoms1-4. HD is caused by the expansion of cytosineadenine-guanine (CAG, translated into glutamine) trinucleotide repeats in the first exon of the human huntingtin (HTT) gene5. Mutant HTT with expanded polyglutamine (polyQ) is widely expressed in the brain and peripheral tissues2,6, but causes selective neurodegeneration that is most prominent in the striatum and cortex of the brain. Although rodent models of HD have been developed, these models do not satisfactorily parallel the brain changes and behavioural features observed in HD patients. Because of the close physiological7, neurological and genetic similarities8,9 between humans and higher primates, monkeys can serve as very useful models for understanding human physiology and diseases10,11. Here we report our progress in developing a transgenic model of HD in a rhesus macaque that expresses polyglutamine-expanded HTT. Hallmark features of HD, including nuclear inclusions and neuropil aggregates, were observed in the brains of the HD transgenic monkeys. Additionally, the transgenic monkeys showed important clinical features of HD, including dystonia and chorea. A transgenic HD monkey model may open the way to understanding the underlying biology of HD better, and to the development of potential therapies. Moreover, our data suggest that it will be feasible to generate valuable non-human primate models of HD and possibly other human genetic diseases.
We injected 130 mature rhesus oocytes with high titre lentiviruses expressing exon 1 of the human HTT gene with 84 CAG repeats (HTT-84Q; Fig. 1c) and lentiviruses expressing the green fluorescent protein (GFP) gene (Fig. 1c), under the control of the human polyubiquitin-C promoter, into the perivitelline space. After fertilization by intracytoplasmic sperm injection, 82% (89 out of 108) of the zygotes developed to the 4-8-cell stage and 30 embryos were transferred to eight surrogates. Of those surrogates, six pregnancies (75%; 6 out of 8) were established and five live newborns (22%, 5 out of 23; rHD-1, rHD-2, rHD-3, rHD-4 and rHD-5) were delivered at full term (Table 1). Among these five newborns, two sets of twins (rHD-1 and rHD-2; rHD-4 and rHD-5) were delivered by caesarean section, and a singleton (rHD-3) was delivered naturally.
Figure 1. Generation of a transgenic model in monkeys of HD.

a, b, The transgenic HD monkeys rHD-1 (left) and rHD-2 (right) are shown. Transmission light image (a) and fluorescent image (b) showing GFP expression in HD monkeys. c, Top panel: lentiviral vector that carries exon 1 of the HTT gene with 84 CAG repeats (pLVU-HTT-84Q). Bottom panel: lentiviral vector that carries the GFP gene (pLVU-G). Arrows indicate the positions of PCR primers; arrowheads denote restriction digest sites. Flap, HIV-flap sequence; GFP, green-fluorescent-protein gene; HTT, huntingtin gene; LTR, long terminal repeat; Ubi, ubiquitin promoter; WPRE, woodchuck post-transcriptional regulatory element. d, e, The presence of transgenes in HD monkeys was confirmed by PCR analysis using primer sets specifically for mutant HTT (top panels) and for the GFP gene (bottom panels). PCR of the cord (C) and placental (P) tissues of all HD monkeys (d), and PCR of different tissues collected from rHD-4 and rHD-5 (e). f, Expression of the transgenic mutant HTT was confirmed by western blot analysis using the placental tissues. Immunostaining was performed using mouse-monoclonal-mEM48 antibody (top panel) and an antibody against γ-tubulin (bottom panel).
Table 1.
Summary of transgenic HD monkeys
| Movement dysfunction |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| rHD | Sex | CAG repeats | HTT copy number | Expression of mutant HTT/GFP | Body weight ratio | Onset/ frequency | Dystonia | Chorea | Swallowing difficulty | Respiratory difficulty |
| F, female; M, male; ND, not determined. | ||||||||||
| 1 | M | 29 | 1 | +/+ | 2.85* | − | − | − | − | − |
| 2 | M | 83 | 1 | +/+ | 2.16* | 1 week/ sporadic | + | + | − | + |
| 3 | F | 84 | 2 | +/+ | 1.11† | 2-3 days/ persistent | ++++ | ++++ | ++++ | ++ |
| 4 | F | 27, 65 | 2 | +/+ | ND | At birth/ persistent | ++ | ++ | ND | ++++ |
| 5 | F | 88 | 4 | +/+ | ND | At birth/ ND | ND | ND | ND | ++++ |
The ratio of body weight at 24 weeks after birth to birth weight.
