

Abstract
Neurofibromas are benign tumors of peripheral nerve that occur sporadically or in patients with the autosomal dominant tumor predisposition syndrome neurofibromatosis type 1 (NF1). Multiple neurofibroma subtypes exist which differ in their site of occurrence, their association with NF1 and their tendency to undergo transformation to become malignant peripheral nerve sheath tumors (MPNSTs), the most common malignancy associated with NF1. Most NF1 patients carry a constitutional mutation of the NF1 tumor suppressor gene. Neurofibromas develop in these patients when an unknown cell type in the Schwann cell lineage loses its remaining functional NF1 gene and initiates a complex series of interactions with other cell types; these interactions may be influenced by aberrant expression of growth factors and growth factor receptors and the action of modifier genes. Cells within certain neurofibroma subtypes subsequently accumulate additional mutations affecting the p19ARF-MDM2-TP53 and p16INK4ARb signaling cascades, mutations of other as yet unidentified genes and amplification of growth factor receptor genes, resulting in their transformation into MPNSTs. These observations have been validated using a variety of transgenic and knockout mouse models that recapitulate neurofibroma and MPNST pathogenesis. A new generation of mouse models is also providing important new insights into the identity of the cell type in the Schwann cell lineage that gives rise to neurofibromas. Our improving understanding of the mechanisms underlying the pathogenesis of neurofibromas and MPNSTs raises intriguing new questions about the origin and pathogenesis of these neoplasms and establishes models for the development of new therapies targeting these neoplasms.
Keywords: neurofibromatosis type 1, neurofibromin, tumor suppressor, animal model, stem cell
SCHWANN CELL NEOPLASMS ASSOCIATED WITH NEUROFIBROMATOSIS TYPE 1
The overwhelming majority of the tumors that arise in the human peripheral nervous system are derived from Schwann cells or their precursors (Urich and Tien, 1998). These neoplasms, which are collectively referred to as peripheral nerve sheath tumors, include neurofibromas, schwannomas and malignant peripheral nerve sheath tumors (MPNSTs). In the general population, neurofibromas and schwannomas typically occur sporadically as solitary lesions. In contrast, patients with the common genetic disease neurofibromatosis type 1 (NF1; OMIM +162200) characteristically develop multiple neurofibromas and are at increased risk for the occurrence of MPNSTs, while individuals with the rarer conditions neurofibromatosis type 2 (NF2; OMIM #101000), familial schwannomatosis (OMIM #162091) and Carney complex syndrome (OMIM #160980) develop multiple schwannomas associated with cranial and peripheral nerves. Over the last decade, a number of important new studies have provided novel insights into the mechanisms underlying the development of NF1-associated peripheral nerve sheath tumors and have clarified the origin of these neoplasms. Consequently, we will concentrate in this review on the pathogenesis of NF1-associated neurofibromas and MPNSTs, taking care to compare and contrast them to their sporadic counterparts where relevant. We refer readers interested in the pathogenesis of NF2-, schwannomatosis- or Carney complex-associated schwannomas to excellent recent reviews that have examined these subjects (Boikis and Stratakis, 2007; Ferner, 2007; McClatchey and Giovannini, 2005; Okada et al., 2007).
Neurofibromatosis type 1 is one of the most common Mendelian diseases occurring in man, affecting approximately 1 in every 3500 newborn infants (Gutmann, 2001; Le and Parada, 2007; Theos and Korf, 2006). This tumor predisposition syndrome is inherited as an autosomal dominant disorder and is completely penetrant. However, the manifestations of NF1 are quite diverse and so, despite its high degree of penetrance, this disease often presents in a widely variable manner even within closely related family members. Abnormalities associated with NF1 can include learning disabilities, bony dysplasias, pigmentary lesions (café-au-lait spots, Lisch nodules and axillary freckling) and the occurrence of a variety of tumor types such as optic gliomas (WHO Grade I pilocytic astrocytomas), higher grade infiltrating astrocytic neoplasms, juvenile myelomonocytic leukemias and pheochromocytomas. The occurrence of multiple neurofibromas, however, is generally considered to be a hallmark of NF1, as indicated by the name of this disorder. These benign peripheral nerve sheath tumors are a major cause of morbidity in NF1 patients, producing distinct clinical problems in different neurofibroma subtypes (see below) and at different locations. Dermal neurofibromas often cause disfigurement, itching and irritation. Although frequently painless, neurofibromas arising in larger nerves can produce pain and neurologic deficits, particularly if they occur at sites prone to compression (e.g., spinal nerve exit zones or weight-bearing portions of the buttocks; Bruce R. Korf, MD, PhD, personal communication). This complication is particularly problematic in children, as large plexiform neurofibromas can compress vital organs in these patients and cause death. Neurofibromas can also demonstrate a remarkable degree of overgrowth, with extreme examples involving entire limbs or body segments and impairing the function of affected body parts. Large neurofibromas also appear to induce inappropriate growth of adjacent soft tissue and bone, producing impairment disproportionate to the size of the neoplasm. It is not currently clear whether these peritumoral responses are the result of enhanced growth factor release from the neurofibroma, cell autonomous effects arising from NF1 haploinsufficiency in soft tissue and bone (Stevenson et al., 2007; Yang et al., 2006a) or a combination of these factors.
Certain neurofibroma subtypes (see below) are also capable of undergoing malignant transformation to give rise to MPNSTs, the most common malignancy associated with NF1. The lifetime risk for NF1 patients to develop an MPNST has been estimated at 8–13% (Evans et al., 2002) and 5.9–10.3% (McCaughan et al., 2007). Retrospective clinical studies suggest that NF1-associated MPNSTs are significantly more aggressive than their sporadically occurring counterparts (Hagel et al., 2007); however, the poorer outcome in NF1 patients may instead indicate that their MPNSTs, which often arise within pre-existing plexiform neurofibromas, are typically diagnosed later (i.e., when more extensive tumor growth is already established) than their sporadic counterparts. At present, surgery is the primary means of treating these highly aggressive sarcomas, regardless of whether they are sporadic or NF1-associated (Carli et al., 2005; Ferner and Gutmann, 2002). If complete surgical resection is not achieved, the therapeutic options open to these patients are limited. Radiotherapy can help control local disease, but has little effect on long term survival (Ferner and O'Doherty, 2002); chemotherapy is typically ineffective (Ferner and Gutmann, 2002) although some studies suggest that it may benefit children with unresectable MPNSTs (Carli et al., 2005). The treatment of MPNSTs is further complicated by their propensity to metastasize; in a series of 62 NF1 patients with MPNSTs and 58 patients with sporadic MPNSTs, 39% and 16% of these cohorts, respectively, developed metastatic disease (Ducatman et al., 1986). The majority of these metastases were pulmonary, with metastases also occurring (in decreasing order of frequency) in soft tissue, bone, liver, the abdominal cavity, adrenal glands, diaphragm, mediastinum, brain, ovaries, kidneys and retroperitoneum. Given these factors, it is not surprising that 5- and 10-year survival rates for patients with MPNSTs are 34 and 23%, respectively (Ducatman et al., 1986). The devastating effects that neurofibromas and MPNSTs have on NF1 patients have thus been a powerful impetus for efforts to establish the origin of these tumors and to identify the molecular abnormalities responsible for their initiation and progression.
PATHOLOGY AND MALIGNANT POTENTIAL OF NF1-ASSOCIATED NEUROFIBROMAS
It is generally agreed that multiple neurofibroma subtypes exist which differ in their location and pattern of growth, their association with NF1 and their potential for malignant transformation. However, clinicians and investigators studying these lesions have often used varying terminologies to describe different types of neurofibromas, with up to seven neurofibroma subtypes defined by some investigators (Riccardi, 1992). For instance, many clinical and basic science investigators broadly classify neurofibromas as either dermal or plexiform variants, with some investigators separating off a third variant, the nodular neurofibroma. Other clinical investigators categorize neurofibromas as cutaneous (i.e., dermal) neurofibromas (Barbarot et al., 2007), subcutaneous neurofibromas (functionally defined as superficially situated tumors in which the skin moves freely over the lesion (Tucker et al., 2005)) or plexiform neurofibromas. Pathologists recognize five neurofibroma subtypes, which are defined using their own distinct terminology (Scheithauer et al., 1999; Scheithauer et al., 2007). As these definitional differences in neurofibroma classification have the potential to confound the interpretation of a wide range of studies (e.g., microarray analyses of different neurofibroma subtypes, interpretation of the phenotype of emerging transgenic mouse models of neurofibroma pathogenesis), it is worthwhile considering our current understanding of the biology of different neurofibroma subtypes as defined by pathologists so as to provide a basis of comparison to the classification schemes used by other investigators studying these peripheral nerve sheath tumors. This consideration is also useful because it highlights a paradox: despite the differences in their clinical behavior, the cellular composition of all neurofibroma subtypes is identical. At present, the basis for the differences in the biological behavior of different neurofibroma subtypes is a major unresolved issue.
The five neurofibroma subtypes described by pathologists are summarized in Table 1. The first of these subtypes, localized cutaneous neurofibromas, are the most common form of neurofibroma and occur as circumscribed, non-encapsulated lesions localized in the dermis and subcutis (Fig. 1A, B) of skin anywhere on the body. 90% of these tumors are solitary, non-NF1 associated lesions; in NF1, these tumors are typically multiple and may be overlain by café-au-lait spots. Localized cutaneous neurofibromas have typically developed in NF1 patients as they begin to approach puberty and continue to accumulate thereafter. These lesions virtually never undergo malignant transformation. Diffuse cutaneous neurofibromas are uncommon lesions that present as ill-defined plaque-like thickenings in children and young adults; 10% occur in NF1 patients. Although dermal, this neurofibroma subtype is capable of giving rise to MPNSTs at a very low frequency (Scheithauer et al., 1999). Localized intraneural neurofibromas (also known as nodular neurofibromas), the second most common neurofibroma subtype, may affect any spinal, cranial or autonomic nerve at any point along the nerve from its root to its smallest branch. Grossly, localized intraneural neurofibromas are evident as segmental fusiform enlargements of nerves. Microscopically, tumor cells within these neoplasms disrupt the normal perineurium delimited fasicular architecture of the nerve, entrapping axons and attenuating the epineurium as the tumor mass expands (Fig. 1C, D). This neurofibroma variant occurs both sporadically and in association with NF1; it is capable of undergoing malignant change in both settings, albeit at a somewhat lower frequency than is seen in plexiform neurofibromas (Scheithauer et al., 1999). Plexiform neurofibromas are neurofibroma variants that occur almost exclusively in NF1 patients and are thought to be congenital. Plexiform neurofibromas involve the same nerves as localized intraneural neurofibromas and are distinguished from the latter by their characteristic plexiform growth pattern, in which tumor cells spread along multiple fascicles of the nerve (Fig. 1E) while preserving the overall fascicular architecture of the nerve within the perineurium. Plexiform neurofibromas have the highest risk for malignant transformation into MPNSTs (Fig. 1F). Finally, massive soft tissue neurofibromas, the least common form of neurofibroma, occur only in NF1 patients. These tumors often contain a plexiform component and cause either massive enlargement of regional soft tissue or localized gigantism of an extremity (hence the earlier designation of this condition as “elephantiasis neuromatosa”). Massive soft tissue neurofibromas can undergo malignant change but, given their rarity, are a less common source of MPNSTs. At present, the reason why some of these neurofibroma subtypes can undergo malignant transformation while others do not is unknown.
Table 1.
Anatomy and Biological Properties of Neurofibroma Subtypes
| Neurofibroma Subtype | Location | NF1 Association | Malignant Potential | Special Features | Within Perineurium? |
|---|---|---|---|---|---|
| Localized cutaneous | Skin (dermis and subcutis) | 90% sporadic; 10% NF1 associated | No | Most common subtype; development and enlargement associated with puberty, pregnancy | No |
| Diffuse cutaneous | Skin (dermis and subcutis) | 90% sporadic; 10% NF1 associated | Yes | Occurs in children and young adults | No |
| Localized intraneural | Cranial, spinal or autonomic nerves | Either sporadic or NF1 associated | Yes | Second most common subtype; produce segmental fusiform enlargement of nerve rather than diffusely enlarging multiple nerve fascicles like a plexiform neurofibroma | No |
| Plexiform | Cranial, spinal or autonomic nerves | Almost exclusively NF1-associated | Yes | Produce plexiform (diffuse) rather than segmental enlargement of nerve | Yes |
| Massive soft tissue | Extremities, larger nerve associated with massive soft tissue enlargement | Solely NF1-associated | Yes | Least common subtype; often contains a plexiform component | No |
Fig. 1.
