

Abstract
Introduction
Intracellular signaling/synthetic pathways are being increasingly extensively characterized. However, while these pathways can be displayed in static diagrams, in reality they exist with a degree of dynamic complexity that is responsible for heterogeneous cellular behavior. Multiple parallel pathways exist and interact concurrently, limiting the ability to integrate the various identified mechanisms into a cohesive whole. Computational methods have been suggested as a means of concatenating this knowledge to aid in the understanding of overall system dynamics. Since the eventual goal of biomedical research is the identification and development of therapeutic modalities, computational representation must have sufficient detail to facilitate this “engineering” process. Adding to the challenge, this type of representation must occur in a perpetual state of incomplete knowledge. We present a modeling approach to address this challenge that is both detailed and qualitative. This approach is termed “dynamic knowledge representation,” and is intended to be an integrated component of the iterative cycle of scientific discovery.
Methods
BioNetGen (BNG), a software platform for modeling intracellular signaling pathways, was used to model the toll-like receptor 4 (TLR-4) signal transduction cascade. The informational basis of the model was a series of reference papers on modulation of (TLR-4) signaling, and some specific primary research papers to aid in the characterization of specific mechanistic steps in the pathway. This model was detailed with respect to the components of the pathway represented, but qualitative with respect to the specific reaction coefficients utilized to execute the reactions. Responsiveness to simulated lipopolysaccharide (LPS) administration was measured by tumor necrosis factor (TNF) production. Simulation runs included evaluation of initial dose-dependent response to LPS administration at 10, 100, 1000, and 10000, and a subsequent examination of preconditioning behavior with increasing LPS at 10, 100, 1000 and 10000 and a secondary dose of LPS at 10000 administered at ∼27 h of simulated time. Simulations of “knockout” versions of the model allowed further examination of the interactions within the signaling cascade.
Results
The model demonstrated a dose-dependent TNF response curve to increasing stimulus by LPS. Preconditioning simulations demonstrated a similar dose-dependency of preconditioning doses leading to attenuation of response to subsequent LPS challenge—a “tolerance” dynamic. These responses match dynamics reported in the literature. Furthermore, the simulated “knockout” results suggested the existence and need for dual negative feedback control mechanisms, represented by the zinc ring-finger protein A20 and inhibitor kappa B proteins (IκB), in order for both effective attenuation of the initial stimulus signal and subsequent preconditioned “tolerant” behavior.
Conclusions
We present an example of detailed, qualitative dynamic knowledge representation using the TLR-4 signaling pathway, its control mechanisms and overall behavior with respect to preconditioning. The intent of this approach is to demonstrate a method of translating the extensive mechanistic knowledge being generated at the basic science level into an executable framework that can provide a means of “conceptual model verification.” This allows for both the “checking” of the dynamic consequences of a mechanistic hypothesis and the creation of a modular component of an overall model directed at the engineering goal of biomedical research. It is hoped that this paper will increase the use of knowledge representation and communication in this fashion, and facilitate the concatenation and integration of community-wide knowledge.
Introduction
Biological systems have a complex, multi-hierarchical structure. The current reductionist approach to basic research has successfully generated copious amounts of information regarding the basic cellular and molecular components of biological systems, and has been able to characterize the relationships between many of these components. However, there are significant hurdles in translating this mechanistic knowledge in a way that effectively captures the dynamic behavior of these systems. This challenge is most evident in attempts to characterize highly interconnected, multiple-feedback sub-systems at the cellular and molecular level. Knowledge of these systems is presently represented primarily in static diagrams that can be overwhelming in their complexity (see Oda-Kitano map of TLR signaling in [1]). Mathematical approaches developed from the study of dynamical systems offer a method by which these static diagrams can be “brought to life.” However, the application of formal mathematical methods often results in considerable abstraction of the system under examination. While this approach can provide elegant descriptions of underlying principles and behaviors (such as the characterization of organ level physiology), it can also lead to models that are too far removed from the actual “messy” details of biology. This gap reduces the efficacy of these models with respect to the engineering goal that drives a great deal of biomedical research. There needs to be a correlation between the level of resolution of the mathematical model of a system and the level of any planned intervention on that system. For example, equation-based models of cardiac function based on the cardiac myocyte contractile response are sufficient to examine various maneuvers affecting cardiac output so long as the effect of the intervention can be reliably characterized in the terms of the contractility equation. However, once the relationship between the intervention and its effect strains linear causality (as is the case in attempts to manipulate cellular interactions at the molecular level), these reduced mathematical models do not have the sufficient resolution to aid in the evaluation and design of potential interventions.
The tension between the elegance of mathematical representation and the mechanistic detail of biological systems has led to the recognition that there must be a middle ground where mathematical tools can be used to examine the dynamics of biological behavior without necessitating removal of too much detail. The areas of “translational systems biology” [2, 3] and “executable biology” [4] have evolved to address this need. In these approaches the application of mathematical methods is not directed at formal mathematical analysis but rather at the development of a computational analog to the biological system [5, 6]. We further extend this concept into an approach we term “dynamic knowledge representation,” in which the goal of model construction is focused less on the model itself than on computationally representing conceptual models that represent either an individual researcher's knowledge, or, more broadly, the research community's state of knowledge. As such, a computational model becomes a dynamic instantiation of a “thought experiment” based upon a particular hypothesis regarding the behavior of a system [7]. By examining the behavior the model and comparing it to real-world observations, the computational models are a means of conceptual model “checking” or verification, such that a researcher can “know what they know.”
With this goal in mind, the structure of biomedical knowledge can be classified to facilitate translation into a computational framework. Since the majority of mechanistic knowledge exists in either the description of biological components (“objects”) or the interactions between these components (“rules”), an object-oriented, rule-based modeling framework is suited the translational role required of dynamic knowledge representation. Although all modeling requires some degree of abstraction, when dealing with complex systems it is difficult, if not impossible, to determine a priori where abstractions can be safely made. Therefore, there is a need to be able to capture the necessary detail of the system such that mechanistic causality can be approximated as directly as possible, and for modeling assumptions to be explicitly communicated.
The need for this type of representation is further evident when dealing with multi-scale manifestations of dynamic behavioral patterns, exemplified in the “condensed” output, that result from highly parallel control systems. It is highly likely that the true richness of biological behavior requires these multiple control points for the “fine tuning” necessary to deal with heterogeneous challenges, a fact that complicates the investigation of these processes. The detailed, qualitative method presented in this paper is intended as a “middle-ground” approach to addressing this dilemma. Some degree of abstraction is necessary to facilitate understanding; the capability for comprehensive representation is necessary to reproduce biology. The solution is a modular approach that allows for constrained and compartmentalized representation of sufficient detail just at the edge of the “fog of complexity,” but in a format that will allow subsequent concatenation of models to capture the richness of biological behavior. Furthermore, the selection of modeling focus should be made to limit, as much as possible, the nonlinear assumptions of causality that arise from concurrently modeling multiple scales of behavior; such types of behavior should be allowed to “emerge” from the instantiation of underlying mechanisms. Therefore, the rationale for this focus is that characterizing the properties of the intracellular control points provides a guide to the state changes of the constitutive actors (cells) in the inflammatory response. These actors in turn determine, via their aggregated behavior, the observed phenomenon at higher levels of biological organization. Understanding the internal mechanisms of state changes, or “reprogramming,” provides a necessary groundwork before attention can be turned to control mechanisms at the intra-extra-cellular interface (i.e. receptor modulation) and beyond (i.e. pro- and anti-inflammatory cytokine milieu). This is of particular importance if potential interventions are to be developed to target these intracellular control points; in such a case (which is inevitable, given the direction of the drug-candidate discovery process) modeling these control points provides a method of assessing the potential effects of these interventions.
Towards this end, we present a software modeling toolkit called BioNetGen (BNG) that fulfills these criteria, and present a demonstration of detailed, qualitative dynamic knowledge representation of the Toll-like receptor 4 (TLR-4) signaling pathway, and its behavior in the face of preconditioning or “tolerance.”
Modeling Method: BioNetGen
BioNetGen is a modeling system that uses graphs to represent molecules and their functional components and rules to describe the interactions between molecules [2, 8]. The software framework contains both a text-based interface, and a visual graphics tool called RuleBuilder with which to construct models. A detailed description of the BioNetGen software and the associated BioNetGen modeling language has been given in Ref [8]. An overview of the rule-based modeling approach and its applications to modeling biological signal transduction has been given in Ref [9].
Although BioNetGen was originally intended for the development of detailed models based on site-specific protein-protein interactions, BioNetGen is used in this manuscript to provide a coarser level of description that is appropriate when site-specific information is not available. Because the mechanistic basis of many of the protein-protein interactions modeled here remain unknown, we have utilized functional descriptions that are intended to represent the current state of available knowledge. Molecular components in many cases represent functional rather than physical states, and rules describe how protein-protein interactions modify the functional states of the involved molecules. This application of BioNetGen to the modeling of TLR-4 signaling can thus be viewed as an exercise in dynamic knowledge representation, which attempts to convert information about the proteins involved into a predictive model of system behavior. The model is dynamic both in the sense that it can predict the temporal behavior of the system and because it can easily be extended or modified to incorporate the ever-expanding base of knowledge about TLR-mediated signaling. BioNetGen provides a particularly convenient and powerful representation of this knowledge base and provides simulation capabilities that permit thorough investigation of system dynamics.