The ratio of body weight at 28 days after birth to birth weight.
All of the transgenic monkeys carried the transgenic mutant HTT and GFP genes. Fig. 1 shows two monkeys (rHD-1 and rHD-2, fraternal twins; Fig. 1a) expressing GFP at 4 weeks of age (Fig. 1b). Transgene integration was confirmed by polymerase chain reaction (PCR) (Fig. 1d, e) and Southern blot analysis (Supplementary Fig. 1). We estimated that rHD-1 and rHD-2 carried a mutant HTT gene in samples of umbilical cord, whereas placental tissue of extraembryonic origin from rHD-1 had two mutant HTT genes of different size (Fig. 1d). Two or more integration sites were identified in rHD-3, rHD-4 and rHD-5 (Table 1 and Supplementary Fig. 1), suggesting that these monkeys may have expressed the transgenic mutant HTT at a higher level and thus showed more severe phenotypes. Owing to the limited access of tissues from the live HD infants, detailed post-mortem analyses of transgene expression were focused on the HD monkeys rHD-4 and rHD-5.
Variations in the repeat length were found in the transgenic monkeys (Table 1). Specifically, rHD-1 carries 29 CAGs whereas the other four monkeys carried CAG repeats ranging from 27 to 88. Among these five newborns, one (rHD-3) survived for 1 month and two (rHD-4 and rHD-5) survived for less than a day. The latter two newborns had respiratory difficulties and showed signs of motor impairment soon after birth. Because of their early death we were unable to perform thorough testing of rHD-4 and rHD-5, and we could not establish whether their symptoms were caused by the mutant HTT gene. rHD-3 showed dystonia and chorea 2 days after birth; rHD-2 displayed mild involuntary motor movement that appeared sporadically at 1 week of age. As expected, rHD-1—which has CAGs in the normal range for non-HD humans—seems to be a normal, healthy monkey.
The expression of transgenic mutant HTT was confirmed by western blot and immunohistochemistry with mEM48, a monoclonal antibody whose reaction with human HTT is enhanced by polyQ expansion12. Western blotting of the placental tissues of all infants demonstrated the presence of oligomeric HTT at high molecular mass (.250 kDa) in the upper portion of a gradient polyacrylamide gel (Fig. 1f). The intensity of mEM48 immunostaining is determined by the polyQ length and expression levels of transgenic HTT. Given that rHD-1 and rHD-2 carry a single copy of transgenic HTT, the more intense mEM48 staining in rHD-2 placental tissue than in rHD-1 tissues supports the CAG repeat analysis showing that rHD-2 carries a larger repeat (83) than rHD-1 (29) (Fig. 1d and Table 1). Consistently, placental tissues from rHD-3, rHD-4 and rHD-5, which have more copy numbers of transgenic HTT with larger repeats (69-88), show much more intense mEM48 immunostaining. Thus, the levels of mutant HTT in the tissues seem to be associated with the early death of monkeys rHD-3, rHD-4 and rHD-5, and the severity of phenotypes seen in infants rHD-2 and rHD-3, whereas a short CAG repeat (29) in rHD-1 did not result in detectable phenotypes (Table 1).
Expression of mutant HTT was also determined by mEM48 western blotting of post-mortem peripheral (Fig. 2a, b) and brain tissues (Fig. 2c, d) of rHD-4 and rHD-5. As seen in western blots of the placental tissues (Fig. 1f), oligomeric mutant HTT is present in the peripheral tissues (Fig. 2a, b) and brain (Fig. 2c, d) of transgenic monkeys but not in non-transgenic or wild-type monkeys, indicating that the oligomeric HTT specifically represents transgenic mutant HTT. Variations in the extent of aggregation were observed in peripheral tissues (Fig. 2a, b), suggesting that aggregation of mutant HTT or its expression levels vary in different tissues. In contrast to peripheral tissues, different brain regions show equivalent levels or extent of transgenic mutant HTT (Fig. 2c, d).
Figure 2. Expression of mutant HTT in HD monkey peripheral tissues and brains.