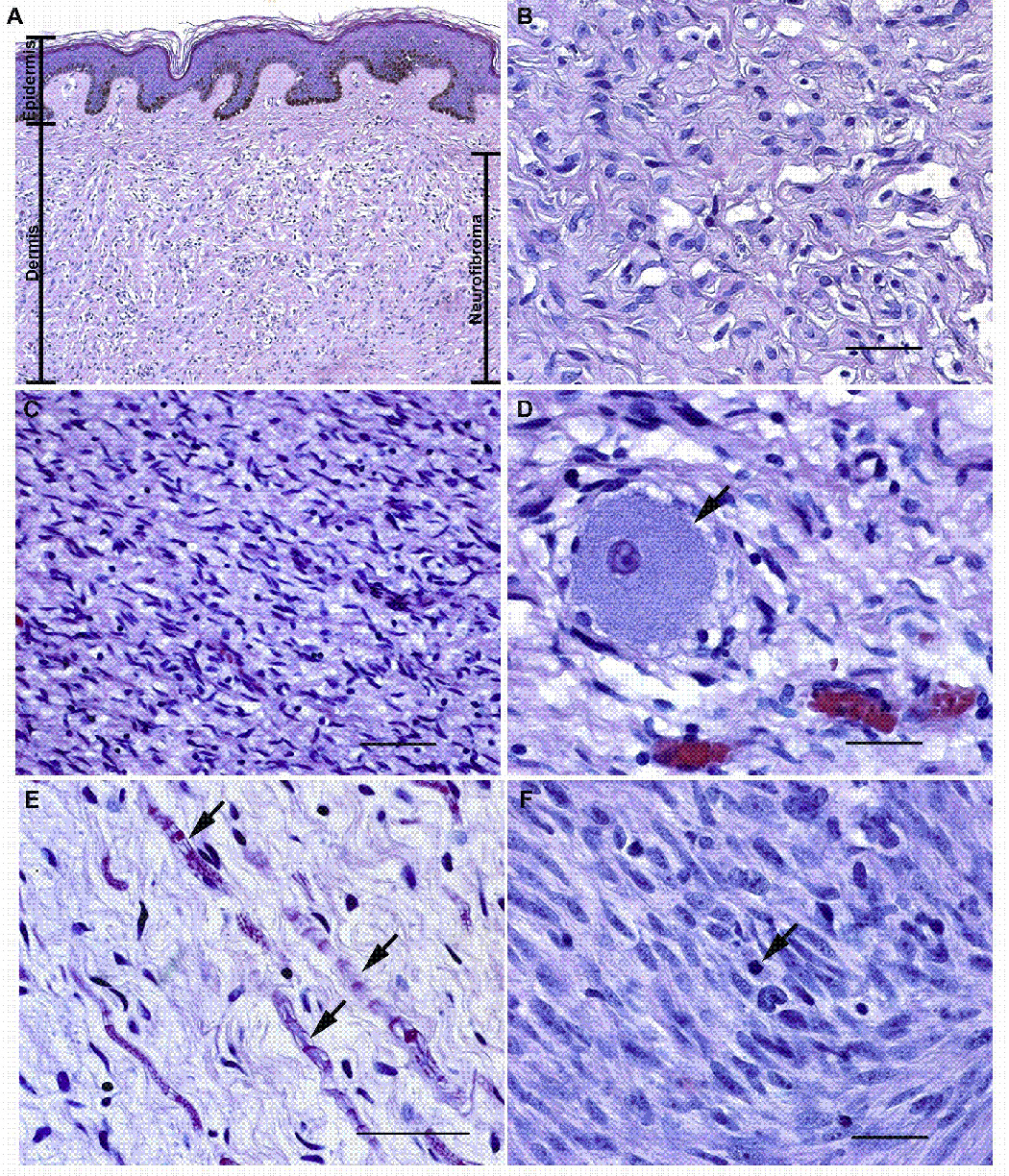
Microscopic anatomy of NF1-associated peripheral nerve sheath tumors. (A, B) Panel A illustrates a low power (10x) view of a localized cutaneous neurofibroma that presented as a pedunculated mass on the left shoulder of a 60 year old woman with NF1. This tumor (indicated by the bar on the right of the panel) spanned almost the entire thickness of the dermis and infiltrated into the underlying subcutis (a layer of loose fibroadipose tissue underlying the dermis; not shown in this image). Note that the tumor does not involve the most superficial portion of the dermis or the overlying epidermis (the maximal extent of the epidermis and the dermis are indicated by bars on the left of the panel). Panel B is a representative hematoxylin and eosin stained high power (40x) field from this densely collagenized tumor. (C, D) Panel C illustrates a low power (20x) field from a localized intraneural neurofibroma that arose in a cervical dorsal spinal nerve root. This tumor infiltrated diffusely through the nerve and associated dorsal root ganglion, destroying the normal fascicular architecture of the nerve as it produced a segmental fusiform enlargement of this structure. Panel D is a higher power view (40x) of this tumor. The arrow indicates a sensory neuron from the dorsal root ganglion that has been entrapped by the growth of the neurofibroma. (E) High power (40x) view of a field within a plexiform neurofibroma resected from a 40 year old woman with NF1. This tumor, like many plexiform neurofibromas, has a looser, more myxoid matrix but is still composed of the same cell types present in the tumors illustrated in A–D. Arrows indicate entrapped axons and their myelin sheaths. (F) An MPNST that arose within a plexiform neurofibroma in an NF1 patient. Note the greater degree of cellularity, atypia and mitotic activity (arrow) in this tumor. Magnification: 40x. Bars in B, D and F: 50 µm. Bar in C: 200 µm. Bar in E: 100 µm.
All five of these neurofibroma subtypes are composed of a complex mixture of cell types (Fig. 2) including Schwann cell-like elements (defined as cells that are immunoreactive for p75 and S100β, these elements are simply referred to as “Schwann cells” by most investigators), mast cells, fibroblasts, vascular elements, perineurial-like cells [defined as cells with the ultrastructural characteristics of perineurial cells (i.e., long thin cytoplasmic processes, numerous pinocytotic vesicles and a discontinuous basal lamina), but lacking the characteristic epithelial membrane antigen immunoreactivity of normal perineurial cells] and cells with features intermediate between the perineurial-like cells and other cell types in the tumor. Neurofibromas also contain a population of CD34-immunoreactive dendritic cells that is distinct from the S100β-positive Schwann-like cells. It was originally suggested that these are an as yet undescribed nerve sheath cell type (Weiss and Nickoloff, 1993). Others, however, have argued that these CD34-positive cells are instead resident tissue macrophages (Cohen et al., 1993). To date, studies rigorously examining the expression of other markers associated with these CD34-positive cells have not been performed and consequently their identity remains unclear. Although it has been suggested that certain histologic features (e.g., differences in patterns of collagen deposition or the presence of structures reminiscent of Wagner-Meissner or pacinian corpuscles) are useful for distinguishing different neurofibroma subtypes, these findings are vague or inconsistently present and thus cannot be reliably used to make such distinctions.
Fig. 2.
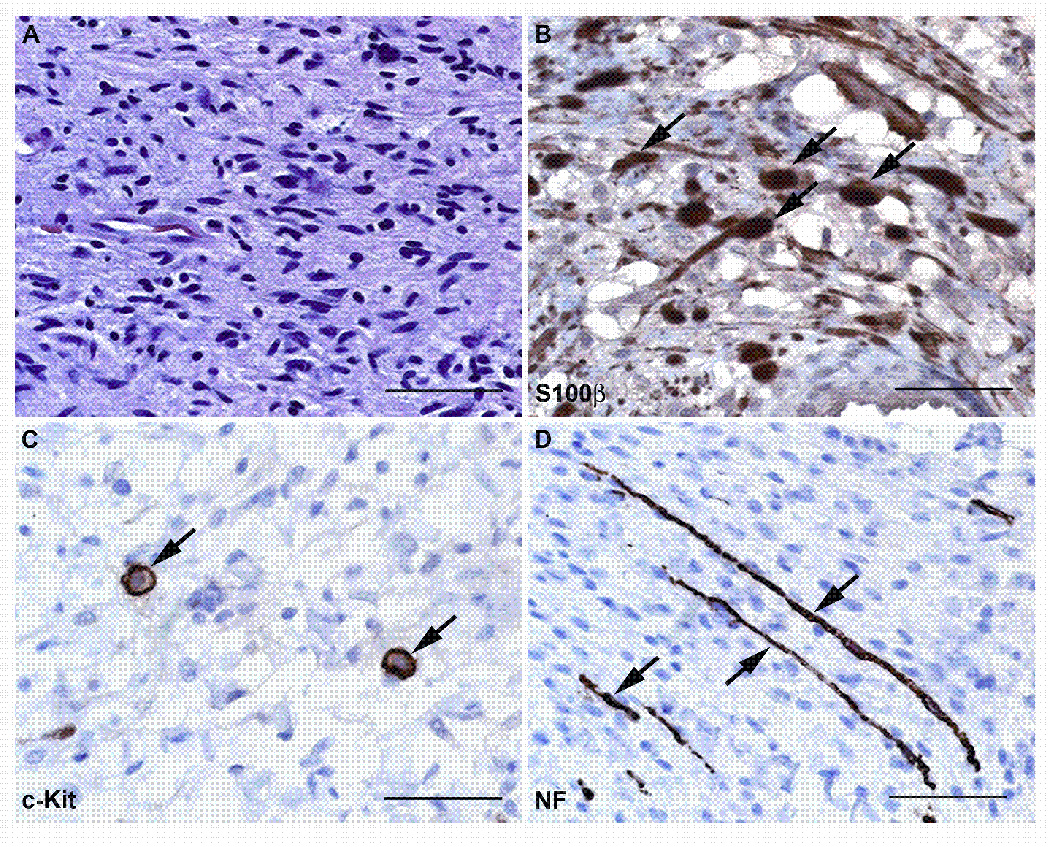
Neurofibromas are composed of a complex mixture of cell types. (A) High power (40x) view of a hematoxylin and eosin stained section from a localized intraneural (nodular) neurofibroma that arose on the 5th lumbar nerve of a 61 year old woman. This neoplasm is composed of a mixture of cell types that can be detected using immunostains for cell-type specific markers, as shown in B–D. (B) Immunoperoxidase stains (brown) for S100β label the Schwann cell-like elements in this neurofibroma. The section has been lightly counterstained with hematoxylin to highlight the nuclei of cells (primarily fibroblasts and perineural-like cells) that do not stain for this antigen. Arrows indicate some of the cells that are intensely S100β immunoreactive. (C) Another section of this tumor immunostained for the mast cell marker c-Kit (CD117). Arrows indicate two cells that show intense membranous immunoreactivity for this membrane tyrosine kinase receptor. (D) The same tumor immunostained for neurofilaments to demonstrate the presence of axons entrapped by the infiltrative growth pattern of this tumor (arrows). Bars in A–D: 50 µm.
Given this cellular heterogeneity, it is understandable that for several decades investigators debated the identity of the primary neoplastic cell type in neurofibromas. At present, however, there is a general consensus that the primary neoplastic element in neurofibromas is derived from the Schwann cell lineage. This consensus is supported by several lines of evidence. Loss of heterozygosity of the NF1 locus is evident in Schwann cells, but not fibroblasts, isolated from neurofibromas (Kluwe et al., 1999; Rutkowski et al., 2000). Neurofibroma-derived Schwann cells, like wild-type Schwann cells, will not proliferate in the presence of lowered growth factor concentrations or grow in an anchorage-independent manner and they interact normally with axons in vitro. However, neurofibroma-derived Schwann cells do demonstrate functional abnormalities as they are capable of invading through basement membranes (Muir, 1995; Sheela et al., 1990) and promote angiogenesis (Sheela et al., 1990). It is also notable that neoplastic cells within at least some MPNSTs, which commonly arise by progression from neurofibromas, have features characteristic of Schwann cells such as immunoreactivity for S100β and partial investment of tumor cells by a basement membrane (Urich and Tien, 1998). The concept that the primary neoplastic cell type in neurofibromas is derived from Schwann cells or their precursors has also been experimentally established by demonstrating that mice with Schwann cell lineage specific ablation of the Nf1 gene develop neurofibromas (Zhu et al., 2002)(also see below for a more extensive discussion of this landmark study and other important transgenic mouse models of neurofibroma pathogenesis). The precise identity of the cell type within the Schwann cell lineage (Schwann cell precursors, immature Schwann cells or mature Schwann cells) that gives rise to neurofibromas remains unresolved, although a growing body of evidence argues that neurofibromas are derived from Schwann cell precursors or immature Schwann cells (see below). As at least some of the NF1-null Schwann cells within neurofibromas have characteristics of more mature Schwann cells, these neoplastic elements can presumably differentiate to some extent and neoplastic cells with varying degrees of differentiation are simultaneously present in neurofibromas. Thus, another major unanswered question is whether MPNSTs originate from neoplastic Schwann cell precursors or another, more or less well differentiated neoplastic element within neurofibromas.
NF1 MUTATIONS AND OTHER ABNORMALITIES CONTRIBUTING TO THE PATHOGENESIS OF NF1-ASSOCIATED NEUROFIBROMAS
At least 95% of NF1 patients carry a constitutional mutation of a tumor suppressor gene located on the long arm of chromosome 17 (17q11.2) together with a functional copy of this same gene (Messiaen et al., 2000). Analyses of family pedigrees suggest that while the mutated gene may be inherited from a parent, new mutations occur at a high rate in the NF1 locus and up to 50% of cases are new mutations (Packer et al., 2002); indeed, NF1 has the highest rate of new mutation of any known single gene disorder (Theos and Korf, 2006). Although some sites within the NF1 gene are more prone to mutation than others (e.g., exons 10a-c, 37), mutations occur throughout this locus (Messiaen et al., 2000). Only 5% of the constitutional mutations in NF1 patients involve total gene deletion (Wimmer et al., 2006), with the majority of mutations resulting from a variety of more subtle changes including nonsense mutations, frameshift mutations, missense mutations (Messiaen et al., 2000) or mutations that produce mRNA splicing defects (Wimmer et al., 2007). As NF1 is a tumor suppressor gene, it is expected that tumorigenesis will occur when a second somatic mutation disrupts the remaining functional copy of this gene. In keeping with this expectation, NF1 loss of heterozygosity (LOH) is evident in Schwann cells isolated from neurofibromas and MPNSTs; fluorescent in situ hybridization (FISH) analyses confirm that NF1 LOH is present in S100β-immunoreactive Schwann cells, but not fibroblasts, in these neoplasms (Kluwe et al., 1999; Legius et al., 1993; Wallace et al., 2000). Neurofibromas contain a mixture of NF1+/− and NF1−/− Schwann cells which can be purified by culturing disaggregated tumor cells in the presence or absence of forskolin (Serra et al., 2000). Examination of the NF1 mutations evident in these two neurofibroma-derived Schwann cell populations has confirmed the presence of a constitutional NF1 mutation in both populations, with a second acquired mutation evident in the NF1−/− Schwann cells (Maertens et al., 2006).