Reference System: TLR-4 and Inflammation
Toll-like receptors (TLR) on inflammatory cells represent a key aspect of the body's initial response to injury and/or infection [1, 10-12]. Activation of these receptors initiates signaling events that lead to an initial predominance of a pro-inflammatory response designed to contain an infectious insult, and also a concurrent compensatory anti-inflammatory response intended to limit the degree of the pro-inflammatory response. The interplay between response and counter-response can be seen at multiple levels of biological organization. It is present with respect to systemic levels of mediators and inflammatory cell populations, within local tissues and individual organs, and also within cells themselves. This multi-scale robustness is a property of biological systems, leading to maintenance of the organism's “integrity of self.” However, it can also make it difficult to parse out the specific effects of targeted interventions from one level to another, as is the case when attempting to translate molecular mechanisms to the clinical level. Given that this is the area of greatest effort in biomedical research, we focus on the intracellular mechanisms of control of TLR signaling. This investigation places particular emphasis on characterizing the phenomenon of tolerance or preconditioning, a cell-level manifestation of biological robustness. It is well recognized that the response of cells to inflammatory stimuli is affected by a past history of exposure to lower levels of that stimuli [11-17]. The dynamics of the preconditioning effect are a dose-dependent manifestation of the pro-inflammatory/positive feedback/amplification and anti-inflammatory/negative feedback/attenuation pattern seen in the inflammatory response as a whole. From a teleological standpoint, such a dynamic of rapid response and subsequent quiescence would be evolutionarily favored as a means for an organism to deal with external threats. Furthermore, there is increasing evidence that this system is involved with dealing with internal threats as well, as the responsiveness of the TLR signaling to endogenous mediators indicating tissue damage suggests the further importance of preconditioning in the “danger signal” hypothesis of acute inflammation [18, 19]. With this hypothesis the inflammatory system is viewed as being subjected to a continual minimal, baseline level of recurrent tissue damage through the course of “normal” existence. Therefore, tight modulation and tolerance is critical to the maintenance of a homeostatic yet responsive inflammatory response.
The TLR-4 pathway is one of the most intensively studied inflammatory signaling pathways [1, 10]. The control of TLR-4 signaling includes both “extracellular” mechanisms, seen in the autocrine and paracrine effects of anti-inflammatory cytokines, such as Interleukin-10 (IL-10) and Transforming growth factor-beta (TGF-beta), and soluble TLR-4 receptor antagonists, and “intrinsic” mechanisms resulting from the intracellular regulation of TLR-4 signaling in response to stimulus to the bacterial product lipopolysaccharide (LPS) [11, 13, 16, 17, 20-23]. For purposes of this paper we focus on the intracellular pathways and intrinsic signaling modulation of the TLR-4 cascade.
Methods
Model Development
A series of review articles on the modulation of TLR-4 signaling was used to identify the components and rules to be included in the BNG model, as well as some more focused research articles to provide mechanistic insight into selected components [1, 10-14, 16, 17, 20-29]. The TLR-4 signaling pathway is an extensive cascade that interacts with multiple synthetic and metabolic pathways, and the intent of this exercise is not to produce a comprehensive model of all aspects of this system (for this description, see [1]). The BNG model presented herein is intended to demonstrate the intracellular feedback effects of a particular sub-system of the TLR-4 signaling pathway and its contribution to LPS tolerance.
Control loops were mechanistically modeled by representing signaling events from stimulus to production of the inhibitory compound (usually implying gene activation and protein synthesis) and the transport of the inhibitory compound back to the signaling cascade. The production of the negative feedback mediator, often a protein, was accounted for by modeling transcriptional and translational events in an abstracted fashion.
For an inhibitory pathway to be included there needed to be at least some information on the causal mechanisms and intermediate components involved in the generation of the inhibitory compounds. For instance, it was not sufficient for the literature to state that “increased expression of the gene for Inhibitor X led to down regulation of TLR-4 signaling:” this statement does not give any insight as to how endogenous Inhibitor X was produced, or what up-regulated gene X in vivo. Also, a statement such as “LPS activation led to increased levels of Inhibitor X” is insufficient due to the absence of at least some mention of the intermediate steps involved in the production of Inhibitor X. This modeling requirement precluded the inclusion of such competitive inhibitors such as a shortened version of myeloid differentiation factor 88 (MyD88), MyD88s, and IL-1-associated kinase (IRAK)-M (an inactive isoform of IRAK-1 and IRAK-4) [11, 13].
Additionally, if possible, complicated molecular events were reduced to a simple state/relationship transition, and therefore were not explicitly modeled. This was done to reduce model complexity, and was utilized when evidence in the literature suggested that a complex, but relatively well-characterized molecular process led to a relatively straightforward event. This was done with a recognition of the level of detail desired for a particular model, and therefore could be a source of model inadequacy if subsequent planned interventions had effects within the abstracted steps. For instance, the interactions associated with tumor-necrosis factor-receptor-associated factor 6 (TRAF-6) involve complicated ubiquitination and de-ubiquitination processes, not only with respect to its activation but also in terms of interactions with its putative inhibitor, the zinc ring-finger protein A20 [11, 20-23]. However, the end result of these events is that TRAF6 is activated via upstream LPS/TLR-4 receptor complexes, and A20, when present, inactivates TRAF6. These interactions were modeled as simple state transitions in this model. If, in the future an intervention should be identified that centered on the ubiquitination steps of the A20-TRAF6 interaction, these steps would need to be further delineated. Because the architecture of the BNG model is modular, extension of the model in this way would be straightforward.
It should be emphasized that these guidelines are subjective in nature, and that the resulting model represents an instantiation of particular conceptual model of TLR-4 signaling, reproducing the subjective process that is the primary method biomedical researchers use to establish the intellectual frameworks for their investigations. Since all modeling (be it conceptual, computational or laboratory-based) is to some degree subjective, we focus on the need to be as explicit as possible with respect to communicating the underlying assumptions of the model. These assumptions are:
Because the emphasis was on intracellular feedback loops, soluble TLR-4 receptor antagonists and paracrine/autocrine effects of cytokines such as IL-10 and TGF-beta were not considered.
Given the intent to focus on causal, mechanistic control loops, A20 and inhibitor kappa B proteins (IκB) are the primary intracellular negative feedback compounds included in this model [11, 13, 20-23, 26].
The nuclear factor kappa-B (NF-κB) activation pathway, via TGF-beta-activated kinase 1 (TAK1) provides the common link between upstream TLR-4 signaling and the production of A20, IκB, and the putative pro-inflammatory output, tumor necrosis factor (TNF) [11, 13].
Although TAK1 activation also leads to signal transduction in the ERK (extracellular signal-regulated kinase), JNK (c-Jun N-terminal kinase), and p38 MAPK (mitogen-activated protein kinase) pathways with the subsequent activation of the nuclear transcription factor AP-1 (activator protein 1), also acknowledged to contribute to pro-inflammatory signaling, the decision was made not to incorporate these pathways because of the lack of clearly identified intracellular negative feedback mediators generated by this pathway [1, 24]. A causal mechanistic relationship between AP-1 and other intracellular modulators of TLR-4 signaling could not be established by the authors' review of the literature, and therefore precluded their incorporation into this model.
The lack of explicit information regarding the potential relationship between these signaling cascades and the production of other intracellular mediators such as toll-interacting proteins (Tollip) and suppressors of cytokine signaling (SOCS) precluded their inclusion in this model [25].
Since mechanistic representation of feedback mechanisms forms a central part of the model, it was necessary to model, at least abstractly, the processes of gene activation, transcription and protein translation. DNA was abstractly represented as a molecular species that contained multiple binding sites representing specific transcriptional domains. Furthermore, as opposed to the concentrations of the other molecular species in the model, there were only 2 copies of DNA included in the model. As a result, the process of transcription represented a significant amplification point in the TLR-4 pathway and its synthetic consequences.
The model was constructed using these guidelines and assumptions, and initially produced using the RuleBuilder interface of BNG. The simulated experiments, however, were implemented using the BNG text interface due to its greater flexibility in setting experimental conditions (see below in Simulations and Results for details). For a more detailed description of setting up experiments using BioNetGen see Ref [8]. A graphical depiction of the components included in the model, and an example of the RuleBuilder interface can be seen in Figure 1. This Figure should give some measure of the complexity of even this admittedly abstract model. The model is available for download at http://bionetgen.org/SCAI-wiki/index.php/Main_Page as a RuleBuilder file, a BioNetGen Language file, a Systems Biology Markup Language (SBML) file [30]. In addition, the SBML file and a MatLab file of this model are available for examination as Supplementary Materials to this text. It should be noted that the BNG engine automatically generates both SBML and MatLab files, should the researcher elect those options. The purpose for providing these outputs is to allow researchers to perform detailed mathematical analysis on the model and its behavior. As such, it is expected that the detailed documentation of the model would occur in the BNGL interface, and the additional files would serve as adjuncts to the primary model file.
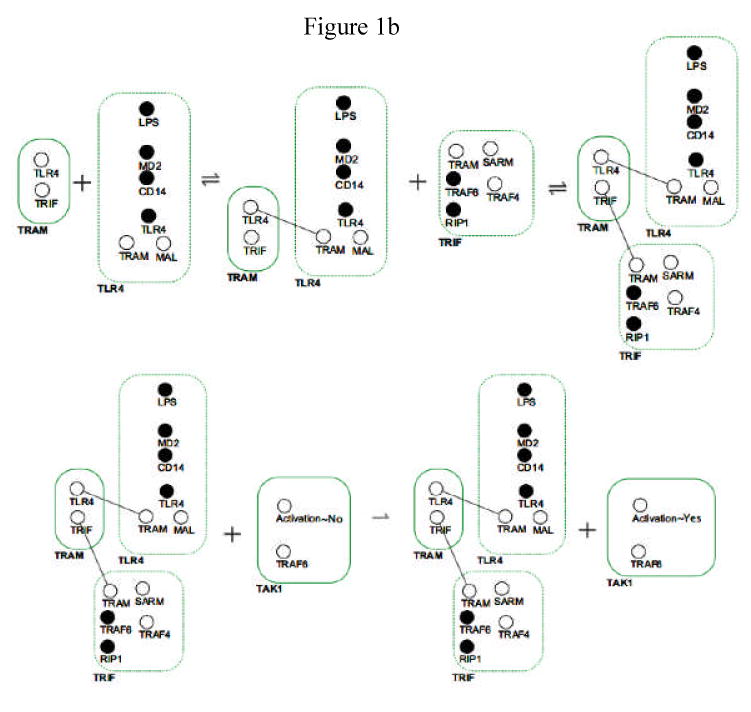
Figure 1A and B. Schematic diagram of the components and functions of the BioNetGen TLR-4 signaling model, and an example of the RuleBuilder interface for a component of the model.