a-d, mEM48 immunoblot of peripheral and brain tissues reveals high-molecular-mass oligomeric HTT (arrow) and soluble HTT products. The blot was also probed with an antibody to γ-tubulin as an internal control. Shown are immunoblots of peripheral tissues collected from monkey rHD-4 (a) and rHD-5 (b), and samples from different brain regions of monkey rHD-4 (c) and rHD-5 (d), with antibody to mEM48 (top panel) and γ-tubulin (bottom panel). WT, wild-type non-transgenic monkey.
Consistent with the western blotting analysis, immunohistochemistry with mEM48 and 1C2—antibodies specific to expanded polyQ domain—showed HTT aggregates or inclusions in the striatum and cortex of rHD-4 and rHD-5 (Fig. 3a, b). Transgenic HTT is widespread in the various brain regions but with no significant difference between regions. Expression of transgenic mutant HTT is extensive in the brain (Fig. 3a, b; upper panel; × 100). At higher magnification (Fig. 3a, b; lower panel; × 630), mutant HTT accumulates in the nuclei and also forms abundant small aggregates that do not seem to be in the cell body. Both antibodies illustrated neuropil aggregates13 located in an array manner, suggesting that they were originally localized in neuronal processes such as axons and dendrites14-19. The size of these neuropil aggregates can be larger than that of axons or dendrites (arrowhead in Fig. 3a, b); this is consistent with neuropil aggregates found in the brains of HD patients13,20 and in HD mouse models15-18 in which neuropil HTT aggregates are associated with axonal degeneration. However, Nissl staining did not reveal any obvious degeneration in the striatum of rHD-5 (data not shown).
Figure 3. Histopathology of HD monkey brain.
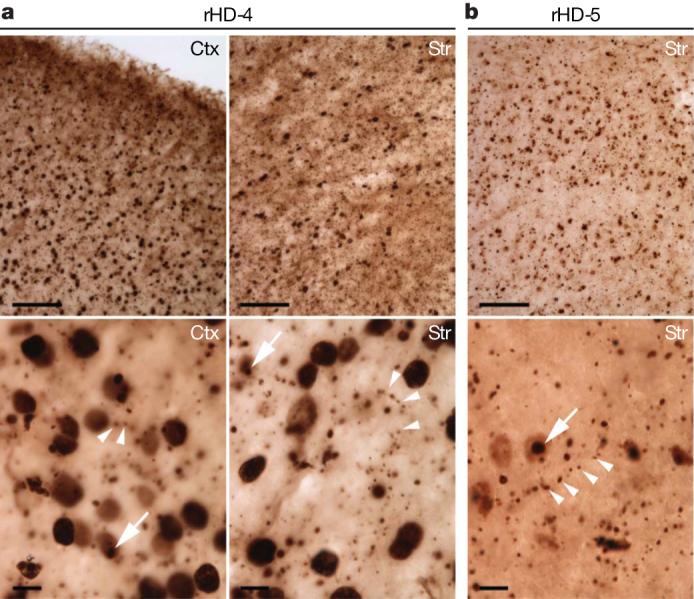
a, b, Brain sections of rHD-4 (a) and rHD-5 (b) were immunostained with mEM48 and 1C2, respectively. Low magnification (upper panels; scale bars, 100 mm) shows the abundant distribution of transgenic mutant HTT in the cortex (Ctx) and striatum (Str). High magnification (bottom panels; scale bars, 10 mm) demonstrates that transgenic mutant HTT is distributed in neuronal nuclei and forms neuropil aggregates (arrowheads). Nuclear inclusions (arrows) are evident in sections stained with mEM48 or 1C2.