The NF1 gene (GeneID: 4763) includes 57 exons and several alternatively spliced exons that span 350 kb of genomic DNA and encode a 2818 amino acid protein known as neurofibromin. Neurofibromin contains a domain (amino acids 1125–1537) homologous to both a mammalian Ras GTPase activating protein (GAP) known as p120RasGAP and the Saccharomyces cerevesiae Ras GAPs IRA1 and IRA2 (Ballester et al., 1990; Xu et al., 1990a); transfection of sequences encoding the neurofibromin Ras GAP-related domain (GRD) into IRA1- and IRA2-deficient yeast rescues their phenotype (Xu et al., 1990b), confirming that the neurofibromin Ras-GRD has functional characteristics similar to IRA1 and IRA2. Immediately adjacent to the neurofibromin Ras-GRD is a bipartite domain (amino acids 1560–1816) consisting of a segment homologous to the yeast Sec14p protein (amino acids 1560–1698) that is linked to a pleckstrin homology domain (amino acids 1715–1816) by a partially helical linker peptide (D'Angelo et al., 2006). Yeast Sec14p is a phosphatidylinositol/phosphatidylcholine transfer protein whose function is to coordinate lipid metabolism and protein transfer from the Golgi complex (Mousley et al., 2006). The neurofibromin Sec14p homology domain has also been shown to specifically bind several glycerophospholipids (Welti et al., 2007). Although the physiologic consequences of the interaction between the neurofibromin Sec14p domain and glycerophospholipids have not been experimentally established, neurofibromin action has been shown to be inhibited by arachidonic acid (Golubic et al., 1992; Han et al., 1991). There is also evidence that neurofibromin can regulate cAMP controlled signaling pathways (Kim et al., 2001; The et al., 1997). However, the identity of the neurofibromin domain that mediates this effect and the function of other portions of the neurofibromin protein are at present unknown.
Given our current limited knowledge of neurofibromin action, it is perhaps understandable that the majority of studies performed to date have focused on the effects neurofibromin exerts on the regulation of Ras proteins. Ras proteins are small GTP-binding proteins (G-proteins) that play key roles in proliferation, survival, migration and other essential cellular functions. These molecules become activated upon binding GTP and are deactivated when a GTPase activity intrinsic to the Ras protein cleaves GTP and converts it to GDP. However, the ability of Ras proteins to “turn off” their activity on their own is limited as the intrinsic GTPase activity of Ras proteins is relatively ineffective. Consequently, GTPase activating proteins (GAPs) such as neurofibromin are required to enhance the intrinsic GTPase activity of Ras proteins and thereby inactivate these small G-proteins. Given this, it would be expected that the neoplastic Schwann cell-like elements within neurofibromas and MPNSTs, in which the NF1 gene have undergone biallelic inactivation, would have increased Ras activation. Consistent with this hypothesis, neurofibroma (Sherman et al., 2000) and MPNST cells (Basu et al., 1992; DeClue et al., 1992) contain levels of GTP-bound Ras proteins higher than those detected in non-neoplastic Schwann cells. Our understanding of the Ras-regulated effector pathways that are critically important for peripheral nerve sheath tumorigenesis is at present limited. However, the PI3 kinase-Akt-TSC2-mTOR-S6 kinase has been implicated as one of these critical effector pathways (Johannessen et al., 2005; Johannessen et al., 2008), suggesting that agents targeting molecules in this cascade (e.g., rapamycin and rapamycin derivatives) may be effective for the treatment of neurofibromas and MPNSTs (Johansson et al., 2008).
In contrast to the NF1−/− Schwann cells within neurofibromas, NF1+/− Schwann cells and other cellular elements in these tumors such as fibroblasts and mast cells still carry an intact copy of the NF1 allele. The functional consequences of NF1 haploinsufficiency in these other cell types remains incompletely understood. However, studies of cells isolated from mice with a targeted null mutation of the Nf1 gene have provided evidence that losing the function of even one Nf1 allele in Schwann cells and other cell types intrinsic to neurofibromas is sufficient to produce abnormalities potentially contributing to neurofibroma pathogenesis. Stem cell factor (SCF, also known as Kit ligand), a growth factor capable of promoting the migration of immature mast cells and their subsequent maturation, activation and survival, is expressed by Schwann cells within neurofibromas (Demitsu et al., 1998; Hirota et al., 1993; Ryan et al., 1994). Further, SCF secretion is evidently enhanced by a loss of neurofibromin as Nf1−/− Schwann cells secrete approximately 6-fold more SCF than Nf1+/− or wild-type Schwann cells (Yang et al., 2003). In turn, Nf1 haploinsufficient mast cells have been found to demonstrate enhanced migration (Yang et al., 2003), proliferation (Ingram et al., 2000; Ingram et al., 2001) and survival (Ingram et al., 2000; Ingram et al., 2001) in response to SCF, suggesting that increased SCF secretion by NF1−/− Schwann cells has an exaggerated effect on NF1+/− mast cells in vivo. Nf1 haploinsufficiency has also been shown to affect the proliferation of Schwann cells (Kim et al., 1995), fibroblasts (Yang et al., 2006b) and keratinocytes (Atit et al., 2000). Observations such as these have led to the hypothesis that an NF1 haploinsufficient microenvironment is required in conjunction with the presence of NF1−/− Schwann cells for the initial pathogenesis of NF1-associated neurofibromas (see below). As patients in the general population are not NF1 haploinsufficient, however, it should be noted that this hypothesis is clearly not relevant to the pathogenesis of sporadic neurofibromas.
As neurofibromin loss leads to increased Ras activity in neurofibromas and MPNSTs, it is logical to suggest that inhibiting Ras activity may be an effective means of treating these peripheral nerve sheath tumors. However, this hypothesis cannot be tested until it is determined which Ras isoforms or combination of Ras isoforms should be targeted. Neurofibromin functions as a GAP for both the classic Ras proteins (H-, N- and K-Ras) and the R-Ras subfamily of small G-proteins (R-Ras, R-Ras2/TC21 and R-Ras3/M-Ras) (Ohba et al., 2000). At present, relatively little is known regarding the contributions each of these Ras isoforms makes to the proliferation, survival and migration of neoplastic Schwann cells. We do know that all three of the classic Ras proteins as well as R-Ras and R-Ras2/TC21 are expressed in wild-type and Nf1−/− Schwann cells (Huang et al., 2004). Further, farnesyltransferase inhibitors, which interfere with the action of H-Ras, while sparing N- and K-Ras, correct the proliferative abnormality observed in Nf1−/− Schwann cells (Kim et al., 1997). In contrast, an H-Ras dominant negative mutant, which is capable of inhibiting the action of all three classic Ras proteins, does not impede the increased levels of migration characteristic of Nf1−/− Schwann cells and it has been found that R-Ras2/TC21 is instead essential for the migration of these glia (Huang et al., 2004). Taken together, these observations would suggest that at least two Ras proteins (H-Ras and R-Ras2) are likely to play important roles in neurofibroma pathogenesis and may be important therapeutic targets; indeed, a phase I clinical trial with the farnesyltransferase inhibitor tipifarnib, which targets H-Ras, has been completed in children with NF1-associated plexiform neurofibromas (Widemann et al., 2006). However, H-Ras and R-Ras2 have not yet been shown to play a similar role in neurofibroma and MPNST cells and some studies indicate that other Ras proteins such as N-Ras are activated in MPNST cells (Mattingly et al., 2006). It is also currently unclear whether inhibiting the action of essential Ras isoforms in other NF1 haploinsufficient cell types within neurofibromas may have a therapeutically useful effect. Information as to which Ras proteins mediate the proliferation, survival and migration of other cell types affected in NF1 is limited, but suggests that this depends on the cell type being examined. For example, K-Ras activation in Nf1−/− astrocytes is important for optic glioma formation (Dasgupta et al., 2005) and loss of K-Ras modulates the excessive proliferation and migration observed in c-Kit stimulated Nf1+/− mast cells (Khalaf et al., 2007). These apparently cell-type dependent roles for different Ras proteins will require that the identity of the Ras isoforms mediating the mitogenesis, proliferation and migration of specific cell types within neurofibromas be empirically determined.
As increased Ras activation is evident in neurofibromas, it might be expected that once the pathogenesis of these neoplasms was initiated, they would demonstrate continuous growth throughout the life of the patient. However, clinical observations indicate that this is not the case. The growth of neurofibromas instead appears to be self-delimiting, with the enlargement of these neoplasms periodically stopping and then restarting. This behavior may be explained in part by a phenomenon known as oncogene-induced senescence, which has been shown to be induced by constitutively active Ras mutants in some, but not all cell types (Serrano et al., 1997). Ablation of neurofibromin expression in human fibroblasts via RNA interference produces an initial activation of Ras and its downstream targets ERK and Akt which is rapidly suppressed; this suppression is accompanied by growth arrest and the expression of senescence-associated β-galactosidase and other markers of senescence (Courtois-Cox et al., 2006). The induction of senescence in these cells is mediated by a global negative feedback signaling program in which the action of molecules enhancing Ras activity (e.g., Ras guanine nucleotide exchange factors, which counter GAP action by activating Ras) is impeded and that of molecules inhibiting Ras activity (RasGAPs; Sprouty proteins, which inhibit the Ras signaling cascade at multiple levels; MAP kinase phosphatases, also known as dual specificity phosphatases (DUSPs)) is enhanced. A subpopulation of neurofibromin-negative cells within dermal neurofibromas from NF1 patients, most of which express markers characteristic of Schwann cell precursors, show similar evidence of senescence (Courtois-Cox et al., 2006). This suggests that oncogene-induced senescence limits the growth of neurofibromas, with periods of growth occurring when tumor cells escape from this process via an as yet unknown mechanism. It is also likely that other processes contribute to the variable growth of neurofibromas. For instance, neurofibroma growth is enhanced during puberty and pregnancy (Dugoff and Sujansky, 1996; Huson et al., 1988; Swapp and Main, 1973), suggesting that increased expression of steroid hormones, growth hormone or other factors during puberty and pregnancy promotes neurofibroma growth. At present, however, the mechanism(s) producing puberty- and pregnancy-associated increases in neurofibroma growth is unknown as findings from a number of laboratories that have examined this hypothesis have been contradictory.
As noted above, neurofibromin acts as a GAP, which terminates Ras signaling. This, however, begs the question of what stimulates Ras activation in the first place. As growth factor signaling is an important means of activating Ras, a number of laboratories have suggested that growth factors, particularly those with mitogenic effects on Schwann cells, or their aberrantly expressed receptors might promote neurofibroma growth (Carroll and Stonecypher, 2005). A number of candidate molecules have been identified. The EGF receptor (EGFR), a membrane tyrosine kinase present only in fibroblasts and perineurial cells in normal nerve, has been found to be expressed by a subpopulation of S100β-immunoreactive cells in neurofibromas (DeClue et al., 2000), suggesting that aberrant expression of this receptor contributes to neurofibroma pathogenesis. Neurofibromas also express multiple class II [glial growth factor (GGF)] and class III (sensory and motor neuron-derived factor) neuregulin-1 isoforms and their erbB receptors (the erbB3 and erbB4 receptors and/or their erbB2 coreceptor) (Stonecypher et al., 2005), platelet-derived growth factor (PDGF)-BB and its β receptor (Kadono et al., 1994; Kadono et al., 2000), hepatocyte growth factor and its c-Met receptor (Fukuda et al., 1998; Rao et al., 1997; Watanabe et al., 2001) and transforming growth factor-β1 and its receptor (Watanabe et al., 2001), all of which have been suggested to have effects on the proliferation of neoplastic Schwann cells. At present, however, a functional role for most of these growth factors and their receptors has not been established in human neurofibromas or transgenic mouse models of neurofibroma pathogenesis (see below, however, for a discussion of their potential function in MPNSTs). The two exceptions are the EGFR and NRG-1, both of which have been expressed in the peripheral nerves of transgenic mice, resulting in the production of informative phenotypes (see below).
Finally, in addition to the influences described above, there is evidence that the pathogenesis of neurofibromas in NF1 patients is influenced by modifying genes. An examination of the occurrence of dermal and plexiform neurofibromas in 175 patients from 48 pedigrees that included 6 pairs of monozygotic twins demonstrated that tumor occurrence had the highest correlation between monozygotic twins (Easton et al., 1993). This correlation was weaker among first degree relatives and even lower among more distant relatives within a pedigree, indicating that modifier genes rather than an NF1 mutation presumably shared among all family members was the major influence on neurofibroma occurrence. The modifier genes that predispose NF1 patients to the development of neurofibromas have not yet been identified. However, the development of MPNST-like neoplasms in mice carrying cis-linked Nf1 and p53 mutations has been shown to be influenced by strain background (Hawes et al., 2007) and by an imprinted locus that epistatically influences two unlinked polymorphic loci known as nerve sheath tumor resistance 1 (Nstr1) and Nstr2 (Reilly et al., 2006).