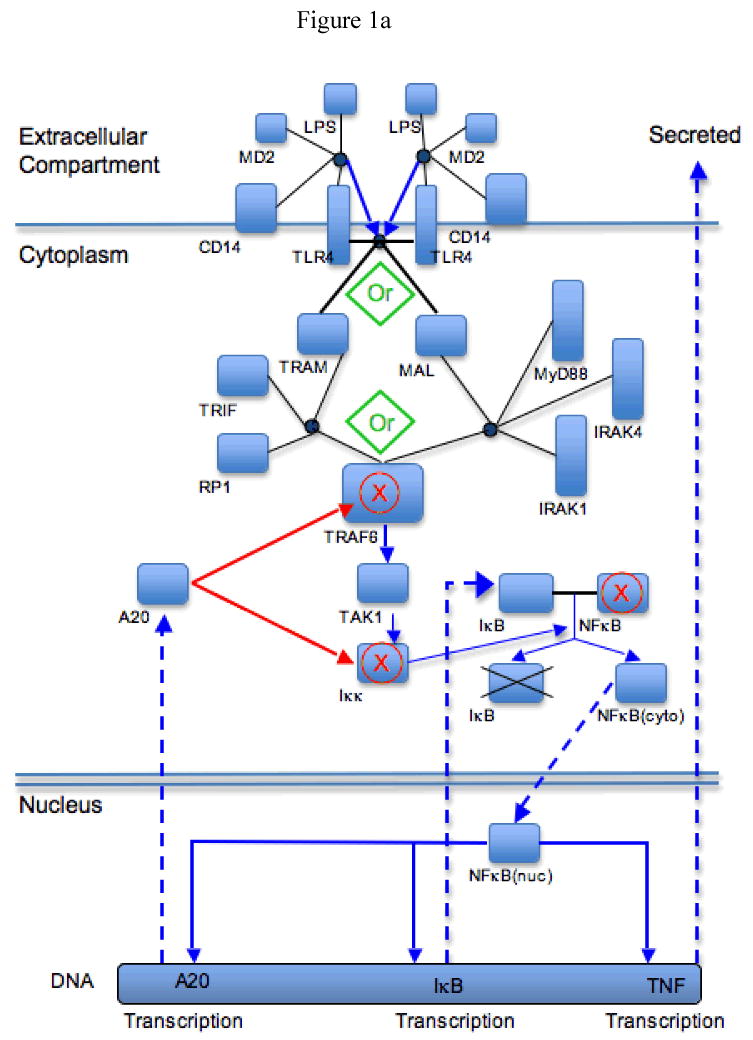
Panel 1A: This figure demonstrates a highly abstracted schematic of the components and interactions of the BNG model. The various states of the molecules are not represented, nor the individual binding sites, and the molecular interactions/reactions are generally depicted using the following legend:
Solid Black Lines: denote molecule-to-molecule binding
Circular Nodes at Convergence of Solid Black Lines: denote complexes
composed of attached molecules
Solid Blue Arrows: denote activation processes
Solid Red Arrows: denote inhibitory processes
Red “X”s”: denote targets of inhibition
Dashed Blue Arrows: denote transport or change in location of molecular species
Also, there are two points where pathways converge/diverge and signals can follow either path. These points can be seen as green “Or”s set in diamonds. These are seen at the divergence of the TLR-4-dimer binding to either MAL or TRAM, and at the convergence of these two pathways as TRAF6 is bound to either of the TRAM-TRIF-RP1 or MAL-MyD88-IRAK1-IRAK4 complexes. The negative feedback components are A20, acting on TAK1 and Iκκ, and IκB, which is constitutively bound to NF-κB.
Panel 1B: This figure demonstrates what the actual RuleBuilder rules look like for a specific section of the above pathway. The region represented is a portion of the TRAM-TRIF-RP1-TRAF6 arm of the signaling pathway, specifically the binding of the TRAM-TRIF-RP1-TRAF6 complex to the TLR-4-dimer and the activation by this complex of TAK1. Molecular species are represented at smoothed rectangles; binding sites are represented as small circles within the molecule. Open binding sites are shown as open circles; darkened circles represent occupied binding sites. The first row demonstrates the rules for binding of theTRIF-RP1-TRAF6 complex to the TLR-4-dimer. The second row shows the activation of TAK1 by the TRAM-TRIF-RP1-TRAF6 complex, this function denoted by the change of the activation site on TAK1 from “Activation∼No” to “Activation∼Yes.”
It should be noted that the resulting model is both qualitative and detailed. It is qualitative in the sense that the rate constants for the various reactions are rough estimates; quantitative details of these reaction rates as they occur in the intracellular milieu are difficult to obtain. It is detailed in terms of the components included in the model, and the causal chain of mechanisms that link those components together. It is our contention that this is an appropriate balance of detail and abstraction, given the pragmatic constraints on obtaining quantitative data, and the type of potential interventions planned (i.e. targeted molecular modulation). The model includes 31 molecule types and 41 reaction rules, which altogether generate a network of 76 possible molecular species and 202 reactions. A total of 97 parameters are used for the initial concentrations of species and the reaction rate constants. All of these have been crudely estimated based on order-of-magnitude ranges for protein expression levels and reaction rates. All proteins involved in the signaling pathway were assumed to be present at a level of 10,000 copies per cell, dissociation rates were given a default value of 0.1 /s, and association rates were set to 0.001 1/molecule 1/s, a value that strongly favors binding over unbinding (the dimensionless equilibrium constant K*NT=100, where K is the equilibrium binding constant and NT is the number of binding partners per cell). After construction of the model, experiments were run using the command-line BNG interface due to the increased flexibility offered in terms of defining experimental conditions.
Simulations and Results
For all of the simulations the initial number of protein copies per cell (set using seed species in the BNG input file) is set to 10,000. The initial number of DNA molecules per cell is set to 2, with each of the DNA strands having multiple promoter binding sites. An initial equilibration simulation was then run in which the LPS stimulus was not present in order to allow the baseline state of the cell to be reached; specifically, this permits formation of the transcriptionally inactive IκB-NF-κB complex. (The BNG text interface and RuleBuilder both allow for this equilibration phase during the experimental setup.) Specific experimental conditions were set by adding various levels of LPS after the equilibration phase.
The first series of simulations consisted of determining the dose response for output of TNF to lipopolysaccharide (LPS) at doses of 10, 100, 1000 and 10000 molecules per cell. In these simulations a single “dose” of LPS at each level was added immediately after the equilibration phase. Next, pre-conditioning behavior was evaluated by re-dosing the model with a high dose of LPS (= 10000) at 100000 seconds (= 27.8 h) following initial preconditioning doses of LPS at 10, 100, 1000 and 10000. The preconditioning simulations added a section of code to the end of the dose response simulations to reproduce the effect of the second LPS dose. The simulation following the initial dose of LPS progressed as before, but at the predetermined interval (= 100000 seconds) the values of the molecular species were saved to record the state of the model at this point. This in turn became the new “initial conditions” for a second dose of LPS (= 100000), with a resulting response following as the model continued on for another 100000 seconds. This second response curve, representing the “post-pre-conditioning” behavior, is presented in Figure 3.
Figure 3. Dose Dependent Preconditioning-Tolerance Effect of TNF response to subsequent challenge of LPS.
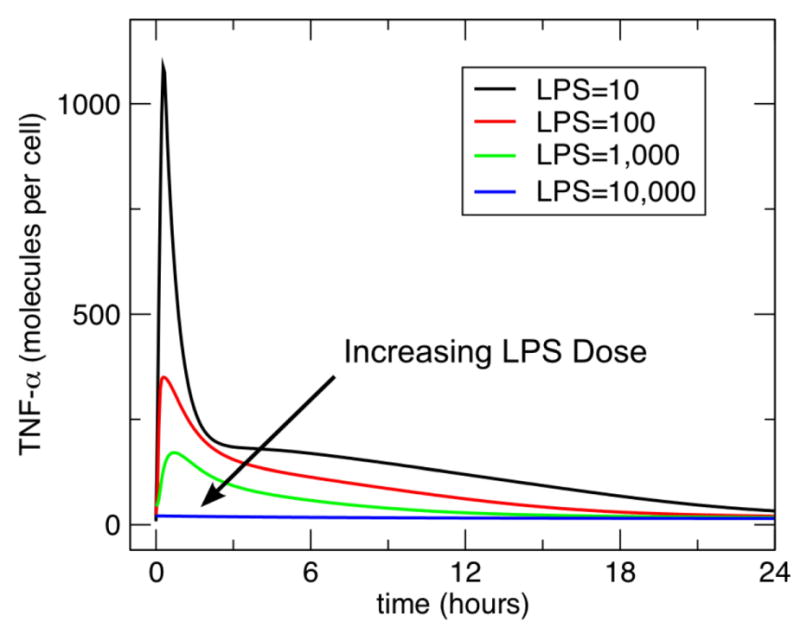
This figure demonstrates the response of the model in term of TNF production following a second administration of LPS = 10000 after initial doses of LPS = 10, 100, 1000 and 10000. The second dose is administered approximately 27 hours after the initial dose. The amplitude of the TNF response at the second LPS challenge decreases as the initial preconditioning dose is increased, suggesting that greater tolerance is seen with higher preconditioning doses of LPS.
The model exhibited the anticipated dose-dependent TNF response to perturbations with increasing levels of LPS at 10, 100, 1000 and 10000. The results of these runs can be seen in Figure 2. Next, the model demonstrated preconditioning behavior in the form of tolerance. These results are presented in Figure 3. With increasing doses of LPS from 10 to 10000 there is increased attenuation of the response of LPS at the dose of 10000. This is the expected behavior based on the reported tolerance response in the literature [27].
Figure 2. Dose Dependent TNF response to single dose of LPS.
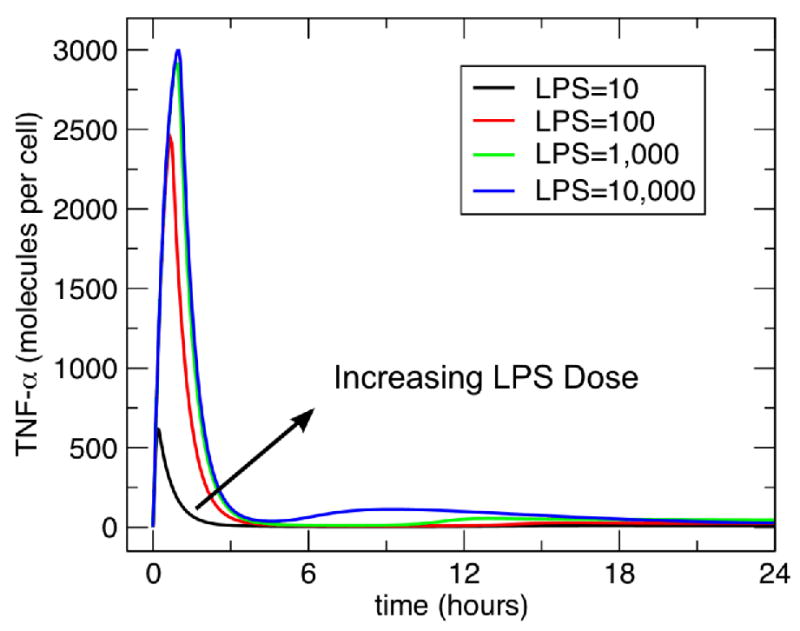
This figure demonstrates the initial response of the model in terms of produced TNF to progressively greater doses of LPS. There is a clear dose-dependent response curve seen to the challenges of LPS = 10, 100, 1000 and 10000.