Variable extents of motor dysfunction were observed in four (rHD-2, rHD-3, rHD-4 and rHD-5) of the five HD monkeys, although we were unable to examine rHD-4 and rHD-5 in detail before their death. rHD-2 and rHD-3 showed different degrees of difficulty in movement coordination and involuntary movement such as chorea and dystonia, whereas rHD-1 has no signs of clinical symptoms (Table 1). On the basis of our preliminary observations, the severity, frequency and onset of the involuntary movements seemed to depend on the length of the CAG repeats and the number of integration sites (Table 1). Monkey rHD-3 survived for 1 month and showed severe chorea, dystonia and difficulty in swallowing. At the time of writing, the monkeys rHD-1 and rHD-2 are 6 months old. Behavioural and cognitive testing and in-depth characterizations are underway; thorough testing, including brain imaging, will be performed periodically throughout development until adulthood. As there is no standard rating scale for HD in monkeys—especially for infants—our evaluation is based on the unified Huntington's disease rating scale, which has been modified for the use in infant monkeys. On the basis of our preliminary assessment using the Huntington's disease primate model rating scale, in which 80 is the highest score reflecting the most severe symptom, rHD-1 has the score of ‘0’ at 2 and 6 months, whereas rHD-2 has the scores 12 and 26, respectively. In addition, rHD-3 had the score of 60 at 1 month of age.
The early death of transgenic HD monkeys carrying a higher copy number of transgenes expressing a small amino-terminal HTT fragment supports the idea that N-terminal mutant HTT fragments are pathogenic. Although detailed and periodically repeated behavioural characterization of the surviving transgenic HD monkeys remains to be done, the infant HD monkeys already showed some features similar to those found in HD patients, including chorea and dystonia. The transgenic HD monkeys provide the opportunity for a wide range of behavioural and cognitive assessments that are identical to or close to the assessments used for human patients. Furthermore, the two male HD monkeys could serve as founders, and their semen can be collected throughout life non-invasively and cryopreserved. Thus, a cohort of HD monkeys could then be established using these semen and reproductive techniques in a reasonable time frame. Our progress in establishing a HD transgenic monkey also bodes well for the generation of non-human primate models of other important neurodegenerative diseases. Establishing such models is invaluable for understanding disease pathogenesis and for the development of early diagnostic and treatment strategies.
METHODS SUMMARY
Lentiviruses carrying exon 1 of the human HTT gene with 84 CAGs (HTT-84Q) and the GFP gene under the control of human polyubiquitin-C promoter were microinjected into the perivitelline space of monkey metaphase-II-arrested oocytes followed by in vitro fertilization, in vitro culture and embryo transfer into surrogate females. Transgenic status of HD monkeys was confirmed by PCR and Southern blot analyses on available tissues. The expression of the mutant HTT gene was confirmed by immunohistochemistry and western blot. The expression of GFP gene was visualized under a Sky-blue II epifluorescent light. Motor impairment was evaluated by the Huntington's diseases primate model rating scale.
Acknowledgements
We thank J. Fanton (deceased), M. Zelinski-Wooten, M. Sparman and D. Wolf for consultation, C. Lois for lentivirus backbone and C. Testa for critical review of the manuscript. We also thank F. Zhang, T. Caspary, S. Warren, J. Greene, T. Wichmann, M. Wilson, S. Chikazawa, K. Gould, L. Walker, K. Layug, E. Strobert, J. Else, J. Ksiazek, K. Strait, F. Stroud, J. Jenkins, J. Cohen, J. Pare, S. Jenkins, K. Paul, S. Lackey, J. Johnson-Ward, the veterinary staff, the animal resource and the endocrine core laboratory at the Yerkes National Primate Research Center. Acryline was provided by NICHD/NIH. All transgenic HD monkeys were housed under the guideline of the IACUC approved procedures and the support of YNPRC Division of Animal Resources. All newborn monkeys were closely monitored by the veterinary staff and infant care personnel. All procedures were approved by YNPRC/Emory Animal Care and Biosafety Committees. The YNPRC is supported by NIH/NCRR. A.W.S.C., S.H.L. and X.J.-L are supported by grants awarded by the NIH.
METHODS
Construction and preparation of lentiviruses carrying the HTT-84Q and GFP genes21
Exon 1 of the human HTT gene with 84 CAGs (HTT-84Q) was inserted into a lentiviral vector, which was regulated by the human polyubiquitin-C promoter (pLVU-HTT-84Q). For GFP lentiviruses, the HTT-84Q gene in the vector was replaced by the GFP gene (pLVU-G). High titre lentiviruses were generated by co-transfection of lentiviral vectors coding for HTT-84Q or GFP, pΔ8.9, and pVSV-G (Invitrogen) into a 293FT packaging cell (Invitrogen). Viruses were then concentrated by ultracentrifugation using the method described previously21.