MUTATIONS INVOLVED IN THE PROGRESSION OF NEUROFIBROMAS TO MALIGNANT PERIPHERAL NERVE SHEATH TUMORS
Biallelic inactivation of NF1 and mutations of numerous additional tumor suppressor genes within the p19ARF-MDM2-TP53 and p16INK4A-Rb signaling cascades have been identified in MPNSTs. Deletions and other mutations resulting in loss of function of the TP53 tumor suppressor gene are one of the more common abnormalities found in MPNSTs (Birindelli et al., 2001; Legius et al., 1994; Menon et al., 1990), with recent series typically reporting TP53 mutations in approximately 75% of MPNSTs (Holtkamp et al., 2007; Upadhyaya et al., 2008). Curiously, biallelic inactivation of the TP53 locus is rarely found in MPNSTs (Lothe et al., 2001), which has led to the suggestion that hemizygous TP53 mutations may suffice for neurofibromas to progress and become MPNSTs (Upadhyaya et al., 2008). The CDKN2A gene encodes both the cell cycle inhibitor p16INK4A, a protein that acts by inhibiting the action of cyclin-dependent kinases 4 (CDK4) and 6 (CDK6), and p19ARF, a protein that binds to and inhibits the ubiquitin ligase MDM2 resulting in stabilization of p53. Deletions of the CDKN2A locus are also common in MPNSTs, being found in about 50% of these neoplasms (Kourea et al., 1999; Nielsen et al., 1999). Expression of the Retinoblastoma (Rb) protein, a molecule that functions to impede cell cycle progression, is lost in 25% of MPNSTs (Mantripragada et al., 2008; Mawrin et al., 2002). Importantly, the abnormalities noted above are not present in neurofibromas. It is therefore thought that the development of neurofibromas and their subsequent progression to become MPNSTs involves a sequential series of tumor suppressor mutations (Fig. 3).
Fig. 3.
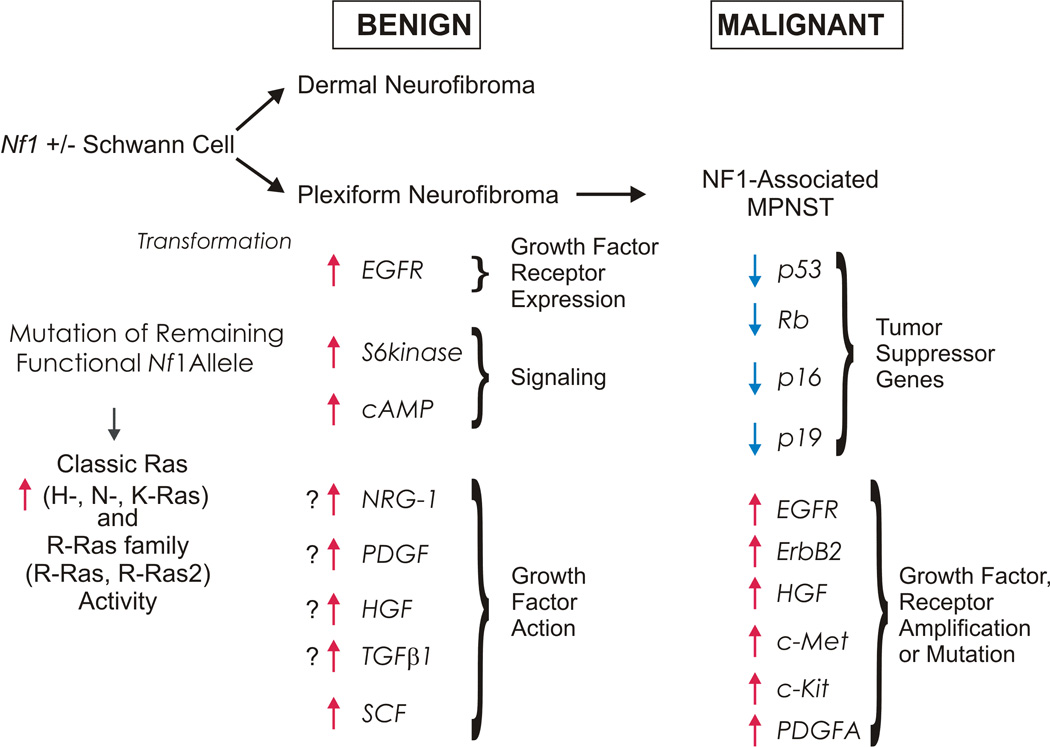
Schematic illustrating some of the multiple factors contributing to the development of neurofibromas in NF1 patients and their subsequent progression to become MPNSTs. The development of neurofibromas is initiated when an NF1 haploinsufficient cell in the Schwann cell lineage loses its remaining functional copy of NF1. This results in increases in the activity of Ras and other signaling molecules; tumorigenesis is enhanced by interactions with other cell types, inappropriate expression of EGFR and possibly the action of other growth factors. Subsequent progression to become an MPNST is associated with loss of additional tumor suppressor genes in the p19ARF-MDM2-TP53 and p16INK4A-Rb signaling cascades. Amplification or mutation of several growth factors receptors also contributes to MPNST pathogenesis.
In addition to the tumor suppressor mutations described above, a growing body of evidence indicates that aberrant signaling by growth factors or growth factor receptors also contributes to MPNST pathogenesis (Fig. 3). Amplification of genes encoding the EGFR (Perry et al., 2002), the neuregulin-1 coreceptor erbB2 (Storlazzi et al., 2006), c-Kit (Holtkamp et al., 2006), hepatocyte growth factor and its c-Met receptor (Mantripragada et al., 2008) as well as amplification (Badache and DeVries, 1998; Holtkamp et al., 2006; Mantripragada et al., 2008) and mutations (Holtkamp et al., 2006) of PDGF receptor-α have all been detected in MPNSTs. In vitro studies with MPNST cell lines support a functional role for the molecules encoded by these amplified genes. The proliferation of MPNST cells grown in the presence of limiting amounts of serum is dependent on exogenous EGF; in higher serum concentrations, these cells do not require exogenous EGF, but their proliferation is still inhibited by small molecular inhibitors and neutralizing antibodies targeting the EGFR (DeClue et al., 2000; Johansson et al., 2008). Pharmacologic inhibitors targeting neuregulin-1 receptors (Stonecypher et al., 2005), c- Kit (Badache et al., 1998) and PDGF receptors (Holtkamp et al., 2006) similarly inhibit MPNST mitogenesis. PDGF (Aoki et al., 2007) and c-Met (Su et al., 2004) have also been implicated in MPNST cell invasion. At present, it is not clear whether all of these growth factors and growth factor receptors are uniformly required for MPNST pathogenesis or whether distinct subsets of MPNSTs exist in which tumorigenesis is dependent on only some of these molecules.
Although the findings supporting the outlined scheme (Fig. 3) are compelling, it is evident that our understanding of this process is incomplete. Like most sarcomas, chromosomal gains, losses and rearrangements in MPNSTs are highly variable (Wallace et al., 2000). These neoplasms commonly have hypodiploid or near-triploid karyotypes, with gains evident on chromosome 7, 8q and 15q and losses that affect multiple chromosomal regions (1p, 9p, 11, 12p, 14q, 17q, 18, 22q, X and Y) (Forus et al., 1995; Lothe et al., 1996; Mechtersheimer et al., 1999; Mertens et al., 1995; Mertens et al., 2000; Plaat et al., 1999; Schmidt et al., 1999; Schmidt et al., 2000; Schmidt et al., 2001). Although some of the affected chromosomal regions encode tumor suppressors previously implicated in MPNST tumorigenesis [e.g., the NF1 locus (17q) and the CDKN2A gene (9p)], others do not, suggesting that we have not yet fully delineated the oncogenes and tumor suppressors relevant to MPNST pathogenesis. Chromosomal gains and losses are also highly variable from tumor to tumor, raising the question of whether there is more than one series of mutations that can lead to MPNST tumorigenesis. This latter point is underscored by the recent demonstration that two MPNST cell lines (YST-1 and STS-26T cells) derived from sporadically occurring MPNSTs have intact and functional NF1 genes (Miller et al., 2006). Paradoxically, however, investigators using microarray analyses to compare the transcriptomes of sporadic and NF1-associated MPNSTs have not found a molecular signature that distinguishes these neoplasms (Holtkamp et al., 2004; Miller et al., 2006; Watson et al., 2004).
TRANSGENIC MOUSE MODELS OF NF1-ASSOCIATED PERIPHERAL NERVE SHEATH TUMORS
Guided by the observations made in human neoplasms, several genetically modified mouse models have been constructed to directly test the role specific tumor suppressor gene mutations and growth factors play in the pathogenesis of NF1-associated peripheral nerve sheath tumor. The identification of the NF1 gene made it possible to construct mice with null mutations of this locus that were expected to recapitulate the clinical features of human NF1, including the development of neurofibromas and MPNSTs. Disappointingly, however, while mice heterozygous for a Nf1 “knockout” mutation (Nf1+/−) were viable and fertile, they developed only a limited subset of the findings seen in human NF1 patients (pheochromocytomas and leukemias) (Jacks et al., 1994). These observations suggested that neurofibromas do not develop in Nf1+/− mice because inactivation of the remaining functional Nf1 allele in murine Schwann cells occurs at a very low frequency. However, the possibility that complete loss of Nf1 in Schwann cells could trigger neurofibroma tumorigenesis could not be examined in mice homozygous for the Nf1 null mutation (Nf1−/− mice) as these animals died in utero as the result of malformations affecting the brain (exencephaly) (Lakkis et al., 1999) and the heart (endocardial cushion defects and double outlet right ventricle) (Brannan et al., 1994; Jacks et al., 1994; Lakkis and Epstein, 1998). Consequently, Nf1−/− embryos were fused with wild-type embryos to produce chimeras that lacked Nf1 in some, but not all cells (Cichowski et al., 1999). These chimeras subsequently developed tumors resembling human neurofibromas within dorsal spinal nerve roots, large nerves in limbs and cranial nerves.
As the tumor cells within neurofibromas arising in Nf1−/−;wild-type chimeras are overwhelmingly derived from the Nf1−/− embryo, these experiments did not definitively establish that cells of the Schwann cell lineage were the primary neoplastic cell type within neurofibromas. Consequently, mice were produced in which the Nf1 gene could be ablated in the Schwann cell lineage via the action of Cre recombinase expressed under the control of the Krox20/Egr2 promoter (Zhu et al., 2002). Mice with Nf1−/− Schwann cells developed microscopic hyperplastic lesions in peripheral nerves, but did not form tumors with the complex mixture of cell types characteristic of human neurofibromas. However, when Nf1 expression was ablated in the Schwann cell lineage in animals that were Nf1 haploinsufficient in all other cell types, it was found that these mice developed neurofibroma-like lesions in multiple cranial nerves and spine roots. As human neurofibromas are similarly composed of NF1−/− Schwann cells intermingled with a variety of other NF1+/− cell types, it was suggested that this indicates that the pathogenesis of neurofibromas requires both NF1-null Schwann cells and an NF1 haploinsufficient tumor environment. As this hypothesis was consistent with other observations such as the enhanced migration observed in NF1+/− Schwann cells and mast cells (see above), it was well received. However, recently some new observations have begun to raise questions as to whether an NF1 haploinsufficient tumor environment is required for neurofibroma tumorigenesis. Of particular note, mice have now been produced in which Nf1 is ablated in the Schwann cell lineage via the use of a Desert hedgehog (Dhh)-Cre driver (Wu et al., 2008) and it has been found that these animals develop dermal and plexiform neurofibromas despite having a tumor microenvironment in which all other cell types have intact Nf1 function. It is also notable that human sporadic neurofibromas undoubtedly develop within a tumor microenvironment in which the non-Schwann cell elements within the neoplasm maintain intact NF1 function.
Transgenic mouse models expressing growth factors or growth factor receptors in peripheral nerve have also been constructed and provide useful insights into the mechanisms underlying the pathogenesis of NF1-associated peripheral nerve sheath tumors and the role aberrant growth factor signaling plays in this process. As inappropriate expression of EGFR is evident in subpopulations of neurofibroma-derived Schwann cells, mice were developed in which a Schwann cell promoter drives expression of human EGFR (Ling et al., 2005). These animals develop prominent peripheral nerve hyperplasia between 2 and 12 months of age that upon microscopic examination demonstrates several characteristics of human neurofibromas including a dissociation of non-myelinating Schwann cells from axons, excess deposition of collagen and an accumulation of mast cells; palpable tumors form infrequently in these mice and backcrossing them to Nf1+/− mice does not worsen their phenotype. Studies in which EGFR was blocked via the use of neutralizing antibody cetuximab (IMC-C255) have demonstrated that the phenotype evident in this mouse model develops during a specific perinatal window, suggesting that a critical event occurring during this period, potentially mast cell recruitment (see below), accounts for the development of features (e.g., Schwann cell-axon dissociation, collagen deposition) characteristic of neurofibromas (Wu et al., 2006). The observation that the potent Schwann cell mitogen neuregulin-1 and its erbB receptors were present in human neurofibromas and MPNSTs provided a similar rationale for the construction of transgenic mice that express a class II neuregulin-1 isoform in peripheral nerve (Huijbregts et al., 2003). These animals develop prominent Schwann cell hyperplasia by 1 month of age that results from increased mitogenesis rather than inhibition of apoptosis. Long-term followup of these neuregulin-1 overexpressing mice has demonstrated that the overwhelming majority of them develop multiple neurofibroma-like neoplasms involving dorsal spinal nerve roots, cranial nerves and the sympathetic nervous system. These mice also develop aggressive malignancies with the histologic, immunohistochemical and ultrastructural characteristics of human MPNSTs and initial analyses of these neoplasms indicate that they accumulate mutations of the same tumor suppressor genes that are affected in human MPNSTs.