The detail of the model allowed the examination of the dynamics of each component of the signaling pathway to permit evaluation as to how those dynamics might contribute to the higher-level phenomenon of tolerant behavior. Initially focus was on the two negative feedback components within the model, A20 and IκB. It is important to note that the production of both of these mediators is dependent upon the forward propagation of the inflammatory signal via NF-κB. The pattern of A20 expression shows the expected dose-dependent pattern in Figure 4, which demonstrates A20 expression dynamics at doses of LPS = 10, 100, 1000 and 10000. This is as would be expected in a classic negative feedback loop for the forward signal of LPS. The dynamics of A20 expression with respect to the response to a second LPS challenge after a preconditioning dose are also consistent with “tolerance,” as seen in Figure 5, which demonstrates A20 response to a second LPS dose of 10000 after preconditioning at LPS = 10, 100, 1000, 10000. Since A20 itself is not significantly produced following preconditioning, this suggests that while A20 is primarily responsible for the attenuation of the initial response to LPS (see below for discussion regarding Figure 8), it cannot be responsible for the “memory” of the system that leads to the tolerance effect. If this is the case, then the primary effector of tolerance must be the only other negative feedback mechanism in the model, IκB.
Figure 4. Dose Dependent A20 response to single dose of LPS.
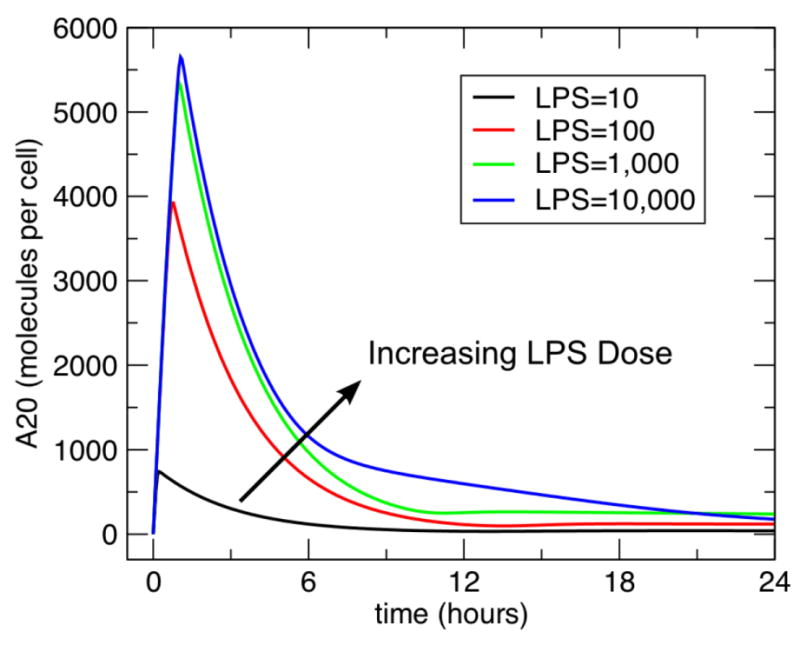
This figure demonstrates the initial response of the model in terms of produced A20 to progressively greater doses of LPS. There is a clear dose-dependent response curve seen to the challenges of LPS = 10, 100, 1000 and 10000. Since A20 is a negative feedback inhibitor of the TLR-4 response to LPS this direct relationship between A20 levels and LPS challenge is as expected.
Figure 5. Dose Dependent Preconditioning-Tolerance Effect of A20 response to subsequent challenge of LPS.
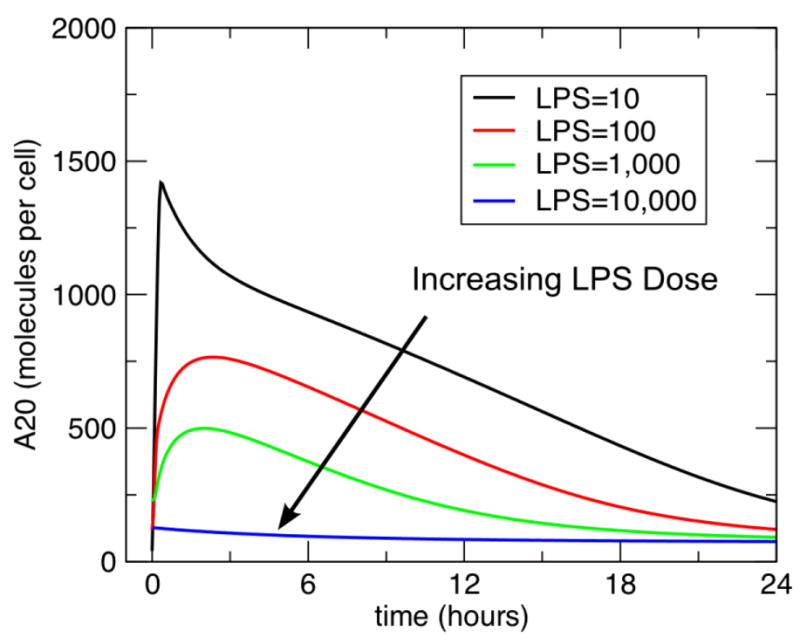
This figure demonstrates the dynamics of A20 in the same conditions used to examine TNF in Figure 3. As with the TNF response, there is an increased tolerance with increasing LPS doses. However, as opposed to the effect of A20 on the initial dose of LPS, the decreasing levels of A20 during the tolerance phase suggests that it cannot be the mechanism by which tolerance occurs.
Figure 8A-E. Dynamics of A20, “Free” IκB, Activated TAK1, Activated Iκκ and Activated-Nuclear NF-κB in response to LPS = 10000 in “wild type” model.
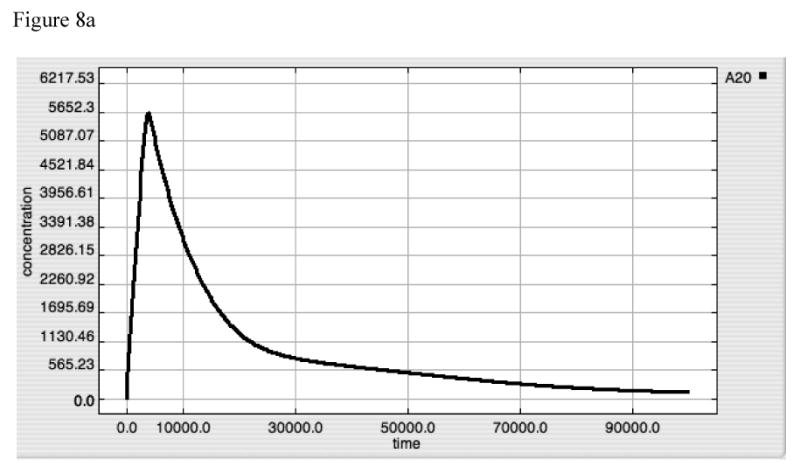
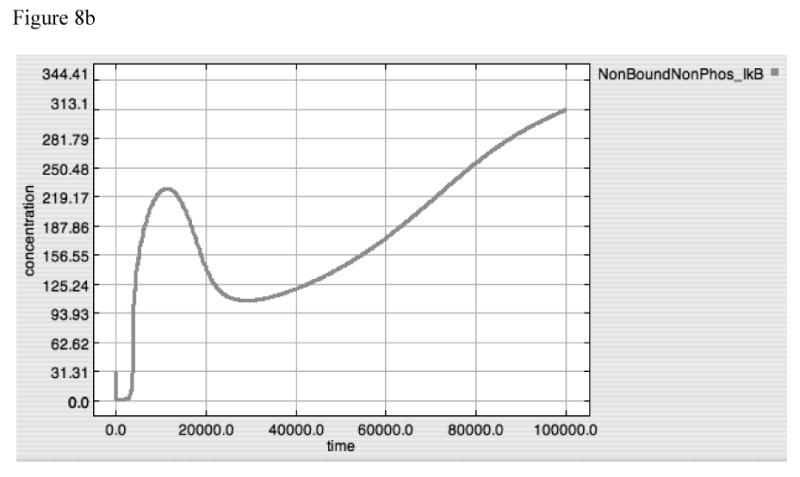
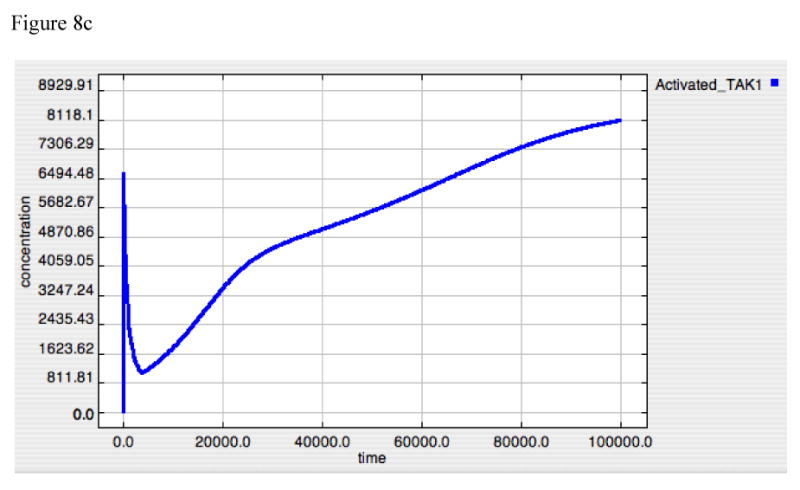
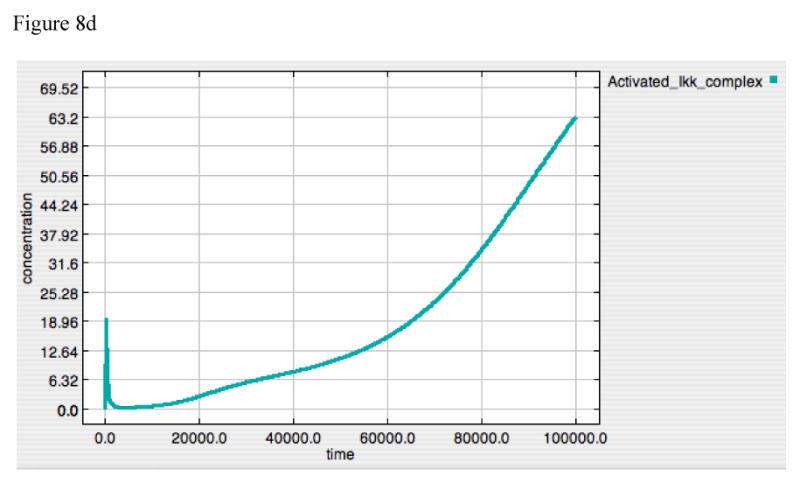
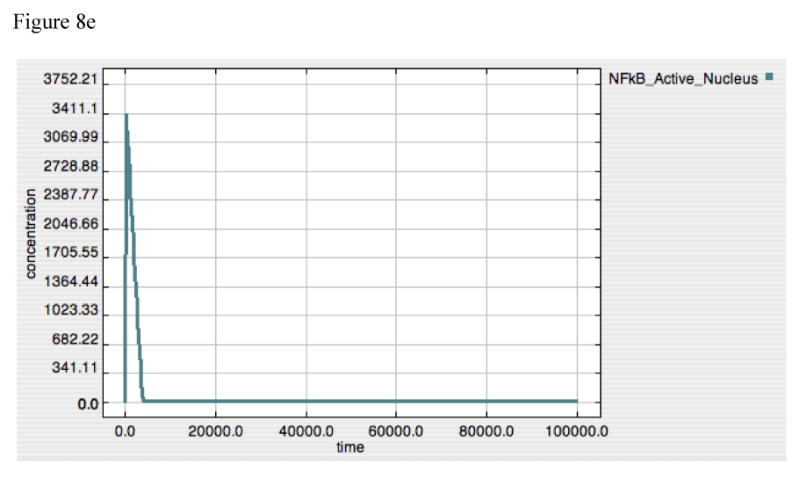
These panels demonstrate the relative effects of A20 (8A) and IκB (8B) on the upstream amplification of the LPS-TLR-4 signal, as seen in the dynamic of activated TAK1 (8C) and Iκκ (8D), and on the downstream effector of gene activation, Active-Nuclear NF-κB (8E). The dynamics of TAK1 and Iκκ have 3 distinct components:
1. The first very sharp up-stroke represents initial activation via the inflammatory signal, prior the production of negative feedback compounds.