Production of rhesus monkey oocytes, embryos and babies
Assisted reproductive techniques in rhesus monkey have been described elsewhere11. In brief, adult females were hormone stimulated and their oocytes were recovered for in vitro fertilization and culture. Embryos at the 4-8-cell stage were selected for embryo transfer based on morphological appearance. Surrogate females at synchronized reproductive cycles were identified based on their hormone profile.
Gene delivery in mature rhesus monkey oocytes
Metaphase-II-arrested oocytes were selected for perivitelline space injection followed by intracytoplasmic sperm injection. Lentiviral solution was loaded into the injection needle by micropipette and the viral solution was injected into the perivitelline space. After virus injection, the oocytes were fertilized by intracytoplasmic sperm injection followed by in vitro culture until embryo transfer at the 4-8-cells stage10,11.
Monitoring GFP expression in infants
Live HD monkey infants were placed under a Sky-blue II epifluorescent light (475 nm; Youlum), and images were captured by a digital camera equipped with an emission filter at an emission wavelength of 520 nm.
PCR analysis
To detect the HTT-84Q gene, ubiquitin forward primer (5′-GAGGCGTCAGTTTCTTTGGTC-3′) and HTT-84Q-R reverse primer (5′-GCTGGGTCACTCTGTCTCTG-3′) was used to yield an 818-base-pair product after amplification of genomic DNA from the HD monkey tissues. However, variations in size resulted because of the number of CAG repeats. Genomic DNA (100 ng) from different tissues was subjected to PCR for 35 cycles at 96 °C for 5 min, 96 °C for 45 s, 62 °C for 45 s, 72 °C for 150 s, and then 72 °C for 7 min. To determine the number of CAG repeats in HD monkeys, the PCR products were sequenced using HD exon 1 forward primer (5′-GGCGACCCTGGAAAAGCTGA-3′). To detect the GFP gene, GFP-F forward primer (5′- TTCAAGGACGACGGCAACTAC-3′) and GFP-R reverse primer (5′-TAGTGGTTGTCGGGCAGCAG-3′) were used for amplification for 35 cycles at 94 °C for 5 min, 94 °C for 30 s, 64 °C for 30 s, 72 °C for 20 s, and then 72 °C for 5 min, yielding a product of 302 bp. DNA from wild-type monkeys was used as the negative control, and plasmid HTT-84Q and GFP were used as the positive controls.
Southern blot analysis
Eight micrograms of genomic DNA were digested overnight using EcoRI, which only cut once within the transgene. The digested genomic DNA was then separated by gel electrophoresis on a 0.8% agarose gel and transferred to Hybond-N+ nylon membranes (Amersham). To determine the number of integration events of the two viruses (HTT-84Q or GFP) in HD monkeys, a subtraction approach was used because both constructs have identical lentiviral backbones and the short fragment of HTT-84Q is not sufficient to distinguish between the mutant HTT and endogenous HTT. We hybridized two identical membranes with a [32P]-labelled probe that specifically binds to the GFP gene and to the flap sequence of the lentiviral vector. The number of integration events of the GFP gene and the flap sequence was calculated by subtracting the number of GFP integration events from the total number of integration events determined by the flap probe. The number of transgenic mutant HTT gene integration events could then be estimated. Integration sites were determined by exposing [32P]-hybridized members to the phosphor screen and scanning using the Typhoon phosphorimager (GE). Plasmid DNA (HTT-84Q and GFP) digested with EcoRI was used as a positive control; genomic DNA from a non-transgenic monkey was used as a negative control.
Western blot analysis
Total proteins were extracted from different tissues and their concentration was determined using the Bradford assay (Pierce). Equal amounts (20-30 μg) of protein extract with loading dye were boiled before loading into 4-15% gradient polyacrylamide gels (Bio-Rad). After electrophoresis, proteins were transferred onto a PVDF membrane (Bio-Rad) using Bio-Rad's transblot followed by blocking in 5% skimmed milk for 2 h. The membrane was incubated with the primary antibodies mouse monoclonal mEM4819 (1:50 dilution) and γ-tubulin (Sigma; 1:2,000 dilution), followed by secondary peroxidase-conjugated antibodies (Jackson ImmunoResearch laboratories) for detecting proteins with an Amersham ECL kit (PerkinElmer).