The frequent coexistence of NF1 and TP53 mutations in human MPNSTs suggests that these mutations cooperate to promote MPNST pathogenesis. To test this hypothesis, two laboratories independently produced mouse models in which Nf1 and p53 null mutations were linked in cis (Cichowski et al., 1999; Vogel et al., 1999). In addition to a background of the neoplasms associated with isolated Nf1 and p53 mutations, these mice developed sarcomas with the histologic and immunohistochemical characteristics of MPNSTs and a well-known MPNST variant [malignant Triton tumors, which are MPNSTs showing rhabdomyosarcomatous (i.e., muscle) differentiation]. An examination of these tumors demonstrated LOH of the remaining functional Nf1 and p53 alleles, consistent with the expectation that these mutations cooperatively promote MPNST pathogenesis. An examination of permanent cell lines derived from these murine tumors showed that the tumor cells expressed a diverse set of markers characteristic of neural crest cells, Schwann cells and myogenic cells (Vogel et al., 1999). This suggested that the MPNST-like neoplasms arising in these mice were derived from a multipotent precursor, a postulate that is now being explored in a new generation of transgenic mouse models (see below).
NEW INSIGHTS INTO THE CELL OF ORIGIN OF NEUROFIBROMAS
Although there is general agreement that the neoplastic cell that gives rise to neurofibromas is derived from the Schwann cell lineage, the question of precisely which cell type within this lineage gives rise to these tumors is an area of active investigation. Recent reports examining this question have attempted to relate the identity of the neurofibroma initiating cell type to known stages in the differentiation of peripheral glia from migrating neural crest cells (Jessen and Mirsky, 2002; Jessen and Mirsky, 2005)(see also the more detailed description of this process provided by Sommer and Woodhoo in this issue). In the trunk of mice (Fig. 4), neural crest cells give rise to BLBP-immunoreactive Schwann cell precursors at embryonic day (E)12–13 (Jessen and Mirsky, 2002; Jessen and Mirsky, 2005) that are capable of differentiating into Schwann cells, myofibroblasts and neurons in vitro (Morrison et al., 1999) and Schwann cells and fibroblasts in vivo (Joseph et al., 2004); given their multipotency, some investigators have referred to these cells as neural crest stem cells (Morrison et al., 1999). Around E13–E15, these Schwann cell precursors in turn give rise to immature Schwann cells, which in the first postnatal weeks differentiate into myelinating or nonmyelinating Schwann cells. Migrating neural crest cells also give rise to boundary cap cells (E10.5–11), a second population with stem cell properties (Aquino et al., 2006; Maro et al., 2004; Stemple and Anderson, 1993; Zorick et al., 1999), which subsequently differentiate to become spinal root glial cells via a series of poorly understood developmental stages. Consequently, there are multiple developmental stages in the Schwann cell lineage which could potentially give rise to the primary neoplastic cell type in neurofibromas.
Fig. 4.
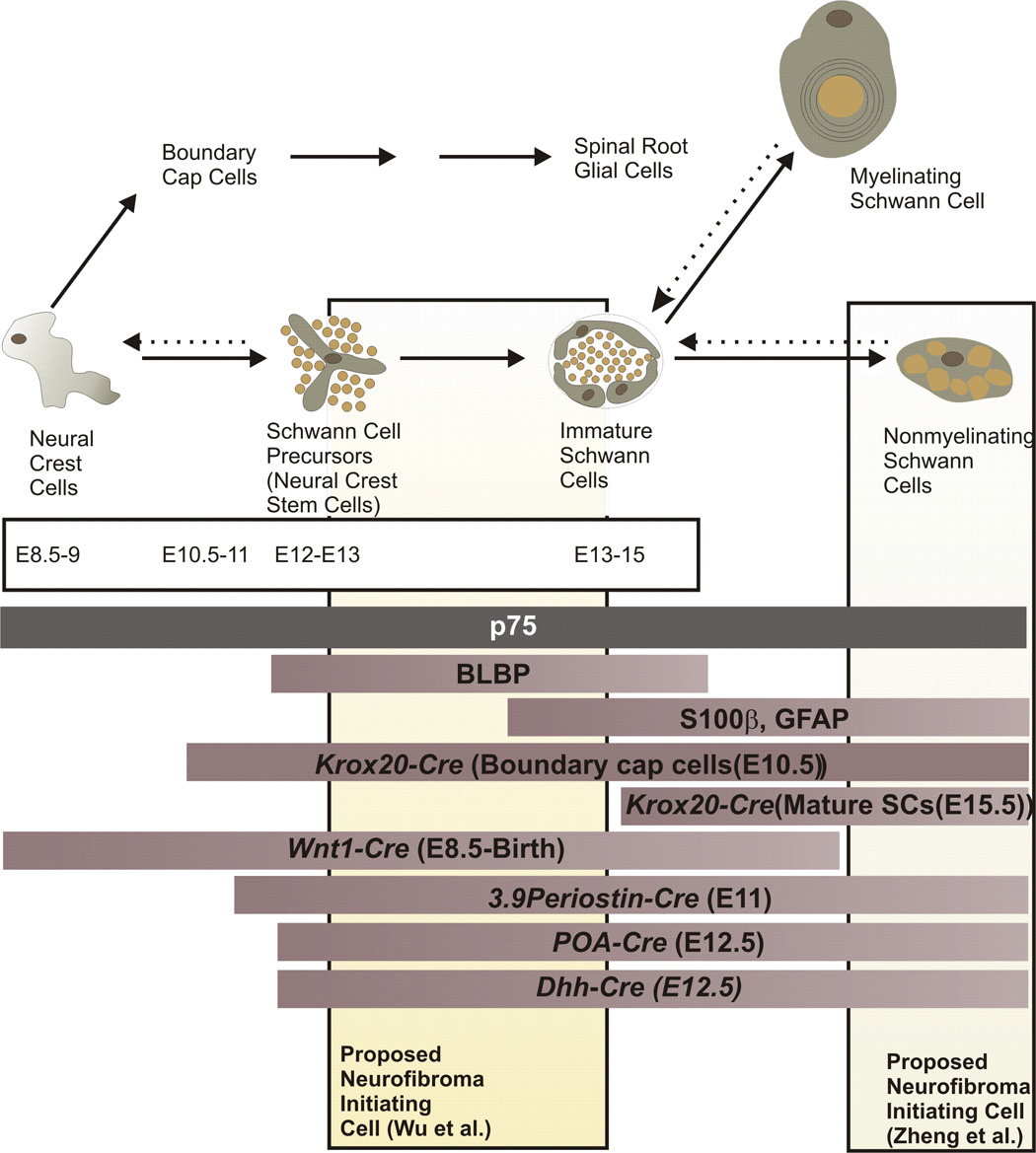
Schematic illustration of the differentiation of neural crest cells and their progeny in the peripheral nervous system, relating this to the expression of markers for various developmental stages and periods when Cre-mediated recombination is evident in the Schwann cell lineage in various mouse models of neurofibroma pathogenesis. Times indicated for the appearance of each cell type (embryonic days 8.5 to 15) refer to the developmental sequence seen in peripheral nerves within the trunk of mice. The three bars immediately beneath the time course (p75; BLBP; S100β, GFAP) indicate the period that markers for specific developmental stages are expressed. The next series of bars indicate the periods in which Cre-mediated recombination is evident in the Schwann cell lineage in Nf1 flox mice in which recombination is driven by the indicated promoters. Wnt1-Cre;Nf1fl/− mice and most 3.9Periostin-Cre;Nf1fl/− mice die at birth (Gitler et al., 2003; Joseph et al., 2008). Nf1 ablation produced by Cre recombinase expression driven by the Krox20, P0A and Dhh promoters results in neurofibroma pathogenesis in adult mice. See the text for a more detailed discussion of the phenotype of these mouse models and the implications this has for the cell of origin of neurofibromas.
Three recent studies agree that Nf1-null neural crest cells are not the source of the primary neoplastic cell type in neurofibromas (Joseph et al., 2008; Wu et al., 2008; Zheng et al., 2008) and that biallelic loss of Nf1 in more differentiated glial cells is instead relevant to the pathogenesis of these neoplasms. However, these studies differ as to precisely which of the more differentiated glial stages give rise to neurofibromas. Morrison and colleagues have reported that Nf1−/− neural crest stem cells (Schwann cell precursors) do not form tumors when grafted in the sciatic nerves of Nf1+/− mice or persist postnatally in regions of the PNS of 3.9Periostin-Cre;Nf1fl/− mice that give rise to neurofibromas in other mouse models (Joseph et al., 2008). Zhu and colleagues have found that ablation of Nf1 expression in Schwann cell precursors in P0A-Cre;Nf1fl/− mice efficiently induces neurofibroma formation, but noted that nonmyelinating Schwann cells in Remak bundles dissociated from axons and proliferated postnatally (Zheng et al., 2008). Considering these observations jointly, these laboratories concluded that non-myelinating Schwann cells in Remak bundles, which do not form until the third postnatal week in rodents (Martin and Webster, 1973; Monk et al., 2008; Webster et al., 1973), give rise to the neoplastic cells within neurofibromas.
When Nf1 is ablated at E12.5 in the late boundary cap cells and Schwann cell precursors of DhhCre;Nf1fl/fl mice, animals develop plexiform neurofibromas as well as dermal neurofibromas (Wu et al., 2008). As in P0A-Cre;Nf1fl/− mice, neurofibroma formation in DhhCre;Nf1fl/fl mice correlates with a disruption of normal axon-glial interactions in Remak bundles, the development of hypertrophy throughout peripheral nerves and an enhancement of mast cell recruitment into these nerves. The hypertrophied peripheral nerves and neurofibromas that develop in DhhCre;Nf1fl/fl mice contain large numbers of BLBP+/S100β+ cells, suggesting that cells similar to immature Schwann cells accumulate in these tissues. Considered together with the fact that the P0A-Cre promoter initially becomes active in Schwann cell precursors [at E11.5 (Zheng et al., 2008)], these findings highlight the potential relevance of Schwann cell precursors to tumorigenesis. Considered together with the presence of large numbers of BLBP+/S100β+ cells in the PNS of DhhCre;Nf1fl/fl mice, these findings suggest that the neurofibroma progenitor cell corresponds to cells at the boundary between Schwann cell precursors and immature Schwann cells; an alternative possibility, namely that neurofibroma progenitors instead arise from late boundary cap cells or their progeny (for which, unfortunately, no definitive markers exist), cannot be excluded (Wu et al., 2008). A resolution of these conflicting views of the origin of the primary neoplastic cell type in neurofibromas will likely require the development of mice in which Nf1 can be inactivated via the use of a transgene in which Cre expression is controlled by a promoter that is active late in glial development or by using an inducible form of Cre.
Recent experiments in which transgenic mice expressing EGFR in peripheral nerve (CNPase-EGFR mice) were used to assess the role wild type mast cells play in the development of nerve pathology (Monk et al., 2008) do not fully support the interpretation that neurofibromas arise from non-myelinating Schwann cells. In this mouse model, axon-Schwann cell interactions in Remak bundles are also disrupted coincident with the recruitment of mast cells into the nerve, which occurs 5–6 weeks postnatally. The disruption of axon-Schwann cell interactions in Remak bundles is apparently dependent on the action of mast cells as genetically ablating mast cell recruitment by crossing CNPase-EGFR mice to c-Kit hypomorphs (the mouse strain W41) prevents the development of peripheral nerve pathology. Further, the development of peripheral nerve pathology is reconstituted when wild-type bone marrow-derived cells are grafted into CNPase-EGFR;W41 mice and pharmacologic stabilization of mast cells by cromolyn administration prevents the development of nerve pathology in CNPase-EGFR mice. These findings suggest the possibility that mast cells are recruited into peripheral nerve by a small number of more primitive cells that are persistently present in Nf1 knockout mice, resulting in a secondary disruption of Remak bundles and dysfunction of non-myelinating Schwann cells.
CONCLUSION
Based on the findings described above, a complex picture has emerged in which the complete loss of neurofibromin expression in the Schwann cell lineage acts cooperatively with other cell types and growth factor signaling to promote the formation of neurofibromas. Other mutations accumulate to promote progression to become MPNSTs. However, major questions remain unanswered regarding such issues as the biological basis for the differing malignant potential of different neurofibroma subtypes, the nature of the growth factor signaling events and modifier genes that influence the formation of neurofibromas, the identity of additional oncogenes and tumor suppressors that contribute to MPNST pathogenesis and the precise identity of the cells in the Schwann cell lineage that give rise to neurofibromas and MPNSTs. Answering questions such as these will undoubtedly enhance our growing understanding of these complex neoplasms and point the way to the development of effective new therapies that can relieve the extensive morbidity and mortality peripheral nerve sheath tumors cause in NF1 patients. Continued cooperation between clinical and basic science investigators seeking to answer these questions is essential if we are to achieve this goal.
ACKNOWLEDGMENTS
SLC is supported by the National Institutes of Health (grants R01 NS048353, R01 CA122804 and P30 NS057098). NR is supported by the National Institutes of Health (R01 NS028840 and R01 CA118032), and the Department of Defense Program on Neurofibromatosis (grants DAMD-17-01-0704 and W81XWH-04-1-0273). We thank Dr. Bruce R. Korf for his insightful comments on this manuscript and discussions of the clinical features of NF1-associated peripheral nerve sheath tumors and Dr. Jon Williams for his contributions to the discussion presented on the cell of origin for neurofibromas.