2. The second very sharp decrement represents the direct negative feedback as both A20 (see Panel 8A) and IκB (see Panel 8B) rise initially. Note that A20 likely has the predominant effect at this point due to the very low amplitude of the IκB spike.
3. The third phase of the response occurs at a transition point between effective suppression of signaling through TRAF6 and failure of this suppression resulting from the progressive degradation of A20; the A20 inhibition of TRAF6 is not an irreversible reaction and the fall of A20 below its equilibrium point allows “recovery” of the TRAF6-mediated activation of TAK1, and subsequently Iκκ. Small amounts of TRAF6 lead to a persistent rise in the levels of both of these compounds.
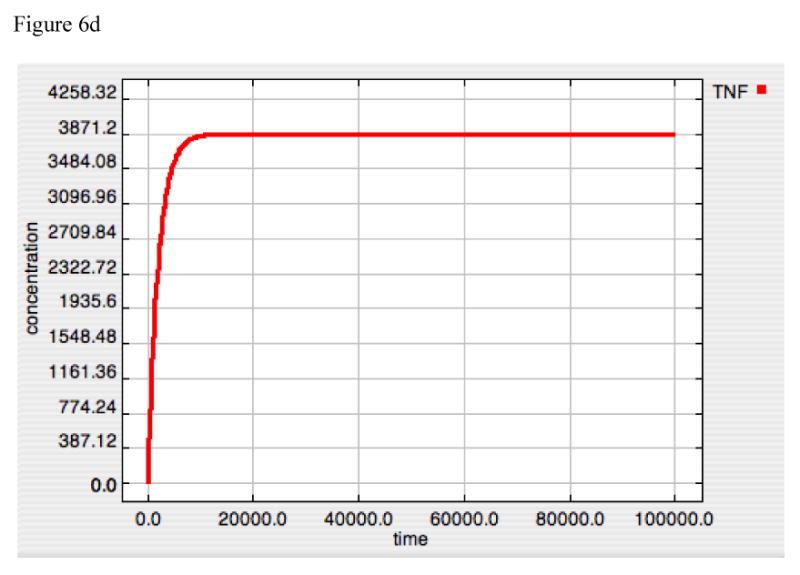
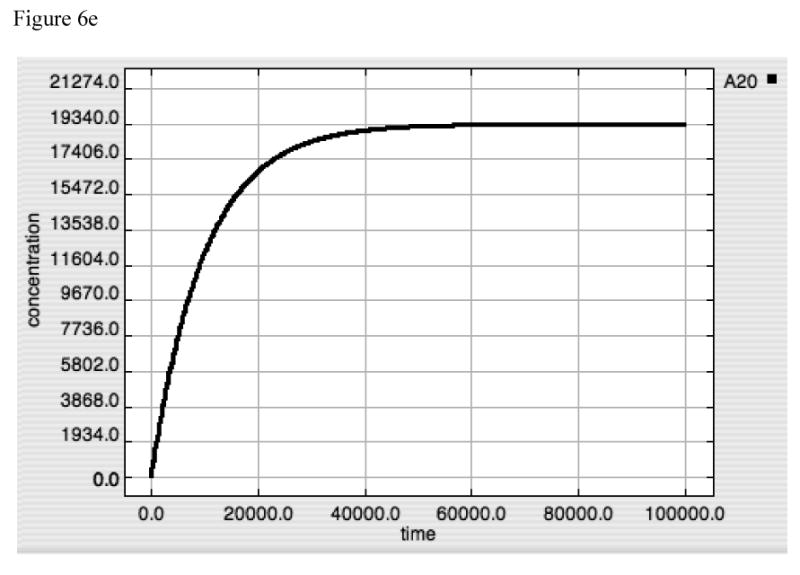
With the need to invoke an additional negative feedback mechanism to generate tolerance, the question arises as to whether tolerance should be treated as an additional behavior to be selected for in order for its evolutionary development? The answer is suggested when experiments mimicking an IκB “knock-out” system are simulated. This is done by shutting off the IκB transcription step, and examining the dynamics in response to LPS = 10000. The results can be seen in Figure 6. Panels 6A, 6B and 6C show the standard system (IκB intact) response to LPS = 10000 of TNF, A20 and Active-Nuclear NF-κB. The response of TNF, A20 and Active-Nuclear NF-κB in the “IκB-knockout” can be seen in Panels 6D, 6E and 6F. What is readily apparent is that in the “IκB-knockout” A20 reaches an equilibrium state where there is enough NF-κB activity to maintain A20 at suppressive levels. However, since NF-κB also produces TNF, TNF production is never completely suppressed (6D). This suggests that a single stimulus-dependent negative feedback loop is insufficient to completely control the forward response. Furthermore, the addition of the second control loop in the form of a dis-inhibition activation mechanism for IκB, which is necessary in order to effectively attenuate an initial stimulus, concurrently produces the tolerance effect.
Figure 6A-F. Dynamics of TNF, A20 and Active-Nuclear NF-κB in response to LPS = 10000 in both “wild-type” and “IκB-knockout” model versions.