Immunohistochemistry of transgenic monkey tissues
Post-mortem brain tissues of transgenic monkeys were fixed in 4% paraformaldehyde overnight, transferred to 30% sucrose, stored at 4 °C, embedded and then cut into 50 μm sections. For mEM48 staining, the sections were incubated with 0.3% hydrogen peroxide for 15 min, blocked for 1 h at room temperature, and incubated with the primary monoclonal antibody mEM48 (1:50) at 4°C overnight. For 1C2 (1:4000 dilution) staining, the tissues were treated with formic acids (88%) for 10 min.
Footnotes
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Supplementary Material
References
- 1.Cummings CJ, Zoghbi HY. Trinucleotide repeats: mechanisms and pathophysiology. Annu. Rev. Genomics Hum. Genet. 2000;1:281–328. doi: 10.1146/annurev.genom.1.1.281. [DOI] [PubMed] [Google Scholar]
- 2.Davies S, Ramsden DB. Huntington's disease. Mol. Pathol. 2001;54:409–413. doi: 10.1136/mp.54.6.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Myers RH, Marans KS, MacDonald ME. In: Huntington's Disease in Genetic Instabilities and Hereditary Neurological Diseases. Wells RD, Warren ST, editors. Academic; San Diego: 1998. pp. 301–324. [Google Scholar]
- 4.Rubinsztein DC. Lessons from animal models of Hungtington's disease. Trends Genet. 2002;18:202–209. doi: 10.1016/s0168-9525(01)02625-7. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald ME, et al. HD research collaborative groups. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntintgton's disease chromosome. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 6.Sharp AH, et al. Widespread expression of Huntington's disease gene (IT15) protein product. Neuron. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- 7.Lane MA. Nonhuman primate models in biogerontology. Exp. Gerontol. 2000;35:533–541. doi: 10.1016/s0531-5565(00)00102-9. [DOI] [PubMed] [Google Scholar]
- 8.King MC, Wilson AC. Evolution at two levels in humans and chimpanzees. Science. 1975;188:107–116. doi: 10.1126/science.1090005. [DOI] [PubMed] [Google Scholar]
- 9.McConkey EH, Varki A. A primate genome project deserves high priority. Science. 2000;289:1295–1296. doi: 10.1126/science.289.5483.1295b. [DOI] [PubMed] [Google Scholar]
- 10.Chan AWS, Chong KY, Martinovich C, Simerly C, Schatten G. Transgenic monkeys produced by retroviral gene transfer into mature oocytes. Science. 2001;291:309–312. doi: 10.1126/science.291.5502.309. [DOI] [PubMed] [Google Scholar]
- 11.Chan AWS, Chong KY, Schatten G. In: Transgenic Animal Technology: A Laboratory Handbook. Pinkert CA, editor. Academic; San Diego: 2002. [Google Scholar]
- 12.Zhou H, et al. Huntingtin forms toxic NH2-terminal fragment complexes that are promoted by the age-dependent decrease in proteasome activity. J. Cell Biol. 2003;163:109–118. doi: 10.1083/jcb.200306038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gutekunst CA, et al. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J. Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bates GP, Mangiarini L, Wanker EE, Davies SW. Polyglutamine expansion and Huntington's disease. Biochem. Soc. Trans. 1998;26:471–475. doi: 10.1042/bst0260471. [DOI] [PubMed] [Google Scholar]
- 15.Lin CH, et al. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum. Mol. Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 16.Li H, et al. Ultrastructural localization and progressive formation of neuropil aggregates in Huntington's disease transgenic mice. Hum. Mol. Genet. 1999;8:1227–1236. doi: 10.1093/hmg/8.7.1227. [DOI] [PubMed] [Google Scholar]
- 17.Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J. Neurosci. 2001;21:8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu ZX, et al. Mutant Huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J. Neurosci. 2003;23:2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Davies SW, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 20.DiFiglia M, et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neuritis in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 21.Yang SH, et al. Enhanced transgenesis by intracytoplasmic injection of envelope-free lentivirus. Genesis. 2007;45:177–183. doi: 10.1002/dvg.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.