REFERENCES
- Aoki M, Nabeshima K, Koga K, Hamasaki M, Suzumiya J, Tamura K, Iwasaki H. Imatinib mesylate inhibits cell invasion of malignant peripheral nerve sheath tumor induced by platelet-derived growth factor-BB. Lab Invest. 2007;87:767–779. doi: 10.1038/labinvest.3700591. [DOI] [PubMed] [Google Scholar]
- Aquino J, Hjerling-Leffler J, Koltzenburg M, Villar M, Ernfors P. In vitro and in vivo differentiation of boundary cap neural crest stem cells into mature Schwann cells. Exp Neurol. 2006;198:438–449. doi: 10.1016/j.expneurol.2005.12.015. [DOI] [PubMed] [Google Scholar]
- Atit RA, Mitchell K, Nguyen L, Warshawsky D, Ratner N. The neurofibromatosis type 1 (Nf1) tumor suppressor is a modifier of carcinogen-induced pigmentation and papilloma formation in C57Bl/6 mice. J Invest Dermatol. 2000;114:1093–1100. doi: 10.1046/j.1523-1747.2000.00994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badache A, DeVries GH. Neurofibrosarcoma-derived Schwann cells overexpress platelet-derived growth factor (PDGF) receptors and are induced to proliferate by PDGF BB. J Cell Physiol. 1998;177:334–342. doi: 10.1002/(SICI)1097-4652(199811)177:2<334::AID-JCP15>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Badache A, Muja N, DeVries GH. Expression of Kit in neurofibromin-deficient human Schwann cells: role in Schwann cell hyperplasia associated with type 1 neurofibromatosis. Oncogene. 1998;17:795–800. doi: 10.1038/sj.onc.1201978. [DOI] [PubMed] [Google Scholar]
- Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- Barbarot S, Nicol C, Volteau C, Le Forestier D, N'Guyen JM, Mansat E, Wolkenstein P, Stalder JF. Cutaneous lesions in neurofibromatosis 1: confused terminology. Br J Dermatol. 2007;157:183–184. doi: 10.1111/j.1365-2133.2007.07903.x. [DOI] [PubMed] [Google Scholar]
- Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–715. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- Birindelli S, Perrone F, Oggionni M, Lavarino C, Pasini B, Vergani B, Ranzani GN, Pierotti MA, Pilotti S. Rb and TP53 pathway alterations in sporadic and NF1-related malignant peripheral nerve sheath tumors. Lab Invest. 2001;81:833–844. doi: 10.1038/labinvest.3780293. [DOI] [PubMed] [Google Scholar]
- Boikis SA, Stratakis CA. Carney complex: the first 20 years. Curr Opin Neurol. 2007;19:24–29. doi: 10.1097/CCO.0b013e32801195eb. [DOI] [PubMed] [Google Scholar]
- Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8:1019–1029. doi: 10.1101/gad.8.9.1019. [DOI] [PubMed] [Google Scholar]
- Carli M, Ferrari A, Mattke A, Zanetti I, Casanova M, Bisogno G, Cecchetto G, Alaggio R, De Sio L, Koscielniak E, Sotti G, Treuner J. Pediatric malignant peripheral nerve sheath tumor: the Italian and German Soft Tissue Sarcoma Cooperative Group. J Clin Oncol. 2005;23:8422–8430. doi: 10.1200/JCO.2005.01.4886. [DOI] [PubMed] [Google Scholar]
- Carroll SL, Stonecypher MS. Tumor suppressor mutations and growth factor signaling in the pathogenesis of NF1-associated peripheral nerve sheath tumors. II. The role of dysregulated growth factor signaling. J Neuropath Exp Neurol. 2005;64:1–9. doi: 10.1093/jnen/64.1.1. [DOI] [PubMed] [Google Scholar]
- Cichowski K, Shih TS, Schmitt E, Santiago S, Reilly K, McLaughlin ME, Bronson RT, Jacks T. Mouse models of tumor development in neurofibromatosis type 1. Science. 1999;286:2172–2176. doi: 10.1126/science.286.5447.2172. [DOI] [PubMed] [Google Scholar]
- Cohen PR, Rapini RP, Farhood AI. Expression of the hematopoietic progenitor cell antigen CD34 in vascular and spindle cell tumors. J Cutan Pathol. 1993;20:15–20. doi: 10.1111/j.1600-0560.1993.tb01243.x. [DOI] [PubMed] [Google Scholar]
- Courtois-Cox S, Genther Williams SM, Reczek EE, Johnson BW, McGillicuddy LT, Johannessen CM, Hollstein PE, MacCollin M, Cichowski K. A negative feedback signaling network underlies oncogene-induced senescence. Cancer Cell. 2006;10:459–472. doi: 10.1016/j.ccr.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Angelo I, Welti S, Bonneau F, Scheffzek K. A novel bipartite phospholipid-binding module in the neurofibromatosis type 1 protein. EMBO Reports. 2006;7:174–179. doi: 10.1038/sj.embor.7400602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-ras in astrocytes. Cancer Res. 2005;65:236–245. [PubMed] [Google Scholar]
- DeClue JE, Heffelfinger S, Benvenuto G, Ling B, Li S, Rui W, Vass WC, Viskochil D, Ratner N. Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J Clin Invest. 2000;105:1233–1241. doi: 10.1172/JCI7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- Demitsu T, Murata S, Kakurai M, Kiyosawa T, Yaoita H. Malignant schwannoma-derived cells support human skin mast cell survival in vitro. J Dermatol Sci. 1998;16:129–134. doi: 10.1016/s0923-1811(97)00038-8. [DOI] [PubMed] [Google Scholar]
- Ducatman BS, Scheithauer BW, Piepgras DG, Reiman HM, Ilstrup DM. Malignant peripheral nerve sheath tumors. A clinicopathologic study of 120 cases. Cancer. 1986;57:2006–2021. doi: 10.1002/1097-0142(19860515)57:10<2006::aid-cncr2820571022>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Dugoff L, Sujansky E. Neurofibromatosis type 1 and pregnancy. Am J Med Genet. 1996;66:7–10. doi: 10.1002/(SICI)1096-8628(19961202)66:1<7::AID-AJMG2>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Easton DF, Ponder MA, Huson SM, Ponder BA. An analysis of variation in expression of neurofibromatosis (NF) type 1 (NF1): evidence for modifying genes. Am J Hum Genet. 1993;53:305–313. [PMC free article] [PubMed] [Google Scholar]
- Evans DGR, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumors in neurofibromatosis 1. J Med Genet. 2002;39:311–314. doi: 10.1136/jmg.39.5.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferner RE. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol. 2007;6:340–351. doi: 10.1016/S1474-4422(07)70075-3. [DOI] [PubMed] [Google Scholar]
- Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumours in neurofibromatosis 1. Cancer Res. 2002;62:1573–1577. [PubMed] [Google Scholar]
- Ferner RE, O'Doherty MJ. Neurofibroma and schwannoma. Curr Opin Neurol. 2002;15:679–684. doi: 10.1097/01.wco.0000044763.39452.aa. [DOI] [PubMed] [Google Scholar]
- Forus A, Weghuis DO, Smeets D, Fodstad O, Myklebost O, van Kessel AG. Comparative genomic hybridization analysis of human sarcomas: I. Occurrence of genomic imbalances and identification of a novel major amplicon at 1q21-q22 in soft tissue sarcomas. Genes Chromosomes Cancer. 1995;14:8–14. doi: 10.1002/gcc.2870140103. [DOI] [PubMed] [Google Scholar]
- Fukuda T, Ichimura E, Shinozaki T, Sano T, Kashiwabara K, Oyama T, Nakajima T, Nakamura T. Coexpression of HGF and c-Met/HGF receptor in human bone and soft tissue tumors. Pathology Int. 1998;48:757–762. doi: 10.1111/j.1440-1827.1998.tb03834.x. [DOI] [PubMed] [Google Scholar]
- Gitler AD, Zhu Y, Ismat FA, Lu MM, Yamauchi Y, Parada LF, Epstein JA. Nf1 has an essential role in endothelial cells. Nat Genet. 2003;33:75–79. doi: 10.1038/ng1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubic M, Roudebush M, Dobrowolski S, Wolfman A, Stacey DW. Catalytic properties, tissue and intracellular distribution of neurofibromin. Oncogene. 1992;7:2151–2159. [PubMed] [Google Scholar]
- Gutmann DH. The neurofibromatoses: when less is more. Hum Mol Genet. 2001;10:747–755. doi: 10.1093/hmg/10.7.747. [DOI] [PubMed] [Google Scholar]
- Hagel C, Zils U, Peiper M, Kluwe L, Gotthard S, Friedrich RE, Zurakowski D, von DA, Mautner VF. Histopathology and clinical outcome of NF1-associated vs. sporadic malignant peripheral nerve sheath tumors. J Neurooncol. 2007;82:187–192. doi: 10.1007/s11060-006-9266-2. [DOI] [PubMed] [Google Scholar]
- Han JW, McCormick F, Macara IG. Regulation of Ras-GAP and the neurofibromatosis-1 gene product by eicosanoids. Science. 1991;252:576–579. doi: 10.1126/science.1902323. [DOI] [PubMed] [Google Scholar]
- Hawes JJ, Tuskan RG, Reilly KM. Nf1 expression is dependent on strain background: implications for tumor suppressor haploinsufficiency studies. Neurogenetics. 2007;8:121–130. doi: 10.1007/s10048-006-0078-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota S, Nomura S, Asada H, Ito A, Morii E, Kitamura Y. Possible involvement of c-kit receptor and its ligand in increase of mast cells in neurofibroma tissues. Arch Pathol Lab Med. 1993;117:996–999. [PubMed] [Google Scholar]
- Holtkamp N, Atallah I, Okuducu A-F, Mucha J, Hartmann C, Mautner VF, Friedrich RE, Mawrin C, von Deimling A. MMP-13 and p53 in the progression of malignant peripheral nerve sheath tumors. Neoplasia. 2007;9:671–677. doi: 10.1593/neo.07304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtkamp N, Okuducu AF, Mucha J, Afanasieva A, Hartmann C, Atallah I, Estevez-Schwarz L, Mawrin C, Friedrich RE, Mautner VF, von Deimling A. Mutation and expression of PDGFRA and KIT in malignant peripheral nerve sheath tumors, and its implications for imatinib sensitivity. Carcinogenesis. 2006;27:664–671. doi: 10.1093/carcin/bgi273. [DOI] [PubMed] [Google Scholar]
- Holtkamp N, Reuss DE, Atallah I, Kuban RJ, Hartmann C, Mautner VF, Frahm S, Friedrich RE, Algermissen B, Pham VA, Prietz S, Rosenbaum T, Estevez-Schwarz L, von Deimling A. Subclassification of nerve sheath tumors by gene expression profiling. Brain Pathol. 2004;14:258–264. doi: 10.1111/j.1750-3639.2004.tb00062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YZ, Rangwala F, Fulkerson PC, Ling B, Reed E, Cox AD, Kamholz J, Ratner N. Role of TC21/R-Ras2 in enhanced migration of neurofibromin-deficient Schwann cells. Oncogene. 2004;23:368–378. doi: 10.1038/sj.onc.1207075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbregts RPH, Roth KA, Schmidt RE, Carroll SL. Hypertrophic neuropathies and malignant peripheral nerve sheath tumors in transgenic mice overexpressing glial growth factor β3 in myelinating Schwann cells. J Neurosci. 2003;23:7269–7280. doi: 10.1523/JNEUROSCI.23-19-07269.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson SM, Harper PS, Compston DA. Von Recklinghausen neurofibromatosis. A clinical and population study in south-east Wales. Brain. 1988;111:1355–1381. doi: 10.1093/brain/111.6.1355. [DOI] [PubMed] [Google Scholar]
- Ingram DA, Hiatt K, King AJ, Fisher L, Shivakumar R, Derstine C, Wenning MJ, Diaz B, Travers JB, Hood A, Marshall M, Williams DA, Clapp DW. Hyperactivation of p21ras and the hematopoietic-specific Rho GTPase, Rac2, cooperate to alter the proliferation of neurofibromin-deficient mast cells in vivo and in vitro. J Exp Med. 2001;194:57–69. doi: 10.1084/jem.194.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingram DA, Yang FC, Travers JB, Wenning MJ, Hiatt K, New S, Hood A, Shannon K, Williams DA, Clapp DW. Genetic and biochemical evidence that haploinsufficiency of the Nf1 tumor suppressor gene modulates melanocyte and mast cell fates in vivo. J Exp Med. 2000;191:181–188. doi: 10.1084/jem.191.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nature Genetics. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. Signals that determine Schwann cell identity. J Anat. 2002;200:367–376. doi: 10.1046/j.1469-7580.2002.00046.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Johnson BW, Williams SM, Chan AW, Reczek EE, Lynch RC, Rioth MJ, McClatchey A, Ryeom S, Cichowski K. TORC1 is essential for NF1-associated malignancies. Curr Biol. 2008;18:56–62. doi: 10.1016/j.cub.2007.11.066. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci USA. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson G, Mahller Y, Collins MH, Kim M-O, Nobukuni T, Perentesis JP, Cripe TP, Lane HA, Kozma S, Thomas G, Ratner N. Effective in vivo targeting of the mTOR pathway in malignant peripheral nerve sheath tumor. Mol Cancer Therapeutics. 2008 doi: 10.1158/1535-7163.MCT-07-2335. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph NM, Mosher JT, Buchstaller J, Snider P, McKeever PE, Lim M, Conway SJ, Parada LF, Zhu Y, Morrison SJ. The loss of Nf1 transiently promotes self-renewal but not tumorigenesis by neural crest stem cells. Cancer Cell. 2008;13:129–140. doi: 10.1016/j.ccr.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph NM, Mukouyana Y, Mosher JT, Jaegle M, Crone SA, Dormand EL, Lee K-F, Meijer D, Anderson DJ, Morrison SJ. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development. 2004;131:5599–5612. doi: 10.1242/dev.01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadono T, Kikuchi K, Nakagawa H, Tamaki K. Expressions of various growth factors and their receptors in tissues from neurofibroma. Dermatology. 2000;201:10–14. doi: 10.1159/000018421. [DOI] [PubMed] [Google Scholar]