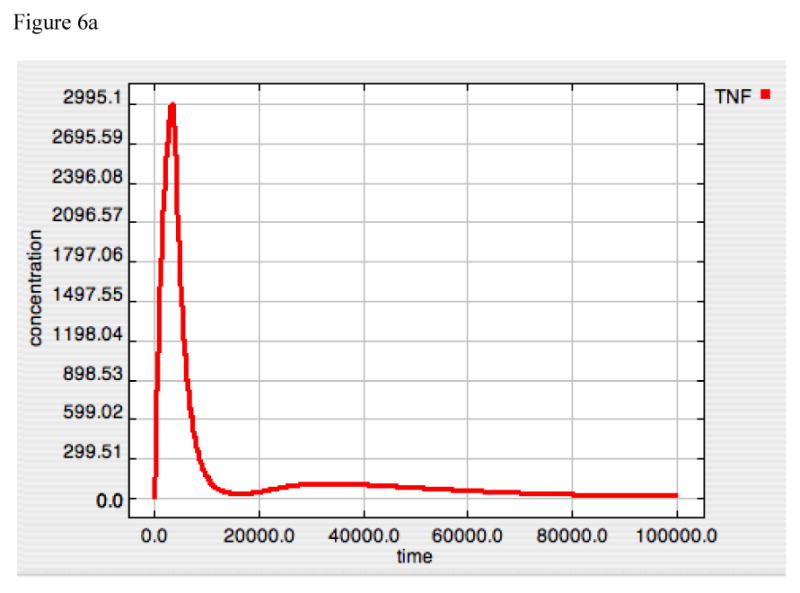
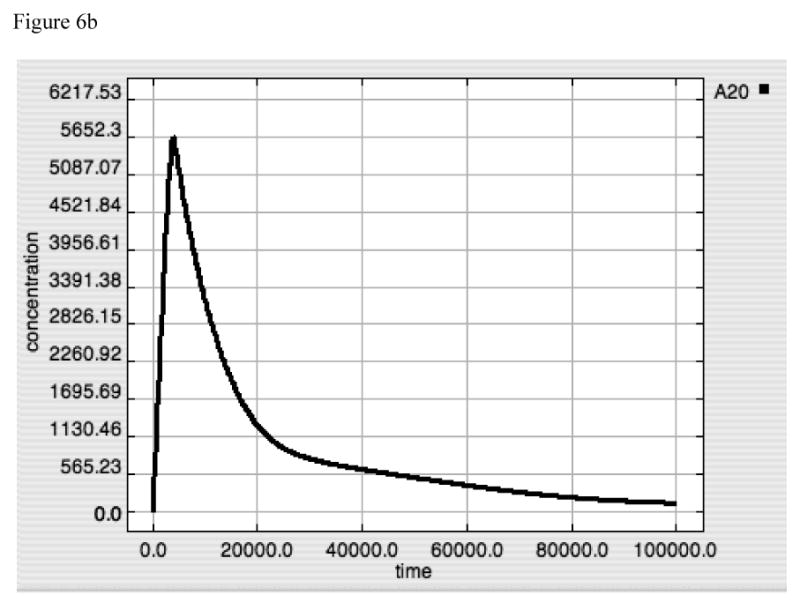
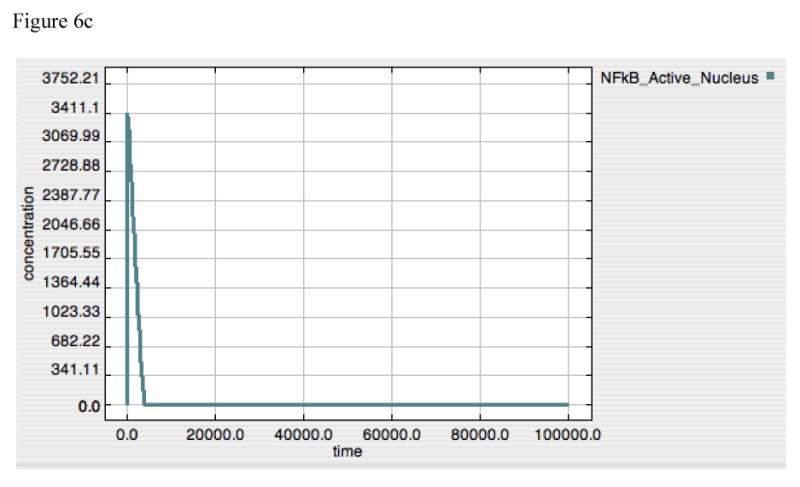
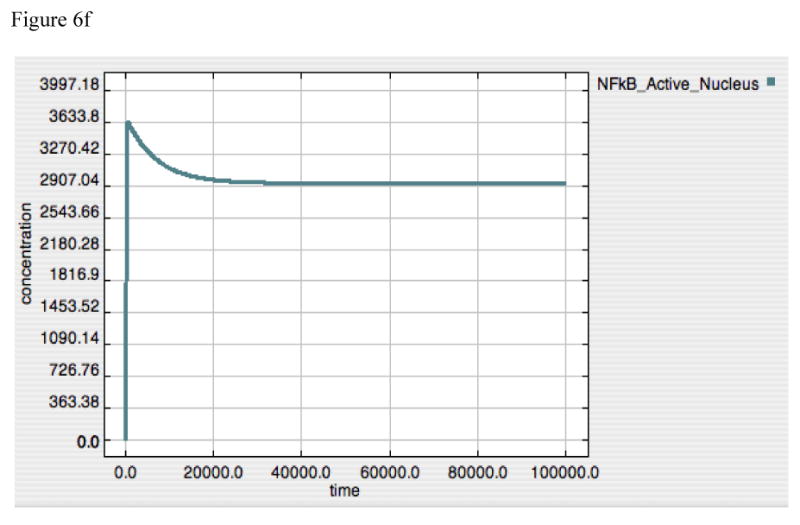
Panels 6A-C demonstrate the responses of TNF (6A), A20 (6B) and Active-Nuclear NF-κB (6C) in the “wild type” intact model. Panels 6A and B reproduce the responses seen above in Figures 2 and 4 for both TNF and A20. Panel 6C demonstrates the rapid activation and subsequent attenuation of Active-Nuclear NF-κB, the necessary activator for both TNF and A20 production.
Panels 6D-F demonstrate the responses of TNF (6D), A20 (6E) and Active-Nuclear NF-κB (6F) in a simulated “IκB-knockout” model. In this model the rate constant for IκB translation was turned to “0” thus preventing the production of any IκB in response to the LPS signal. As can be seen in Figure 6D and E, the rates of production of both TNF and A20 rise with the initiation of the LPS signal and then plateau, denoting an equilibrium point between production and set degradation. The reason for this can be seen in Panel 6F, where Active-Nuclear NF-κB rises, decreases as A20 exerts its negative feedback effect, but then plateaus as well in an equilibrium state. The development of this type of equilibrium state is consistent with the fact that the inhibitory effect of A20 requires its continued production via signal propagation leading to Active-Nuclear NF-κB. Since Active-Nuclear NF-κB also leads to TNF production, TNF production is never shut off in this model.
The importance of the dis-inhibition mechanism of IκB regulation of NF-κB can be seen in “A20 knockout” simulations represented in Figure 7, where Panels 7A, 7B and 7C demonstrate TNF, free IκB and Active-Nuclear NF-κB at LPS = 10000. As opposed to the dynamics of A20 alone, IκB by itself is able to attenuate TNF production, albeit at a lower level that in conjunction with A20, and without the secondary rise in TNF production starting at ∼20000 seconds. NF-κB activity is almost completely (but not completely) suppressed as IκB levels continue to rise (Figure 7B and 7C). The fact that IκB levels do not appear to reach equilibrium likely represents an artifact in the model, but one that, fortuitously, results in realistic behavior, as will be explained in the Discussion. For the moment, however, the model appeared to function “realistically” insomuch that the recovery from the inflammatory signal resulted in the non-perturbed state of completely bound and inhibited NF-κB complex.
Figure 7A-C. Dynamics of TNF, “Free” IκB and Active-Nuclear NF-κB in response to LPS = 10000 in “A20-knockout” model.
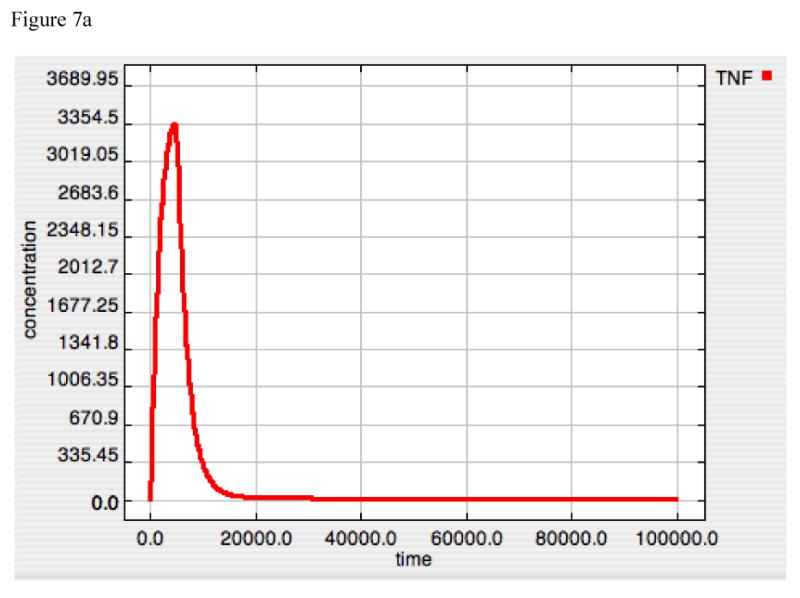
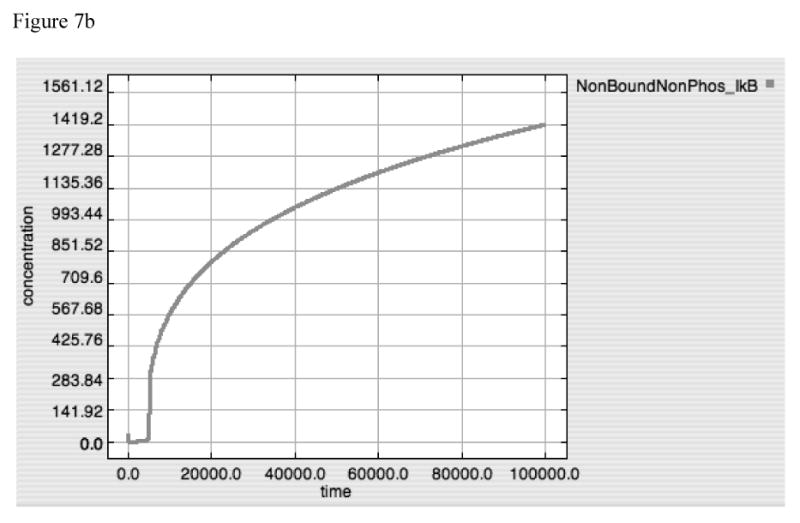
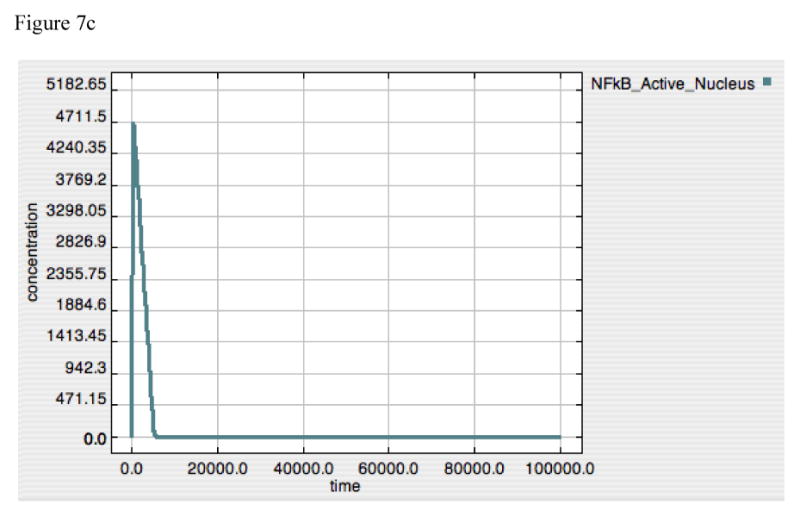
These figures demonstrate the effect of shutting off A20 production (accomplished in a similar fashion as in Figure 6D-F by setting the A20 Translation rate constant to “0”) on TNF (7A), “Free” IκB (= IκB that is free to bind to NF-κB and inactivate it) (7B) and Active-Nuclear NF-κB (7C). As opposed to the A20 dynamics, IκB appears to be able to directly attenuate the TNF response to LPS, albeit with a slightly higher peak value (compared to Panel 6A) and the loss of a second “bump” in TNF production seen at Time ∼20000 sec. The general similarity between Panels 6A and 7A would seem to suggest the predominant role of IκB in determining the overall dynamics of control of NF-κB activation. The dynamics of “Free” IκB inversely mirror the levels of Active-Nuclear NF-κB, as “Free” IκB appears to move towards a limit, while Active-Nuclear NF-κB approaches but never quite reaches 0. This may represent an artifact of the model, as the dynamic equilibrium state of IκB in a non-perturbed state is not included in this model.