- Kadono T, Soma Y, Takehara K, Nakagawa H, Ishibashi Y, Kikuchi K. The growth regulation of neurofibroma cells in neurofibromatosis type-1: increased responses to PDGF-BB and TGF-beta 1. Biochem Biophys Res Comm. 1994;198:827–834. doi: 10.1006/bbrc.1994.1118. [DOI] [PubMed] [Google Scholar]
- Khalaf WF, Yang FC, Chen S, White H, Bessler W, Ingram DA, Clapp DW. K-ras is critical for modulating multiple c-kit-mediated cellular functions in wild-type and Nf1+/− mast cells. J Immunol. 2007;178:2527–2534. doi: 10.4049/jimmunol.178.4.2527. [DOI] [PubMed] [Google Scholar]
- Kim HA, Ling B, Ratner N. Nf1-deficient mouse Schwann cells are angiogenic and invasive and can be induced to hyperproliferate: reversion of some phenotypes by an inhibitor of farnesyl protein transferase. Mol Cell Biol. 1997;17:862–872. doi: 10.1128/mcb.17.2.862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HA, Ratner N, Roberts TM, Stiles CD. Schwann cell proliferative responses to cAMP and Nf1 are mediated by cyclin D1. J Neurosci. 2001;21:1110–1116. doi: 10.1523/JNEUROSCI.21-04-01110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HA, Rosenbaum T, Marchionni MA, Ratner N, DeClue JE. Schwann cells from neurofibromin deficient mice exhibit activation of p21ras, inhibition of cell proliferation and morphological changes. Oncogene. 1995;11:325–335. [PubMed] [Google Scholar]
- Kluwe L, Friedrich R, Mautner VF. Loss of NF1 allele in Schwann cells but not fibroblasts derived from an NF1-associated neurofibroma. Genes Chromosomes Cancer. 1999;24:283–285. doi: 10.1002/(sici)1098-2264(199903)24:3<283::aid-gcc15>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Kourea HP, Orlow I, Scheithauer BW, Cordon-Cardo C, Woodruff JM. Deletions of the INK4A gene occur in malignant peripheral nerve sheath tumors but not in neurofibromas. Am J Pathol. 1999;155:1855–1860. doi: 10.1016/S0002-9440(10)65504-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakkis MM, Epstein JA. Neurofibromin modulation of ras activity is required for normal endocardial-mesenchymal transformation in the developing heart. Develop. 1998;125:4359–4367. doi: 10.1242/dev.125.22.4359. [DOI] [PubMed] [Google Scholar]
- Lakkis MM, Golden JA, O'Shea KS, Epstein JA. Neurofibromin deficiency in mice causes exencephaly and is a modifier for Splotch neural tube defects. Develop Biology. 1999;212:80–92. doi: 10.1006/dbio.1999.9327. [DOI] [PubMed] [Google Scholar]
- Le LQ, Parada LF. Tumor microenvironment and neurofibromatosis type 1: connecting the GAPs. Oncogene. 2007;26:4609–4616. doi: 10.1038/sj.onc.1210261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legius E, Dierick H, Wu R, Hall BK, Marynen P, Cassiman JJ, Glover TW. TP53 mutations are frequent in malignant NF1 tumors. Genes Chromosomes Cancer. 1994;10:250–255. doi: 10.1002/gcc.2870100405. [DOI] [PubMed] [Google Scholar]
- Legius E, Marchuk DA, Collins FS, Glover TW. Somatic deletion of the neurofibromatosis type 1 gene in a neurofibrosarcoma supports a tumour suppressor gene hypothesis. Nature Genetics. 1993;3:122–125. doi: 10.1038/ng0293-122. [DOI] [PubMed] [Google Scholar]
- Ling BC, Wu J, Miller SJ, Monk KR, Shamekh R, Rizvi TA, Decourten-Myers G, Vogel KS, DeClue JE, Ratner N. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell. 2005;7:65–75. doi: 10.1016/j.ccr.2004.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lothe RA, Karhu R, Mandahl N, Mertens F, Saeter G, Heim S, Boresen-Dale AL, Kallioniemi OP. Gain of 17q24-qter detected by comparative genomic hybridization in malignant tumors from patients with von Recklinghausen's neurofibromatosis. Cancer Res. 1996;56:4778–4781. [PubMed] [Google Scholar]
- Lothe RA, Smith-Sorensen B, Hektoen M, Stenwig AE, Mandahl N, Saeter G, Mertens F. Biallelic inactivation of TP53 rarely contributes to the development of malignant peripheral nerve sheath tumors. Genes Chromosomes Cancer. 2001;30:202–206. [PubMed] [Google Scholar]
- Maertens O, Brems H, Vandesompele J, De Raedt T, Heyns I, Rosenbaum T, De Schepper S, De Paepe A, Mortier G, Janssens S, Speleman F, Legius E, Messiaen L. Comprehensive NF1 screening on cultured Schwann cells from neurofibromas. Hum Mutat. 2006;27:1030–1040. doi: 10.1002/humu.20389. [DOI] [PubMed] [Google Scholar]
- Mantripragada KK, Spurlock G, Kluwe L, Chuzhanova N, Ferner RE, Frayling IM, Dumanski JP, Guha A, Mautner V, Upadhyaya M. High-resolution DNA copy number profiling of malignant peripheral nerve sheath tumors using targeted microarray-based comparative genomic hybridization. Clin Cancer Res. 2008;14:1015–1024. doi: 10.1158/1078-0432.CCR-07-1305. [DOI] [PubMed] [Google Scholar]
- Maro G, Vermeren M, Voiculescu O, Melton L, Cohen J, Charnay P, Topilko P. Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nat Neurosci. 2004;7:930–938. doi: 10.1038/nn1299. [DOI] [PubMed] [Google Scholar]
- Martin JR, Webster HD. Mitotic Schwann cells in developing nerve: their changes in shape, fine structure, and axon relationships. Dev Biol. 1973;32:432. doi: 10.1016/0012-1606(73)90251-0. [DOI] [PubMed] [Google Scholar]
- Mattingly RR, Kraniak JM, Dilworth JT, Mathieu P, Bealmear B, Nowak JE, Benjamins JA, Tainsky MA, Reiners JJ., Jr The mitogen-activated protein kinase/extracellular signal-regulated kinase kinase inhibitor PD184352 (CI-1040) selectively induces apoptosis in malignant schwannoma cell lines. J Pharm Exp Therap. 2006;316:456–465. doi: 10.1124/jpet.105.091454. [DOI] [PubMed] [Google Scholar]
- Mawrin C, Kirches E, Boltze C, Dietzmann K, Roessner A, Schneider-Stock R. Immunohistochemical and molecular analysis of p53, RB, and PTEN in malignant peripheral nerve sheath tumors. Virchows Arch. 2002;440:610–615. doi: 10.1007/s00428-001-0550-4. [DOI] [PubMed] [Google Scholar]
- McCaughan JA, Holloway SM, Davidson R, Lam WWK. Further evidence of the increased risk for malignant peripheral nerve sheath tumour from a Scottish cohort of patients with neurofibromatosis type 1. J Med Genet. 2007;44:463–466. doi: 10.1136/jmg.2006.048140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClatchey AI, Giovannini M. Membrane organization and tumorigenesis--the NF2 tumor suppressor, Merlin. Genes Dev. 2005;19:2265–2277. doi: 10.1101/gad.1335605. [DOI] [PubMed] [Google Scholar]
- Mechtersheimer G, Otano-Joos M, Ohl S, Benner A, Lehnert T, Willeke F, Moller P, Otto HF, Lichter P, Joos S. Analysis of chromosomal imbalances in sporadic and NF1- associated peripheral nerve sheath tumors by comparative genomic hybridization. Genes Chromosomes Cancer. 1999;25:362–369. [PubMed] [Google Scholar]
- Menon AG, Anderson KM, Riccardi VM, Chung RY, Whaley JM, Yandell DW, Farmer GE, Freiman RN, Lee JK, Li FP, Barker DF, Ledbetter DH, Kleider A, Martuza RL, Gusella JF, Seizinger BR. Chromosome 17p deletions and p53 gene mutations associated with the formation of malignant neurofibrosarcomas in von Recklinghausen neurofibromatosis. Proc Natl Acad Sci USA. 1990;87:5435–5439. doi: 10.1073/pnas.87.14.5435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens F, Dal Cin P, de Wever I, Fletcher CDM, Mandahl N, Mitelman F, Rosai J, Rydholm A, Sciot R, Tallini G, van den Berghe H, Vanni R, Willen H. Cytogenetic characterization of peripheral nerve sheath tumours: a report of the CHAMP study group. J Pathol. 2000;190:31–38. doi: 10.1002/(SICI)1096-9896(200001)190:1<31::AID-PATH505>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Mertens F, Rydholm A, Bauer HF, Limon J, Nedoszytko B, Szadowska A, Willen H, Heim S, Mitelman F, Mandahl N. Cytogenetic findings in malignant peripheral nerve sheath tumors. Int J Cancer. 1995;61:793–798. doi: 10.1002/ijc.2910610609. [DOI] [PubMed] [Google Scholar]
- Messiaen LM, Callens T, Mortier G, Beysen D, Vandenbroucke I, Van Roy N, Speleman F, De Paepe A. Exhaustive mutation analysis of the NF1 gene allows identification of 95% of mutations and reveals a high frequency of unusual splicing defects. Human Mutation. 2000;15:541–555. doi: 10.1002/1098-1004(200006)15:6<541::AID-HUMU6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Miller SJ, Rangwala F, Williams J, Ackerman P, Kong S, Jegga AG, Kaiser S, Aronow BJ, Frahm S, Kluwe L, Mautner V, Upadhyaya M, Muir D, Wallace M, Hagen J, Quelle DE, Watson MA, Perry A, Gutmann DH, Ratner N. Large-scale molecular comparison of human Schwann cells to malignant peripheral nerve sheath tumor cell lines and tissues. Cancer Res. 2006;66:2584–2591. doi: 10.1158/0008-5472.CAN-05-3330. [DOI] [PubMed] [Google Scholar]
- Monk KR, Wu J, Williams JP, Finney BA, Fitzgerald ME, Filippi M-D, Ratner N. Mast cells can contribute to axon-glial dissociation and fibrosis in peripheral nerve. Neuron Glia Biology. 2008;3:1–12. doi: 10.1017/S1740925X08000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison SJ, White PM, Zock C, Anderson DJ. Prospective identification, isolation by flow cytometry, and in vivo self-renewal of multipotent mammalian neural crest stem cells. Cell. 1999;96:737–749. doi: 10.1016/s0092-8674(00)80583-8. [DOI] [PubMed] [Google Scholar]
- Mousley CJ, Tyeryar KR, Ryan MM, Bankaitis VA. Sec14p-like proteins regulate phosphoinositide homeostasis and intracellular protein and lipid trafficking in yeast. Biochem Soc Trans. 2006;34:346–350. doi: 10.1042/BST0340346. [DOI] [PubMed] [Google Scholar]
- Muir D. Differences in proliferation and invasion by normal, transformed and NF1 Schwann cell cultures are influenced by matrix metalloproteinase expression. Clin Exp Metastasis. 1995;13:303–314. doi: 10.1007/BF00133486. [DOI] [PubMed] [Google Scholar]
- Nielsen GP, Stemmer-Rachamimov AO, Ino Y, Moller MB, Rosenberg AE, Louis DN. Malignant transformation of neurofibromas in neurofibromatosis 1 is associated with CDKN2A/p16 inactivation. Am J Pathol. 1999;155:1879–1884. doi: 10.1016/S0002-9440(10)65507-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba Y, Mochizuki N, Yamashita S, Chan AM, Schrader JW, Hattori S, Nagashima K, Matsuda M. Regulatory proteins of R-Ras, TC21/R-Ras2, and M-Ras/R-Ras3. J Biol Chem. 2000;275:20020–20026. doi: 10.1074/jbc.M000981200. [DOI] [PubMed] [Google Scholar]
- Okada T, You L, Giancotti FG. Shedding light on Merlin's wizardry. Trends Cell Biol. 2007;17:222–229. doi: 10.1016/j.tcb.2007.03.006. [DOI] [PubMed] [Google Scholar]
- Packer RJ, Gutmann DH, Rubinstein A, Viskochil D, Zimmerman RA, Vezina G, Small J, Korf B. Plexiform neurofibromas in NF1: Toward biologic-based therapy. Neurology. 2002;58:1461–1470. doi: 10.1212/wnl.58.10.1461. [DOI] [PubMed] [Google Scholar]
- Perry A, Kunz SN, Fuller CE, Banerjee R, Marley EF, Liapis H, Watson MW, Gutmann DH. Differential NF1, p16, and EGFR patterns by interphase cytogenetics (FISH) in malignant peripheral nerve sheath tumor (MPNST) and morphologically similar spindle cell neoplasms. J Neuropath Exp Neurol. 2002;61:702–709. doi: 10.1093/jnen/61.8.702. [DOI] [PubMed] [Google Scholar]
- Plaat BE, Molenaar WM, Mastik MF, Hoekstra HJ, te Meerman GJ, van den Berg E. Computer-assisted cytogenetic analysis of 51 malignant peripheral-nerve-sheath tumours: sporadic vs. neurofibromatosis-type-1-associated malignant schwannomas. Int J Cancer. 1999;83:171–178. doi: 10.1002/(sici)1097-0215(19991008)83:2<171::aid-ijc5>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Rao UN, Sonmez-Alpan E, Michalopoulos GK. Hepatocyte growth factor and c-MET in benign and malignant peripheral nerve sheath tumors. Hum Pathol. 1997;28:1066–1070. doi: 10.1016/s0046-8177(97)90060-5. [DOI] [PubMed] [Google Scholar]
- Reilly KM, Broman KW, Bronson RT, Tsang S, Loisel DA, Christy ES, Sun Z, Diehl J, Munroe DJ, Tuskan RG. An imprinted locus epistatically influences Nstr1 and Nstr2 to control resistance to nerve sheath tumors in a neurofibromatosis type 1 mouse model. Cancer Res. 2006;66:62–68. doi: 10.1158/0008-5472.CAN-05-1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccardi VM. Neurofibromatosis: phenotype, natural history and pathogenesis. Baltimore and London: Johns Hopkins University Press; 1992. [Google Scholar]
- Rutkowski JL, Wu K, Gutmann DH, Boyer PJ, Legius E. Genetic and cellular defects contributing to benign tumor formation in neurofibromatosis type 1. Hum Mol Genet. 2000;9:1059–1066. doi: 10.1093/hmg/9.7.1059. [DOI] [PubMed] [Google Scholar]
- Ryan JJ, Klein KA, Neuberger TJ, Leftwich JA, Westin EH, Kauma S, Fletcher JA, DeVries GH, Huff TJ. Role for the stem cell factor/KIT complex in Schwann cell neoplasia and mast cell proliferation associated with neurofibromatosis. J Neurosci Res. 1994;37:415–432. doi: 10.1002/jnr.490370314. [DOI] [PubMed] [Google Scholar]
- Scheithauer BW, Louis DN, Hunter S, Woodruff JM, Antonescu CR. Neurofibroma. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of Tumours of the Central Nervous System. Lyon, France: International Agency for Research on Cancer; 2007. pp. 156–157. [Google Scholar]
- Scheithauer BW, Woodruff JM, Erlandson RA. Neurofibroma. In: Scheithauer BW, Woodruff JM, Erlandson RA, editors. Tumors of the Peripheral Nervous System. Washington, D.C.: Armed Forces Institute of Pathology; 1999. pp. 177–218. [Google Scholar]
- Schmidt H, Taubert H, Meye A, Wurl P, Bache M, Bartel F, Holzhausen HJ, Hinze R. Gains in chromosomes 7, 8q, 15q and 17q are characteristic changes in malignant but not in benign peripheral nerve sheath tumors from patients with von Recklinghausen's disease. Cancer Lett. 2000;155:181–190. doi: 10.1016/s0304-3835(00)00426-2. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Taubert H, Wurl P, Bache M, Bartel F, Holzhausen HJ, Hinze R. Cytogenetic characterization of six malignant peripheral nerve sheath tumors: comparison of karyotyping and comparative genomic hybridization. Cancer Genet Cytogenet. 2001;128:14–23. doi: 10.1016/s0165-4608(01)00393-4. [DOI] [PubMed] [Google Scholar]
- Schmidt H, Wurl P, Taubert H, Meye A, Bache M, Holzhausen HJ, Hinze R. Genomic imbalances of 7p and 17q in malignant peripheral nerve sheath tumors are clinically relevant. Genes Chromosomes Cancer. 1999;25:205–211. [PubMed] [Google Scholar]
- Serra E, Rosenbaum T, Winner U, Aledo R, Ars E, Estivill X, Lenard H-G, Lazaro C. Schwann cells harbor the somatic NF1 mutation in neurofibromas: evidence of two different Schwann cell subpopulations. Hum Mol Genet. 2000;9:3055–3064. doi: 10.1093/hmg/9.20.3055. [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Sheela S, Riccardi VM, Ratner N. Angiogenic and invasive properties of neurofibroma Schwann cells. J Cell Biol. 1990;111:645–653. doi: 10.1083/jcb.111.2.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman LS, Atit R, Rosenbaum T, Cox AD, Ratner N. Single cell Ras-GTP analysis reveals altered Ras activity in a subpopulation of neurofibroma Schwann cells but not fibroblasts. J Biol Chem. 2000;275:30740-20745. doi: 10.1074/jbc.M001702200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemple D, Anderson D. Lineage diversification of the neural crest: In vitro investigations. Dev Biol. 1993;159:12–23. doi: 10.1006/dbio.1993.1218. [DOI] [PubMed] [Google Scholar]
- Stevenson DA, Moyer-Mileur LJ, Murray M, Slater H, Sheng X, Carey JC, Dube B, Viskochil DH. Bone mineral density in children and adolescents with neurofibromatosis type 1. J Pediatr. 2007;150:83–88. doi: 10.1016/j.jpeds.2006.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stonecypher MS, Byer SJ, Carroll SL. Activation of the neuregulin-1/erbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene. 2005;24:5589–5605. doi: 10.1038/sj.onc.1208730. [DOI] [PubMed] [Google Scholar]
- Storlazzi CT, Brekke HR, Mandahl N, Brosjo O, Smeland S, Lothe RA, Mertens F. Identification of a novel amplicon at distal 17q containing the BIRC5/SURVIVIN gene in malignant peripheral nerve sheath tumors. J Pathol. 2006;209:492–500. doi: 10.1002/path.1998. [DOI] [PubMed] [Google Scholar]
- Su W, Gutmann DH, Perry A, Abounader R, Laterra J, Sherman LS. CD44-independent hepatocyte growth factor/c-Met autocrine loop promotes malignant peripheral nerve sheath tumor cell invasion in vitro. Glia. 2004;45:297–306. doi: 10.1002/glia.10340. [DOI] [PubMed] [Google Scholar]
- Swapp GH, Main RA. Neurofibromatosis in pregnancy. Br J Dermatol. 1973;88:431–435. doi: 10.1111/j.1365-2133.1973.tb15445.x. [DOI] [PubMed] [Google Scholar]
- The I, Hannigan GE, Cowley GS, Reginald S, Zhong Y, Gusella JF, Hariharan IK, Bernards A. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science. 1997;276:791–794. doi: 10.1126/science.276.5313.791. [DOI] [PubMed] [Google Scholar]
- Theos A, Korf BR. Pathophysiology of neurofibromatosis type 1. Ann Intern Med. 2006;144:842–849. doi: 10.7326/0003-4819-144-11-200606060-00010. [DOI] [PubMed] [Google Scholar]
- Tucker T, Wolkenstein P, Revuz J, Zeller Jq, Friedman JM. Association between benign and malignant peripheral nerve sheath tumors in NF1. Neurology. 2005;65:205–211. doi: 10.1212/01.wnl.0000168830.79997.13. [DOI] [PubMed] [Google Scholar]
- Upadhyaya M, Kluwe L, Spurlock G, Monem B, Majounie E, Mantripragada K, Ruggieri M, Chuzhanova N, Evans DG, Ferner R, Thomas N, Guha A, Mautner V. Germline and somatic NF1 gene mutation spectrum in NF1-associated malignant peripheral nerve sheath tumors (MPNSTs) Hum Mutat. 2008;29:74–82. doi: 10.1002/humu.20601. [DOI] [PubMed] [Google Scholar]
- Urich H, Tien RD. Tumors of the cranial, spinal and peripheral nerve sheaths. In: Bigner DD, McLendon RE, Bruner JM, editors. Russell and Rubinstein's Pathology of Tumors of the Nervous System. New York: Oxford University Press, Inc; 1998. pp. 141–193. [Google Scholar]
- Vogel KS, Klesse LJ, Velasco-Miguel S, Meyers K, Rushing EJ, Parada LF. Mouse tumor model for neurofibromatosis type 1. Science. 1999;286:2176–2179. doi: 10.1126/science.286.5447.2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace MR, Rasmussen SA, Lim IT, Bray BA, Zori RT, Muir D. Culture of cytogenetically abnormal Schwann cells from benign and malignant NF1 tumors. Genes Chromosomes Cancer. 2000;27:117–123. [PubMed] [Google Scholar]
- Watanabe T, Oda Y, Tamiya S, Masuda K, Tsuneyoshi M. Malignant peripheral nerve sheath tumour arising within neurofibroma. An immunohistochemical analysis in the comparison between benign and malignant components. J Clin Pathol. 2001;54:631–636. doi: 10.1136/jcp.54.8.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson MA, Perry A, Tihan T, Prayson RA, Guha A, Bridge J, Ferner R, Gutmann DH. Gene expression profiling reveals unique molecular subtypes of neurofibromatosis type 1-associated and sporadic malignant peripheral nerve sheath tumors. Brain Pathol. 2004;14:297–303. doi: 10.1111/j.1750-3639.2004.tb00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster HD, Martin R, O'Connell MF. The relationships between interphase Schwann cells and axons before myelination: a quantitative electron microscopic study. Dev Biol. 1973;32:401–416. doi: 10.1016/0012-1606(73)90250-9. [DOI] [PubMed] [Google Scholar]
- Weiss SW, Nickoloff BJ. CD-34 is expressed by a distinctive cell population in peripheral nerve, nerve sheath tumors, and related lesions. Am J Surg Pathol. 1993;17:1039–1045. doi: 10.1097/00000478-199310000-00009. [DOI] [PubMed] [Google Scholar]
- Welti S, Fraterman S, D'Angelo I, Wilm M, Scheffzek K. The sec14 homology module of neurofibromin binds cellular glycerophospholipids: mass spectrometry and structure of a lipid complex. J Mol Biol. 2007;366:551–562. doi: 10.1016/j.jmb.2006.11.055. [DOI] [PubMed] [Google Scholar]
- Widemann BC, Salzer WL, Arceci RJ, Blaney SM, Fox E, End D, Gillespie A, Whitcomb P, Palumbo JS, Pitney A, Jayaprakash N, Zannikos P, Balis FM. Phase I trial and pharmacokinetic study of the farnesyltransferase inhibitor tipifarnib in children with refractory solid tumors or neurofibromatosis type I and plexiform neurofibromas. J Clin Oncol. 2006;24:507–516. doi: 10.1200/JCO.2005.03.8638. [DOI] [PubMed] [Google Scholar]
- Wimmer K, Roca X, Beiglbock H, Callens T, Etzler J, Rao AR, Krainer AR, Fonatsch C, Messiaen L. Extensive in silico analysis of NF1 splicing defects uncovers determinants for splicing outcome upon 5' splice-site disruption. Hum Mutat. 2007;28:599–612. doi: 10.1002/humu.20493. [DOI] [PubMed] [Google Scholar]
- Wimmer K, Yao S, Claes K, Kehrer-Sawatzki H, Tinschert S, De Raedt T, Legius E, Callens T, Beiglbock H, Maertens O, Messiaen L. Spectrum of single- and multiexon NF1 copy number changes in a cohort of 1,100 unselected NF1 patients. Genes Chromosomes Cancer. 2006;45:265–276. doi: 10.1002/gcc.20289. [DOI] [PubMed] [Google Scholar]
- Wu J, Crimmins JT, Monk KR, Williams JP, Fitzgerald ME, Tedesco S, Ratner N. Perinatal epidermal growth factor receptor blockade prevents peripheral nerve disruption in a mouse model reminiscent of benign World Health Organization grade I neurofibroma. Am J Pathol. 2006;168:1686–1696. doi: 10.2353/ajpath.2006.050859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Williams JP, Rizvi TA, Kordich JJ, Witte D, Meijer D, Stemmer-Rachamimov AO, Cancelas JA, Ratner N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell. 2008;13:105–116. doi: 10.1016/j.ccr.2007.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R, Weiss R. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990a;62:599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, White R, Weiss R, Tamanoi F. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell. 1990b;63:835–841. doi: 10.1016/0092-8674(90)90149-9. [DOI] [PubMed] [Google Scholar]
- Yang FC, Chen S, Robling AG, Yu X, Nebesio TD, Yan J, Morgan T, Li X, Yuan J, Hock J, Ingram DA, Clapp DW. Hyperactivation of p21ras and PI3K cooperate to alter murine and human neurofibromatosis type 1-haploinsufficient osteoclast functions. J Clin Invest. 2006a;116:2880–2891. doi: 10.1172/JCI29092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F-C, Chen S, Clegg T, Li X, Morgan T, Estwick SA, Yuan J, Khalaf W, Burgin S, Travers J, Parada LF, Ingram DA, Clapp DW. Nf1+/− mast cells induce neurofibroma like phenotypes through secreted TGF-β signaling. Hum Mol Genet. 2006b;15:2421–2437. doi: 10.1093/hmg/ddl165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F-C, Ingram DA, Chen S, Hingtgen CM, Ratner N, Monk KR, Clegg T, White H, Mead L, Wenning MJ, Williams DA, Kapur R, Atkinson SJ, Clapp DW. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/− mast cells. J Clin Invest. 2003;112:1851–1861. doi: 10.1172/JCI19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Chang L, Patel N, Yang J, Lowe L, Burns DK, Zhu Y. Induction of abnormal proliferation by nonmyelinating Schwann cells triggers neurofibroma formation. Cancer Cell. 2008;13:117–128. doi: 10.1016/j.ccr.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1: Schwann cell origin and role of tumor environment. Science. 2002;296:920–922. doi: 10.1126/science.1068452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorick T, Syroid D, Brown A, Gridley T, Lemke G. Krox-20 controls SCIP expression, cell cycle exit and susceptibility to apoptosis in developing myelinating Schwann cells. Development. 1999;126:1397–1406. doi: 10.1242/dev.126.7.1397. [DOI] [PubMed] [Google Scholar]