The further consequences of the dual control nature of the model can be seen by examining the differential dynamics of the various signaling components at points of control and amplification. Figure 8 presents with the response to an initial LPS = 10000, showing the dynamics of the negative effectors, A20 (Panel 8A), IκB (Panel 8B), the first two amplification points of the signaling cascade, TAK1 (Panel 8C) and Iκκ (Panel 8D) activation, and the latter control target, Active-Nuclear NF-κB (Panel 8E). TAK1 and Iκκ are the points where A20 exercises its negative feedback effects, while IκB suppresses Active-Nuclear NF-κB. The dynamics of TAK1 and Iκκ can be seen to have 3 distinct components:
The first very sharp up-stroke represents initial activation via the inflammatory signal, prior the production of negative feedback compounds.
The second very sharp decrement represents the direct negative feedback as both A20 (see Panel 8A) and IκB (see Panel 8B) rise initially. Note that A20 likely has the predominant effect at this point due to the very low amplitude of the IκB spike.
The third phase of the response occurs at a transition point between effective suppression of TRAF6 and failure of this suppression resulting from the progressive degradation of A20; the A20 inhibition of TRAF6 is not an irreversible reaction and the fall of A20 below its equilibrium point allows “recovery” of the TRAF6-mediated activation of TAK1, and subsequently Iκκ. Since these are amplification points, small amounts of TRAF6 lead to a persistent rise in the levels of both of these compounds. The persistent rise of both TAK1 and Iκκ is an artifact resulting from the fact that baseline degradation of these compounds is not explicitly modeled in the current simulation.
These dynamics suggest an upstream role, both mechanistically and temporally, for A20 in suppression of TLR-4 signaling. This effect, however, is lost as A20 production is concurrently decreased below the point at which it can effectively suppress ongoing TRAF6-mediated TAK1-Iκκ activation. In fact, this is a necessity, as the inhibition by IκB of NF-κB activity needed to attenuate the TNF response (demonstrated in the discussion above regarding IκB knockout and Figure 6) also attenuates the expression of A20. On the other hand, the effective restoration of inhibitory levels of IκB requires the loss of NF-κB suppression by A20. It is these levels of IκB that confer “memory” to the system in the form of tolerance.
As mentioned above, there is a persistent increase in the levels of TAK1 activation, Iκκ activation, and IκB expression in the current model. All these phenomena result from extremely low, persistent levels of a generative compound (TRAF6 in the case of TAK1/Iκκ, and Active-Nuclear NF-κB in the case of IκB) coupled with the lack of modeling the baseline state as a dynamic equilibrium. In the absence of a stimulus-independent mechanism for their removal, these compounds “build up” as long as there are even extremely low levels of their generative compounds present. While this is recognized as an artifact of the current model, we believe that it does not invalidate our conclusions about the relative roles of the two negative feedback control mechanisms or the need for both pathways to effectively process the initial signal and bring about tolerance.
Discussion
The greatest progress in biomedical research in the last half century has been in the investigation and characterization of the molecular machinery of cellular behavior. In some ways, this reductionist, analytical endeavor reached its culmination with the sequencing of the human genome. However, as with all important scientific achievements, the completion of the human genome project ended up generating more questions than it answered, namely, how does it all fit together when the machinery actually runs? Unfortunately, to a great degree, the biomedical research community remains embedded in the same reductionist paradigm. It is becoming increasingly clear that perhaps one aspect of the scientific endeavor, the “taking apart” analytical component, has reached its limit, and it is now time to turn to developing the integrative synthetic aspect of science [31, 32]. The process of synthesis has always been part of science; it is the integration of data and formulation of hypotheses that occurs in the mind of a researcher. However, thus far it has also remained a process that primarily relies upon intuition and insight. Admirable as those capabilities may be, they are increasingly challenged by the complexity of the systems they are asked to act upon. There appears to have been a plateau in the efficiency of being able to infer “causality” between events at the molecular level and the behaviors at the organism level, as noted in the U.S. Food and Drug Administration's “Critical Path” statement [33]. Given the complex, multi-hierarchical structure of biological systems this should not seem surprising. But it is only relatively recently that formal synthetic methods, mostly based on mathematical and computational approaches, have been applied to this translational challenge [2-4, 32, 34].
This translational approach hinges on two realizations: first, that there is no current substitute for human intuition for the formulation of integrative hypotheses (i.e. conceptual models), and second, what is lacking is the ability to test or “check” these conceptual models before attempting to implement them in the real world. The first realization means that there is no “free lunch;” i.e. the application of brute force computational power to massive reams of data is unlikely to result in meaningful mechanistic insight, as this type of analysis only leads to correlation, not causation. On the other hand, the latter realization has been present in the engineering community for half a century, represented by the transition from prototyping to simulation as a means of product development. The application of the engineering paradigm to biomedical research thus mandates the development of tools that can dynamically represent a researcher's knowledge (expressed as a conceptual model) to allow visualization of what is believed to occur is actually allowed to occur in the form of a simulation. This is termed “conceptual model verification,” and is a critical step in the process of understanding the behavior of dynamical systems. In addition to allowing a researcher to “know what they know,” the dynamic instantiation of a conceptual model may also lead to the realization that enough current knowledge exists to explain behaviors that may seemingly call out for more components.
The behavior of the model presented herein gives examples of all these benefits of translational knowledge instantiation. The BNG/RuleBuilder interface readily allows the translation of a conceptual model of TLR-4 signal transduction (represented in diagrams) into a computational model. The BNG TLR-4 model is both highly detailed in terms of its represented components and qualitative in the representation of the interactions between those components. The execution of the BNG TLR-4 model produces recognizable behavior in terms of responsiveness to varying stimulus (LPS), and in terms of preconditioning behavior manifest as tolerance. Furthermore, examination of the behavior of the model also provides insight into how the various control mechanisms of TLR-4 signaling lead to the observed behaviors. The structure of the model (as derived from the literature) pointed to an area of interest with respect to the negative feedback actions of IκB and A20, the differences between their mechanisms and sites of action, and the dynamic consequences of those differences. The first important fact is that both A20 and IκB form classic negative feedback mediators in so much as they depend upon an initial forward propagation of the perturbation signal for their production (facilitated via NF-κB). However, the means by which either influences its negative feedback effect differs. A20 acts directly on two points centered on the key signal amplification point of the TLR-4 signaling pathway: reducing TAK1 activation by de-ubiquitination of TRAF6, and deactivating activated Iκκ. These represent relatively “upstream” points of the TLR-4 pathway (see Figure 1A). IκB, on the other hand, acts farther “downstream”: binding and inactivating NF-κB and decoupling NF-κB from DNA, points distal to the key amplification step at TAK1/Iκκ.
Model behavior suggests that the presence of these two control mechanisms leads to a dual pattern of negative feedback, where A20 is an “early” negative switch that attenuates the initial signal propagation, whereas IκB represents the “memory” component of the system responsible for the tolerance behavior. The question arises that if IκB alone can accomplish the general response of initial signal attenuation, what additional role does the A20 pathway have? The answer lies in the “fine tuning” that these systems are likely to evolve under the selection pressure to maintain robustness. The presence of multiple signal amplification points in the cascade and the fact that negative feedback equilibrium dynamics alone are not capable of completely suppressing levels of upstream activation events require multiple levels of control to effectively modulate the response. The implication with respect to the development of therapies targeting intracellular signaling is that parallel control mechanisms may make the cellular response relatively impervious to the modulation of individual steps in the signaling pathway.
Future versions of this model will include the baseline equilibrium dynamics in the pre-perturbed state, the omission of which creates artifacts in the current model for TAK1, Iκκ and IκB dynamics. Also, there are implications regarding the need to be a “closed” as possible when modeling cellular signaling; too many times the life-cycles of inhibitory compounds do not receive the sufficient attention and are therefore cannot be effectively included in this type of mechanistic model. The exclusion of these inhibitors, however, does not obviate the need to represent signaling pathways with some relatively high degree of detail. For instance, in this model only the explicit representation of the multiple amplification and feedback points in the pathway could give insight into the true dynamics of the system. A simple feedback/saturation model might be able to “reproduce” the general dynamics of the system, but to do so would provide scant insight into the actual behavior of the system.
Furthermore, we believe that this detail is important in that sufficient detail be included at a “uni-scalar level” prior to invoking multi-scale mechanisms to explain a particular behavior. For instance, the current model is “longitudinally” detailed; each step in the closed-loop pathway is represented and is able to generate the richness of behavior observed at a much higher levels. All this is done without invoking parallel pathways or autocrine/paracrine effects, or stochastic cellular population dynamics.
It should be noted that these types of conclusions remain speculative. The dynamics of TAK1/Iκκ response, early attenuation by A20, and the “memory” effect of IκB with respect to preconditioning may not be “correct.” However, they are “emergent” behaviors derived from instantiating what is a relatively canonical representation of TLR-4 signaling. Validation of the model will come when a researcher confirms these behaviors in the lab. However, in turn, this type of computational result may suggest that these are the experiments that need to be done. Thus, it should be emphasized that the place for these computational models are as adjuncts to more traditional laboratory research; there exists an iterative loop of discovery and validation between these two methodologies, between analysis and synthesis [31, 35, 36]. Drawing on the experience of physics, these computational models can be viewed as forming the “theoretical” arm of biomedical research, as a way to speculate and create “thought experiments” that can, in turn, help guide and direct wet lab experimental efforts. Furthermore, the explicit representation of conceptual models in this fashion can be used as a means of communicating and comparing hypotheses and their underlying knowledge. It is hoped that the increased use and distribution of modeling methods such as BNG, will lead to their increased integration into the general research process.
Supplementary Material
Supplementary Materials: The RuleBuilder, BioNetGen language and SBML files of the model presented in this manuscript can all be downloaded from http://bionetgen.org/SCAI-wiki/index.php/Main_Page. In addition, the SBML file and a MatLab file of the model are available as supplements to this manuscript.
Acknowledgments
This work was supported in part by the National Institute of Disability Rehabilitation Research (NIDRR) Grant H133E070024 and the National Institute of General Medical Sciences (NIGMS) Grant GM076570. J.R.F. also acknowledges support from the Department of Computational Biology at the University of Pittsburgh School of Medicine.
References
- 1.Oda K, Kitano H. A comprehensive map of the toll-like receptor signaling network. Molecular systems biology. 2006;2:0015. doi: 10.1038/msb4100057. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.An G, Faeder J, Vodovotz Y. Translational Systems Biology: Introduction of an Engineering Approach to the Pathophysiology of the Burn Patient. J Burn Care Res. 2008;29(2):277–285. doi: 10.1097/BCR.0b013e31816677c8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vodovotz Y, Csete M, Bartels J, Chang S, An G. Translational systems biology of inflammation. PLoS computational biology. 2008;4(4):e1000014. doi: 10.1371/journal.pcbi.1000014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fisher J, Henzinger TA. Executable cell biology. Nature biotechnology. 2007;25(11):1239–1249. doi: 10.1038/nbt1356. [DOI] [PubMed] [Google Scholar]
- 5.Yan L, Hunt CA, Ropella GE, Roberts MS. In silico representation of the liver-connecting function to anatomy, physiology and heterogeneous microenvironments. Conf Proc IEEE Eng Med Biol Soc; 2004. pp. 853–856. [DOI] [PubMed] [Google Scholar]
- 6.Hunt CA, Ropella GE, Yan L, Hung DY, Roberts MS. Physiologically based synthetic models of hepatic disposition. Journal of pharmacokinetics and pharmacodynamics. 2006;33(6):737–772. doi: 10.1007/s10928-006-9031-3. [DOI] [PubMed] [Google Scholar]
- 7.An G. Introduction of an agent-based multi-scale modular architecture for dynamic knowledge representation of acute inflammation. Theoretical biology & medical modelling. 2008;5(1):11. doi: 10.1186/1742-4682-5-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Faeder JR, Blinov ML, Hlavacek WS. Rule-based modeling of biochemical systems with BioNetGen. In: Maly IV, editor. Methods in Molecular Biology: Systems Biology. Totowa, NJ: Humana Press; 2008. [DOI] [PubMed] [Google Scholar]
- 9.Hlavacek WS, Faeder JR, Blinov ML, Posner RG, Hucka M, Fontana W. Rules for modeling signal-transduction systems. Sci STKE. 2006;2006(344):re6. doi: 10.1126/stke.3442006re6. [DOI] [PubMed] [Google Scholar]
- 10.Warren HS. Toll-like receptors. Critical care medicine. 2005;33(12 Suppl):S457–459. doi: 10.1097/01.ccm.0000185504.39347.5d. [DOI] [PubMed] [Google Scholar]
- 11.Lang T, Mansell A. The negative regulation of Toll-like receptor and associated pathways. Immunology and cell biology. 2007;85(6):425–434. doi: 10.1038/sj.icb.7100094. [DOI] [PubMed] [Google Scholar]
- 12.Kawai T, Akira S. TLR signaling. Cell death and differentiation. 2006;13(5):816–825. doi: 10.1038/sj.cdd.4401850. [DOI] [PubMed] [Google Scholar]
- 13.Banerjee A, Gerondakis S. Coordinating TLR-activated signaling pathways in cells of the immune system. Immunology and cell biology. 2007;85(6):420–424. doi: 10.1038/sj.icb.7100098. [DOI] [PubMed] [Google Scholar]
- 14.Li CH, Wang JH, Redmond HP. Bacterial lipoprotein-induced self-tolerance and cross-tolerance to LPS are associated with reduced IRAK-1 expression and MyD88-IRAK complex formation. Journal of leukocyte biology. 2006;79(4):867–875. doi: 10.1189/jlb.0905505. [DOI] [PubMed] [Google Scholar]
- 15.Kitano H, Oda K. Robustness trade-offs and host-microbial symbiosis in the immune system. Molecular systems biology. 2006;2:0022. doi: 10.1038/msb4100039. 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han J, Ulevitch RJ. Limiting inflammatory responses during activation of innate immunity. Nature immunology. 2005;6(12):1198–1205. doi: 10.1038/ni1274. [DOI] [PubMed] [Google Scholar]
- 17.Fan H, Cook JA. Molecular mechanisms of endotoxin tolerance. Journal of endotoxin research. 2004;10(2):71–84. doi: 10.1179/096805104225003997. [DOI] [PubMed] [Google Scholar]
- 18.Kaczorowski DJ, Mollen KP, Edmonds R, Billiar TR. Early events in the recognition of danger signals after tissue injury. Journal of leukocyte biology. 2008;83(3):546–552. doi: 10.1189/jlb.0607374. [DOI] [PubMed] [Google Scholar]
- 19.Matzinger P. The danger model: a renewed sense of self. Science (New York, NY. 2002;296(5566):301–305. doi: 10.1126/science.1071059. [DOI] [PubMed] [Google Scholar]
- 20.Lowe EL, Doherty TM, Karahashi H, Arditi M. Ubiquitination and de-ubiquitination: role in regulation of signaling by Toll-like receptors. Journal of endotoxin research. 2006;12(6):337–345. doi: 10.1179/096805106X118915. [DOI] [PubMed] [Google Scholar]
- 21.Beyaert R, Heyninck K, Van Huffel S. A20 and A20-binding proteins as cellular inhibitors of nuclear factor-kappa B-dependent gene expression and apoptosis. Biochemical pharmacology. 2000;60(8):1143–1151. doi: 10.1016/s0006-2952(00)00404-4. [DOI] [PubMed] [Google Scholar]
- 22.Heyninck K, Beyaert R. A20 inhibits NF-kappaB activation by dual ubiquitin-editing functions. Trends in biochemical sciences. 2005;30(1):1–4. doi: 10.1016/j.tibs.2004.11.001. [DOI] [PubMed] [Google Scholar]
- 23.Lipniacki T, Paszek P, Brasier AR, Luxon BA, Kimmel M. Stochastic regulation in early immune response. Biophysical journal. 2006;90(3):725–742. doi: 10.1529/biophysj.104.056754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Banerjee A, Gugasyan R, McMahon M, Gerondakis S. Diverse Toll-like receptors utilize Tpl2 to activate extracellular signal-regulated kinase (ERK) in hemopoietic cells. Proceedings of the National Academy of Sciences of the United States of America; 2006. pp. 3274–3279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang G, Ghosh S. Negative regulation of toll-like receptor-mediated signaling by Tollip. The Journal of biological chemistry. 2002;277(9):7059–7065. doi: 10.1074/jbc.M109537200. [DOI] [PubMed] [Google Scholar]
- 26.Hu X, Yee E, Harlan JM, Wong F, Karsan A. Lipopolysaccharide induces the antiapoptotic molecules, A1 and A20, in microvascular endothelial cells. Blood. 1998;92(8):2759–2765. [PubMed] [Google Scholar]
- 27.Wheeler DS, Lahni PM, Denenberg AG, Poynter SE, Wong HR, Cook JA, Zingarelli B. Induction of Endotoxin Tolerance Enhances Bacterial Clearance and Survival in Murine Polymicrobial Sepsis. Shock. 2008 doi: 10.1097/shk.0b013e318162c190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mollen KP, Levy RM, Prince JM, Hoffman RA, Scott MJ, Kaczorowski DJ, Vallabhaneni R, Vodovotz Y, Billiar TR. Systemic inflammation and end organ damage following trauma involves functional TLR-4 signaling in both bone marrow-derived cells and parenchymal cells. Journal of leukocyte biology. 2008;83(1):80–88. doi: 10.1189/jlb.0407201. [DOI] [PubMed] [Google Scholar]
- 29.Marshall JC. Sepsis: rethinking the approach to clinical research. Journal of leukocyte biology. 2008;83(3):471–482. doi: 10.1189/jlb.0607380. [DOI] [PubMed] [Google Scholar]
- 30.Hucka M, Finney A, Sauro HM, Bolouri H, Doyle JC, Kitano H, Arkin AP, Bornstein BJ, Bray D, Cornish-Bowden A, et al. The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics (Oxford, England) 2003;19(4):524–531. doi: 10.1093/bioinformatics/btg015. [DOI] [PubMed] [Google Scholar]
- 31.Tjardes T, Neugebauer E. Sepsis research in the next millennium: concentrate on the software rather than the hardware. Shock. 2002;17(1):1–8. doi: 10.1097/00024382-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 32.An G. Agent-based computer simulation and sirs: building a bridge between basic science and clinical trials. Shock. 2001;16(4):266–273. doi: 10.1097/00024382-200116040-00006. [DOI] [PubMed] [Google Scholar]
- 33.Administration USFaD, editor. Innovation or Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products. 2004. [Google Scholar]
- 34.Vodovotz Y, Clermont G, Chow C, An G. Mathematical models of the acute inflammatory response. Current opinion in critical care. 2004;10(5):383–390. doi: 10.1097/01.ccx.0000139360.30327.69. [DOI] [PubMed] [Google Scholar]
- 35.An G. In silico experiments of existing and hypothetical cytokine-directed clinical trials using agent-based modeling. Critical care medicine. 2004;32(10):2050–2060. doi: 10.1097/01.ccm.0000139707.13729.7d. [DOI] [PubMed] [Google Scholar]
- 36.Vodovotz Y, Clermont G, Hunt CA, Lefering R, Bartels J, Seydel R, Hotchkiss J, Ta'asan S, Neugebauer E, An G. Evidence-based modeling of critical illness: an initial consensus from the Society for Complexity in Acute Illness. Journal of critical care. 2007;22(1):77–84. doi: 10.1016/j.jcrc.2006.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Materials: The RuleBuilder, BioNetGen language and SBML files of the model presented in this manuscript can all be downloaded from http://bionetgen.org/SCAI-wiki/index.php/Main_Page. In addition, the SBML file and a MatLab file of the model are available as supplements to this manuscript.